Back to Journals » OncoTargets and Therapy » Volume 13
Identification of a Functional ceRNA Network to Explore Potential Biomarkers for Hepatocellular Carcinoma
Authors Jiang Z, Zhang Y, Liu X, Liang J, Qiu G, Zhu X, Chen J, Li L ![]()
Received 27 August 2020
Accepted for publication 11 November 2020
Published 1 December 2020 Volume 2020:13 Pages 12341—12355
DOI https://doi.org/10.2147/OTT.S278912
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Nicola Silvestris
Zhijun Jiang,1,* Yu Zhang,1,* Xinyu Liu,1 Jingchen Liang,2 Guanhua Qiu,2 Xiaoqi Zhu,2 Jie Chen,1 Lequn Li1
1Department of Hepatobiliary Surgery, Guangxi Medical University Cancer Hospital, Nanning, People’s Republic of China; 2Department of Ultrasound, Guangxi Medical University Cancer Hospital, Nanning, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lequn Li; Jie Chen Department of Hepatobiliary Surgery
Guangxi Medical University Cancer Hospital, Nanning, People’s Republic of China
Tel +86 771 533 0855
Fax +86 771 531 2000
Email [email protected]; [email protected]
Purpose: To establish a novel circRNA–miRNA–mRNA network associated with the poor prognosis of hepatocellular carcinoma (HCC).
Materials and Methods: Quantitative real-time PCR was used to verify the differentially expressed circRNA. Moreover, the competing endogenous RNA networks were established using bioinformatics methods. Meanwhile, the prognostic value and potential mechanism of ceRNA network in hepatocellular carcinoma (HCC) were analyzed.
Results: This work found that circ_0130911 was highly expressed in HCC tissues and early recurring HCC. Further, we effectively constructed a ceRNA network. The ceRNA network regulated by circ_0130911 might influence the prognosis of HCC by regulating cell cycle-related pathways.
Conclusion: The ceRNA network proposed here can be used as a novel biomarker for the prognosis of HCC, thereby providing new insights for the targeted therapy of HCC.
Keywords: circRNAs, ceRNAs, biomarker, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is one of the most prevalent malignant tumors with its incidence and mortality rates ranking 6th and 4th respectively across the globe.1,2 Due to the current limitation of medical technology, HCC patients in the Asia-Pacific region are primarily treated via radical resection.3 Nonetheless, after radical resection of HCC, nearly 70% of patients still develop recurrence of HCC. As a consequence, the overall treatment level of HCC and the improvement of the survival rate of HCC patients remain severely affected.2 Therefore, it is necessary to identify the risk factors and potential mechanisms related to the early recurrence of HCC after radical resection geared towards providing novel treatment approaches for controlling the early recurrence and metastasis of HCC.
Circular RNAs (circRNAs) are a type of single-stranded endogenous non-coding RNA (ncRNA) with a covalently closed-loop structure. The ncRNA on the other hand is produced by reverse splicing and exhibits a circular structure that cannot be easily degraded by RNA exonuclease. Therefore, it has a more stable structure compared to linear RNA.4,5 Notably, circRNAs are widely distributed in cells and have tissue-specific expressive characteristics. At the same time, circRNAs might participate in miRNAs sponge, epithelial-mesenchymal transition, and tumorigenesis.6–9 Thus, the dysregulation of circRNAs expression might influence the development of cancer. For instance, circSHKBP1 (hsa_circ_0000936) regulates the HUR-VEGF pathway to inhibit HSP90 degradation by sponging miR-582-3p, thereby promoting the progression of gastric cancer.10 Also, recent studies have shown that circ0003998 regulates the expression of FOSL2 and PCBP1-CD44v6 through sponge miR-143-3p to participate in the process of epithelial-mesenchymal transition of HCC.11 Moreover, circPTK2 (hsa_circ_0005273) is highly expressed in colorectal cancer and positively correlated with tumor growth and metastasis. Based on various studies, circPTK2 could promote EMT of colorectal cancer cells by physically binding to the phosphorylation sites Ser38, Ser55, and Ser82 of vimentin thereby improving the metastasis of colorectal cancer. Therefore, circPTK2 has been predicted to become a potential target for the treatment of colorectal cancer metastasis.12
In this study, 3 early recurrence and 3 early recurrence-free cases were selected for circRNA high-throughput sequencing in the preliminary work of our group. As a result, hsa_circ_0130911 was highly expressed in patients with early recurrence of HCC. Through qRT-PCR and bioinformatics, we found that the expression of circ_0130911 was upregulated in HCC tissues, and positively correlated with tumor growth and metastasis. Besides, the expression of circ_0130911 was positively correlated with lower mediocre survival rates and poorer tumor-free survival in HCC patients. Additionally, a complete ceRNA network was constructed to predict the potential mechanism of circ_0130911. This work thus provides a strong foundation for the study of circRNAs related to the early recurrence of HCC. However, further experiments should be conducted to explore the specific regulatory mechanisms.
Materials and Methods
circRNA High-Throughput Sequencing
We selected 3 early recurrence cases and 3 early recurrence-free HCC tissues for circRNA high-throughput sequencing to detect the expression of circRNA.
Specimen of HCC Tissues
A total of 106 pairs of primary HCC tissues and 106 matched normal liver tissue samples used in this study were provided by the Affiliated Tumor Hospital of Guangxi Medical University. The diagnosis and cut-off values of clinical indicators for primary hepatocellular carcinoma were based on the Chinese guidelines for the diagnosis and treatment of primary HCC.13 First, 106 tissues from patients were removed during liver resection, frozen in liquid nitrogen, and stored at −80°C before RNA isolation. After the operation, all patients were followed up via outpatient review, inpatient review, and telephone follow-up. The follow-up date ended in May 2018. Table 1 records the clinical characteristics of 106 patients. The Ethics Committee of the Affiliated Tumor Hospital of Guangxi Medical University approved the study, and all 106 patients signed the relevant informed consent.
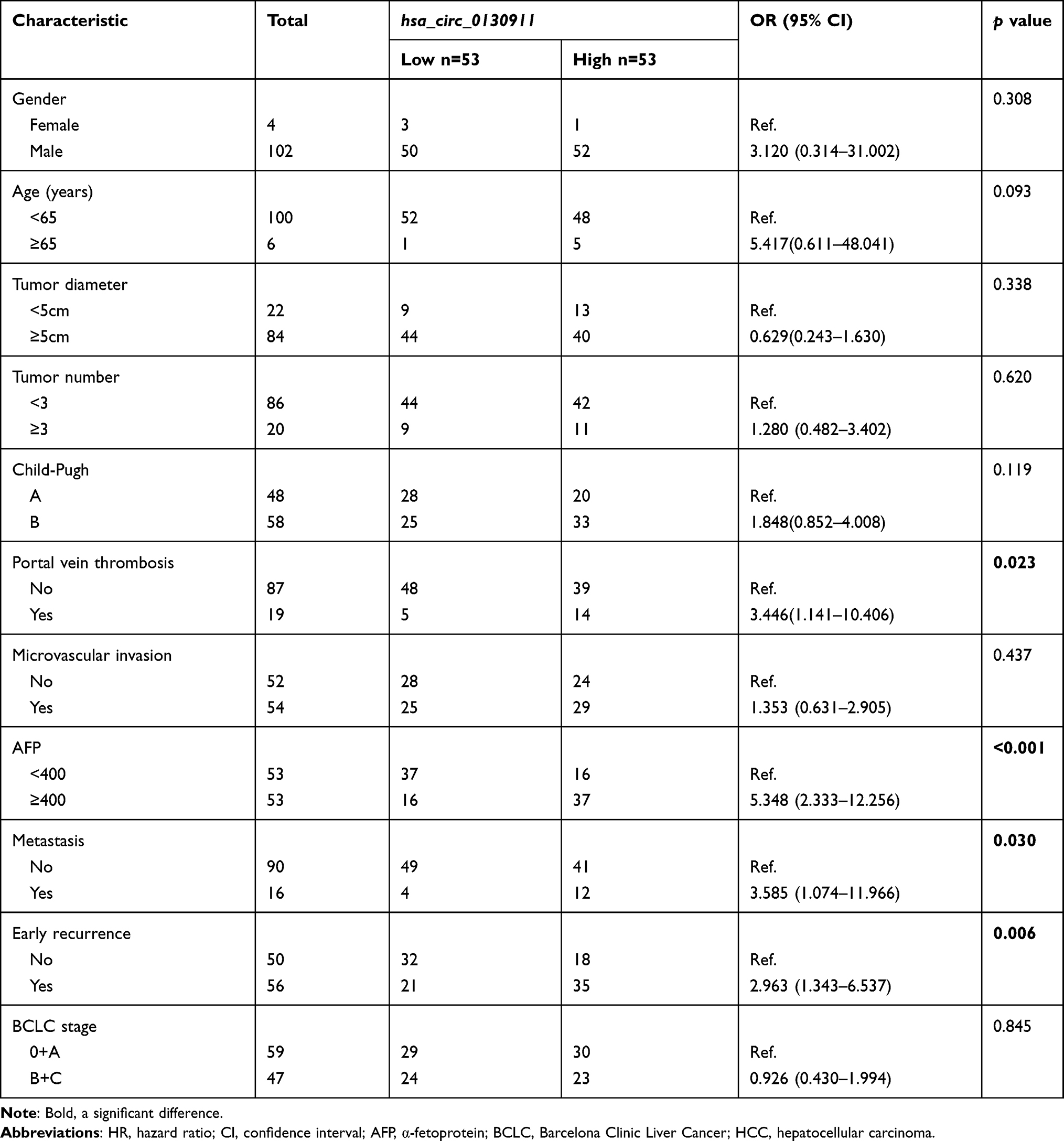 |
Table 1 A Comparison Between circ_0130911 Expression and Clinicopathological Characteristics in HCC Patients |
Quantitative Real-Time PCR
The total RNA of 106 pairs of HCC tissues was extracted using TRIzol reagent (Invitrogen, USA). PrimeScript RT reagent Kit with gDNA Eraser kit (Takara, Japan) was used to reverse transcribed™ into cDNA, while FastStart Universal SYBR Green Master (ROX) (Roche, Germany) was used for real-time quantitative polymerase chain reaction (qRT-PCR) analysis. The expression level of circRNA was calculated by the 2−ΔΔCT method (standardized with β-Actin). The primers for circ_0130911 were 5ʹ-CCTTGGAGGCTGAGTGG −3ʹ and 5ʹ-CTTCATCACCTGCTTACTTTC −3ʹ. The primers for β-Actin were 5ʹ- TGCGTGACATTAAGGAGAAG −3ʹ and 5ʹ- GTCAGGCAGCTCGTAGCTCT −3ʹ.
Prediction of circRNA–miRNA–mRNA Interaction Network
Notably, Cancer-Specific CircRNAs Database (CSCD)14 understands the structural model of circ_0130911. The circbank15 was used to predict the sponge miRNA of circ_0130911. Unlike the normal liver tissue samples, the differentially down-regulated miRNAs (DEmiRNAs) in HCC were identified by OncomiR and miRCancer. Besides, the intersection of miRNA predicted in circbank and miRNA in OncomiR16 and miRCancer17 was considered as the target molecule of circ_0130911 in HCC. Of note, mRNA was predicted by miRTarBase database.18 The GEPIA database19 was used to determine the differentially up-regulated mRNAs in HCC and normal liver tissues. The intersection of mRNA in miRTarBase and GEPIA database was considered as the target gene of circ_0130911 in HCC. Finally, Cytoscape software 3.7.2 was used to visualize the CircRNA-miRNA-mRNA network.
GO and KEGG Pathway Enrichment Analysis
The clusterProfiler software package in R was used to perform GO annotation and KEGG pathway analysis on the selected mRNA and the most frequently changed neighboring genes.
Construction and Analysis of Protein-Protein Interaction (PPI) Network and Hub Gene
The PPI networks were established using String database20 and Cytoscape software 3.7.2 visualization. The Hub genes were identified according to the top ten node degree (the number of genes related to the target gene). Oncomine database21 and HPA database22 verified the expression of the selected 10 Hub genes at the transcription and translation levels. The influence of the 10 Hub genes on the prognosis of HCC were verified by the GEPIA database. Using the survivalROC package in R to draw ROC curve and calculate AUC to evaluate the risk value of Hub genes for the prognosis of HCC. Cox P<0.05 was considered to statistically significant.
Gene Set Enrichment Analysis (GSEA)
A total of 10 Hub genes were grouped based on their high and low expressions in the TCGA-LIHC gene set. With the GSEA 4.0.3 software via the Java platform, we derived the “https://software.broadinstitute.org/gsea/downloads.jsp” to evaluate the potential biological functions that might be involved. The enriched signaling pathways with FDR < 0.25 or nominal p < 0.05 were defined as statistically significant.
Statistical Analysis
SPSS 22.0 statistical software was used for all statistical analyses, while GraphPad Prism 8 was used to draw graphs. The chi-square test was used to evaluate the relationship between circ_0130911 and clinicopathological characteristics. Moreover, the Cox regression model was applied for single factor and multivariate analysis. The Kaplan-Meier method was used to draw OS and DFS curves, and the Log rank test was used to evaluate their statistical significance. With a P value of less than 0.05 (P<0.05), the difference was considered statistically significant. Notably, bioinformatics analysis was mostly performed through the above-mentioned bioinformatics tools and R language. Genes or miRNAs with |log2(fold change)| >1 and P<0.05 were considered statistically significant.
Results
circRNA High-Throughput Sequencing Analysis
In the sequencing results, based on the analysis of the relationship between the expression of the HCC tissues with 3 early recurrence and 3 early recurrence-free, and statistical correlation analysis. We found circ_0130911 showing a trend of highest expression in HCC tissues with early recurrence (Supplement Table 1).
Structural Characteristics of circ_0130911
The results of the circbase database revealed that circ_0130911 originated from exons 43 to 50 of the UTRN gene, located at chr6: 144,858,717–144,898,424, and finally formed a mature sequence of 1246 nt in length (Figure 1). The CSCD database showed that circ_0130911 comprised the structure of microRNA response elements (MREs), suggesting that circ_0130911 potentially regulates key circRNA of HCC development through sponge miRNA.
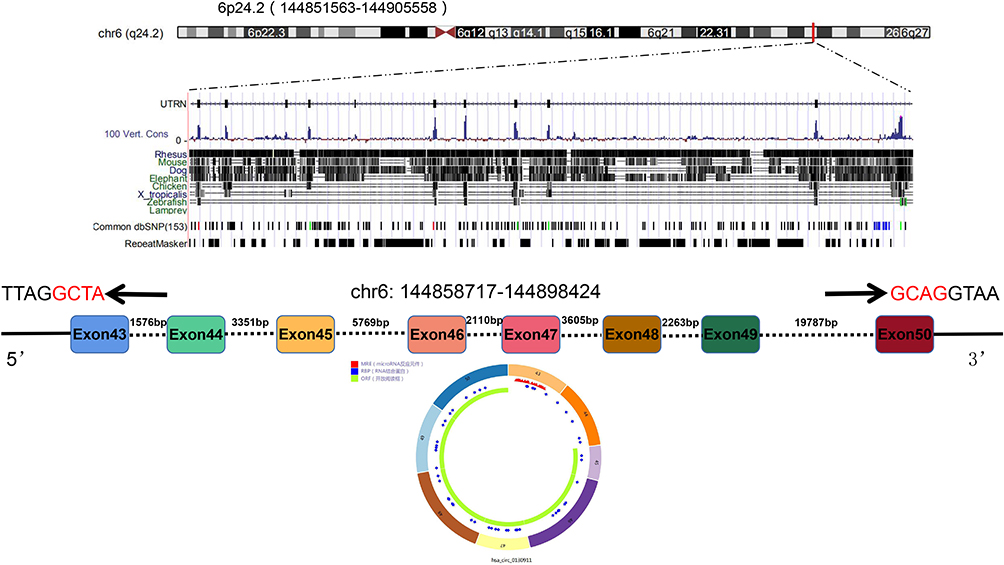 |
Figure 1 The schematic diagram showing the genomic locus of circ_0130911 in the UTRN gene. Structure patterns of circ_0130911 based on the CSCD database. |
Up-Regulation of circ_0130911 in HCC
qRT-PCR technology was used to detect circ_0130911 in 106 pairs of HCC tissues and corresponding adjacent tissues. Statistical analysis using Mann–Whitney U rank-sum test showed that the relative expression of circ_0130911 in HCC tissues was significantly higher compared to that in adjacent tissues (p<0.05, Figure 2A); circ_0130911 was relatively expressed in cancer tissues of patients with early recurrence of HCC. The expression level of circ_0130911 was significantly higher compared to that of cancer tissues in patients with early HCC without recurrence (p<0.05, Figure 2B). This suggests that circ_0130911 might be closely related to the early recurrence of HCC after surgery.
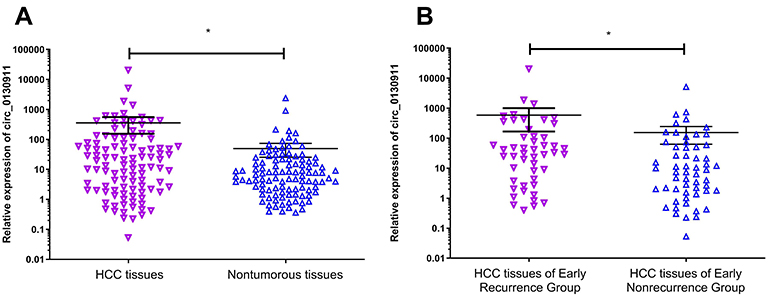 |
Figure 2 (A) The expression level of circ_0130911 in HCC tissues is higher than that in corresponding adjacent nontumorous tissues. (B) The expression of circ_0130911 in the HCC tissues of the early recurrence group is higher compared to that in HCC tissues of the early nonrecurrence group (*p<0.05). |
The Relationship Between circ_0130911 Level in HCC and Clinical Characteristics of Patients
Based on the median relative expression of circ_0130911 in HCC tissues, HCC patients were subdivided into circ_0130911 high expression group (53 cases) and low expression group (53 cases). Chi-square test was used to analyze the relationship between circ_0130911 high expression group and low expression group and clinicopathological characteristics of HCC patients. Consequently, the high expression level of circ_0130911 was significantly related to portal vein tumor thrombus, alpha-fetoprotein, metastasis, and early tumor recurrence (Table 1).
The Relationship Between circ_0130911 Level and Poor Prognosis of HCC Patients
Single-factor and multi-factor COX regression analysis was applied for all the above indicators. The results showed that the high expression of circ_0130911 was an independent risk factor for death and recurrence of HCC patients. In terms of overall survival rate, the HR of the circ_0130911 high expression group was 2.295 (95% CI 1.314–4.011, p=0.008, Table 2) whereas, in terms of the tumor-free survival rate, the HR of the circ_0130911 high expression group was 2.260 (95% CI 1.313) −3.889, p=0.005, Table 3).
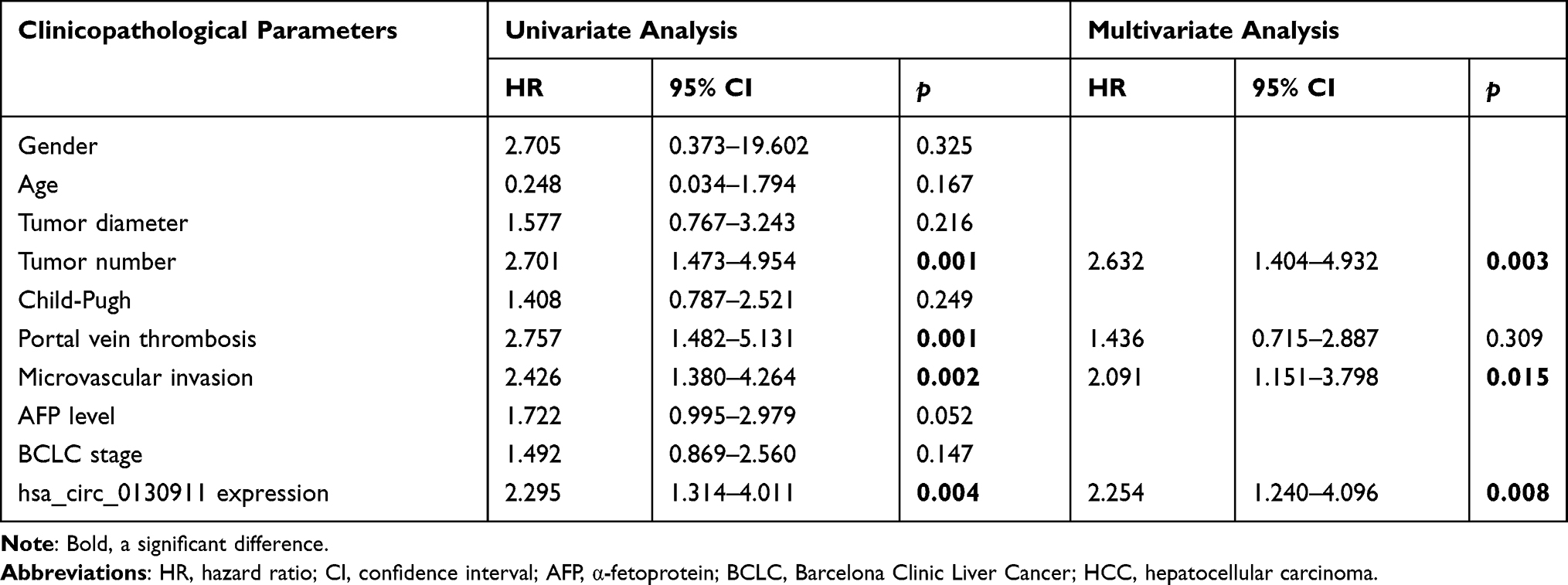 |
Table 2 Univariate and Multivariate Cox Regression Analyses of Overall Survival |
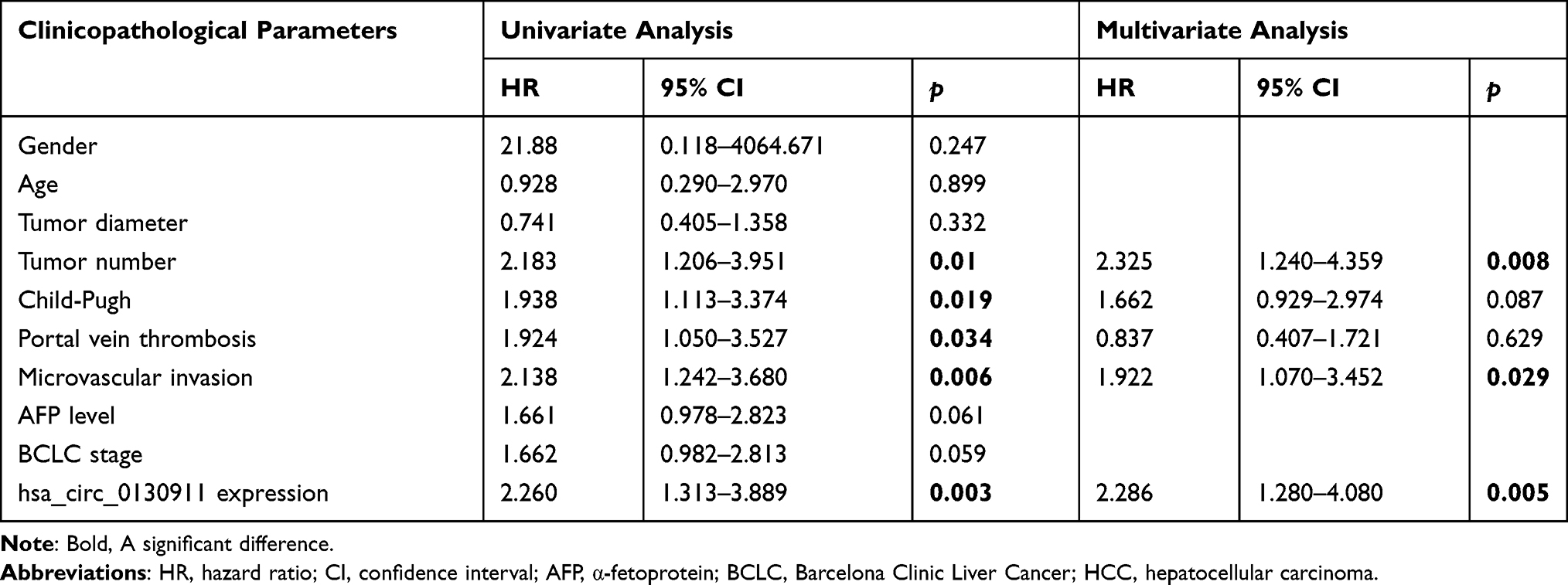 |
Table 3 Univariate and Multivariate Cox Regression Analyses of Disease-Free Survival |
Kaplan-Meier statistics showed that the overall survival rate of patients in the circ_0130911 high expression group (p=0.002, Figure 3A) and the tumor-free survival rate of early (within one year) recurrence (p=0.002, Figure 3B) were both significantly lower compared to the low expression group. This indicates that the higher the content of circ_0130911 in HCC tissue, the worse the prognosis of HCC patients, and the higher the possibility of early recurrence.
 |
Figure 3 (A) The high level of circ_0130911 expression in HCC tissue indicates that the overall survival rate of the patients is worse. (B) The high level of circ_0130911 expression in HCC tissue indicates that the disease-free survival rate of the patient is worse. |
Prediction and Construction of ceRNA Network
Based on the ceRNA theory, circRNA regulates the up-regulation of related target genes by competitively binding miRNA response elements with miRNA. Therefore, this study established an HCC-related ceRNA network through the following steps. First, circRNA-miRNA interaction pairs were predicted in the circbank database, and DEmiRNAs that were differentially down-regulated in HCC were screened based on OncomiR and miRCancer databases. After crossing the 3 data sets, 7 down-regulated DEmiRNAs were selected including, hsa-miR-590-3p, hsa-miR-15a-5p, hsa-miR-23c, hsa-miR-29a-3p, hsa-miR-424 −5p, hsa-miR-497-5p, and hsa-miR-592 (Figure 4A). Then, 1442 target genes of 7 DEmiRNAs were predicted through the miRTarBase database. Based on the GEPIA database, this study selected 1481 genes that were differentially and highly expressed in HCC. After the intersection, 113 highly expressed DEmRNAs were screened out (Figure 4B). Finally, based on the DEcircRNA-DEmiRNA pair and DEmiRNA-DEmRNA pair, a circRNA-miRNA-mRNA network was constructed. Cytoscape v3.7.2 was used to visualize the ceRNA network (Figure 4C).
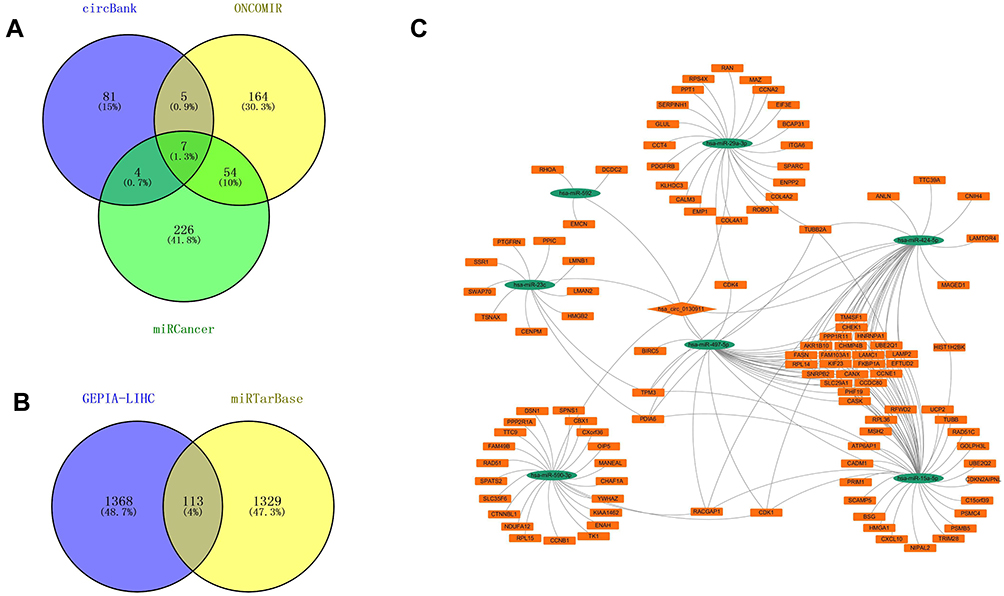 |
Figure 4 (A) Venn diagram of downregulated DEmiRNAs of circ-0130911. (B) Venn diagram of upregulated target genes of circ-0130911. (C) A circRNA-miRNA-mRNA regulatory network. The network comprises one circRNAs, 7 miRNAs, and 113 genes. CircRNAs, miRNAs, and mRNAs are respectively represented by diamond ovals and rectangles. |
Functional Analysis of mRNAs
To better understand the potential biological functions of circ_0130911 in the development of HCC, GO (Figure 5A) and KEGG (Figure 5B) pathway enrichment analyses were performed on 113 target genes in the established ceRNA network. GO biological process (BP) analysis showed that up-regulated target genes were implicated in nuclear division, organelle fission, and mitotic nuclear division. For cellular component (CC) analysis, target genes were significantly enriched in the chromosomal region, focal adhesion, and cell-substrate junction. Additionally, the molecular function (MF) analysis for these DEmRNAs included cadherin binding, cell adhesion molecule binding, and single-stranded DNA binding.
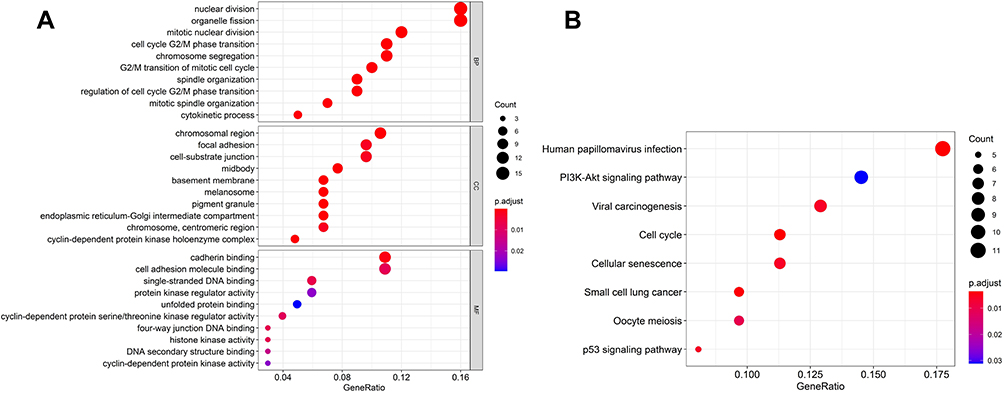 |
Figure 5 Functional enrichment analysis of the ceRNA network. (A) Results of GO analysis, including BP, CC, and MF; (B) Results of KEGG biological process analysis. The horizontal axis represents the gene ratio. The vertical axis represents lots of the GO terms or KEGG pathways. The size and color of a point represent the number and P-value of the pathway, respectively. |
Moreover, the enrichment results of KEGG showed that DEmRNAs exhibit a clear relationship with various cancer-related pathways, such as the PI3K-Aktsignaling pathway, Cell cycle, Cellular senescence, and p53 signaling pathway which were closely related to the ceRNA network. The significant GO enrichment terms and KEGG pathways were shown in bubble maps using the ggplot2. R package with P<0.05. The results revealed that the constructed ceRNA network was related to multiple pathways regulating the growth of HCC.
Construction of PPI Network and Hub Gene
The STRING database was used to analyze the relationship between the 113 DEmRNAs in the ceRNA network and constructed a PPI network with 78 and 318 edges on the node. Cytoscape 3.7.2 was used to visualize the PPI network (Figure 6A). The top 10 Hub genes were identified using the cytoHubba plugin, including CDK1, CCNA2, CCNB1, PPP2R1A, CHEK1, KIF23, BIRC5, OIP5, RAD51, RACGAP1 (Figure 6B). Interestingly, an interactive network was also formed between these Hub genes. Then, Cytoscape was used to built the circRNA-miRNAs-Hubs gene networks (Figure 6C).
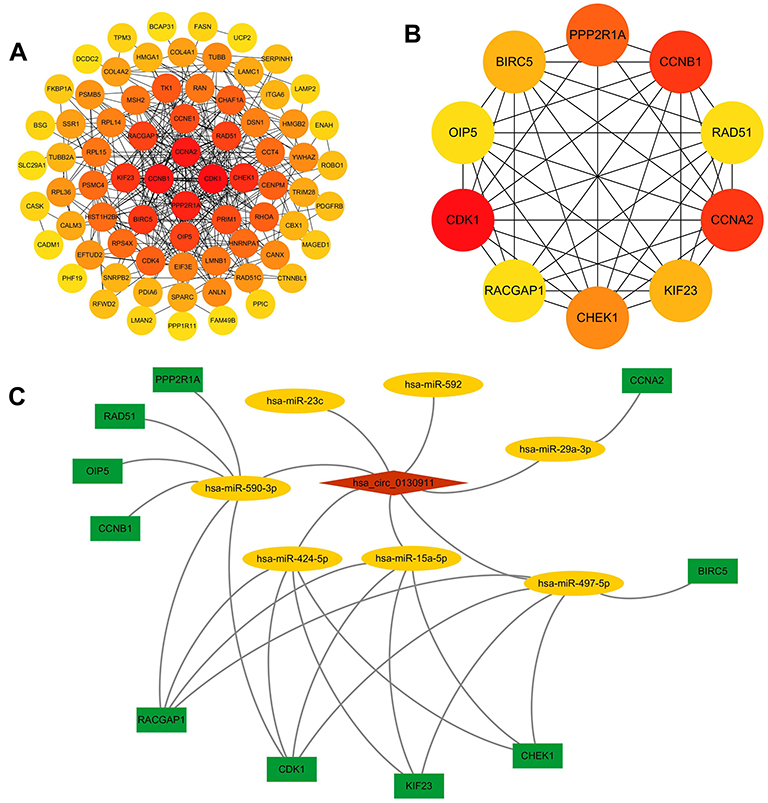 |
Figure 6 (A) PPI network of 113 target genes of circ-0130911 in HCC. (B) 10 Hub-genes (CDK1, CCNA2, CCNB1, PPP2R1A, CHEK1, KIF23, BIRC5, OIP5, RAD51, RACGAP1) in the PPI network. (C) The circRNA-miRNA-Hub-genes axes. |
Identification of Hub Genes
To further understand the biological functions of these 10 Hub genes in HCC, the Oncomine database was used to detect the expression of the Hub genes in HCC. Results suggested that the level of Hub genes in HCC tissues was significantly higher compared to that in normal tissues. Moreover, the Human Protein Atlas database showed the difference between the actual expression of CDK1, CCNA2, CCNB1, PPP2R1A, CHEK1, KIF23, BIRC5, OIP5, RAD51, and RACGAP1 in normal liver tissue and HCC through immunohistochemistry (Figure 7A–J). The patient data of the IHC are listed in Table 4. Subsequently, the GEPIA database was used to evaluate the impact of these Hub genes on the prognosis of HCC. As a result, elevated expressions of CDK1, CCNA2, CCNB1, PPP2R1A, CHEK1, KIF23, BIRC5, OIP5, RAD51, and RACGAP1 suggested a shorter survival time (Figure 8A–J) and tumor-free survival (Figure 9A–J).
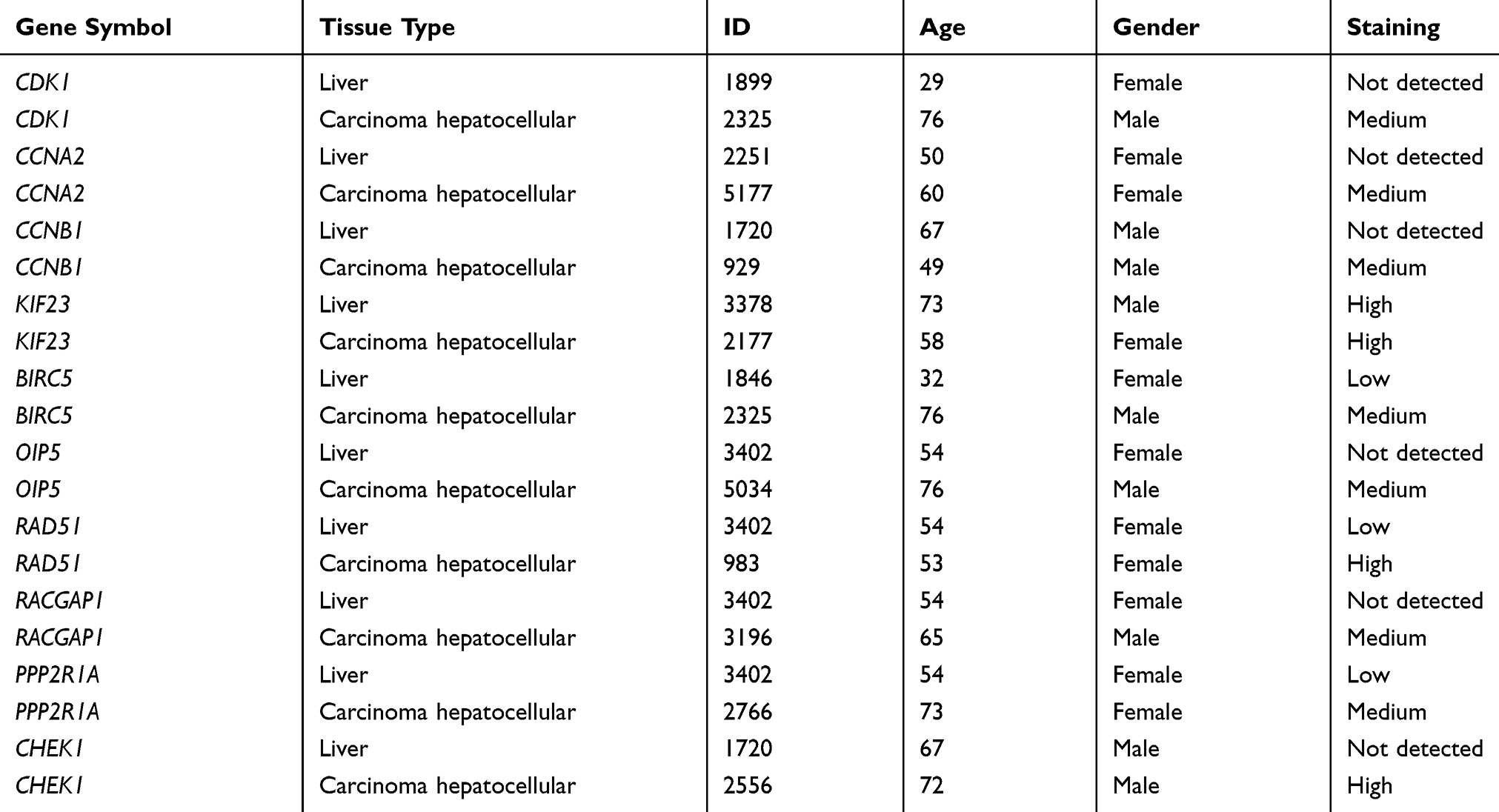 |
Table 4 Clinical Information of IHC in the Human Protein Atlas Database |
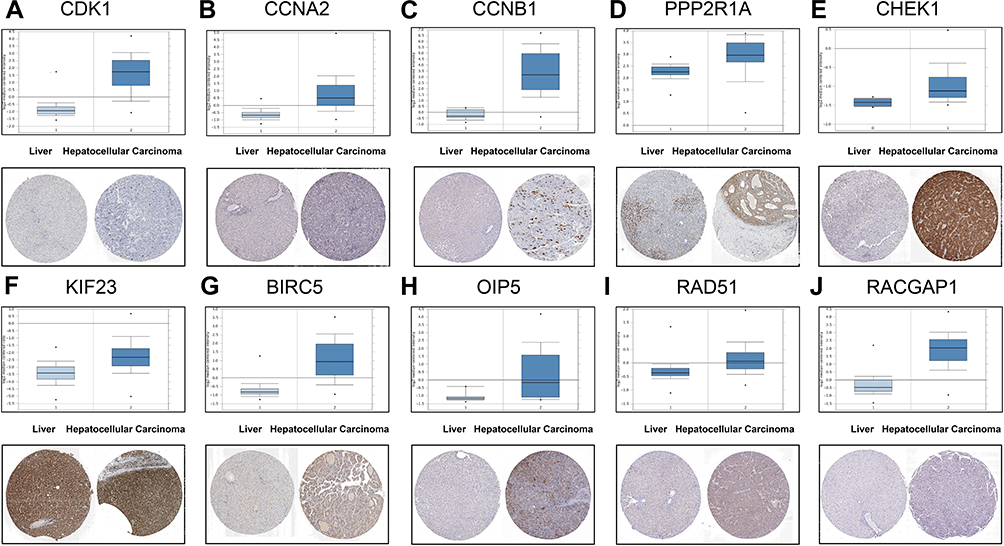 |
Figure 7 Validating the expression of 10 Hub genes on transcriptional and translational level by the Oncomine database and HPA database (immunohistochemistry). (A) CDK1. (B) CCNA2. (C) CCNB1. (D) PPP2R1A. (E) CHEK1. (F) KIF23. (G) BIRC5. (H) OIP5. (I) RAD51. (J) RACGAP1. |
 |
Figure 8 Association of expressions among 10 upregulated Hub genes and the overall survival of patients with HCC. (A) CDK1. (B) CCNA2. (C) CCNB1. (D) PPP2R1A. (E) CHEK1. (F) KIF23. (G) BIRC5. (H) OIP5. (I) RAD51. (J) RACGAP1. |
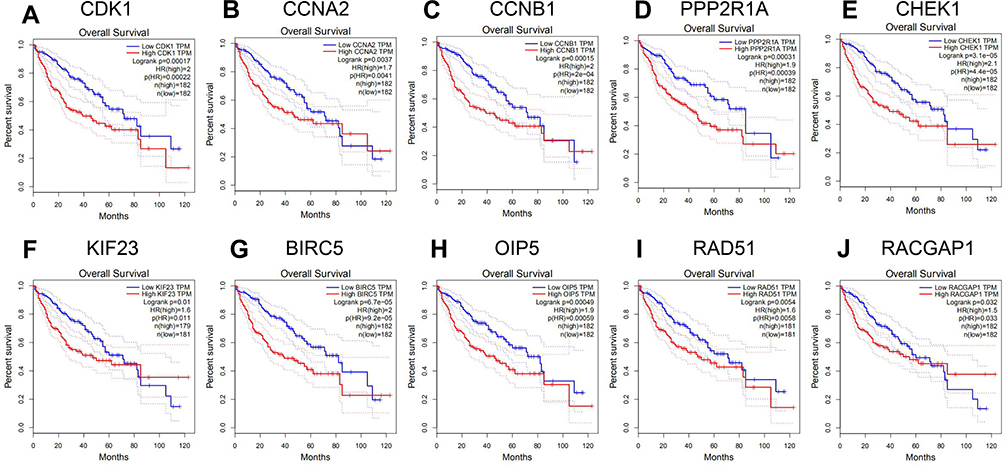 |
Figure 9 Association of expressions among 10 upregulated Hub genes and the disease-free survival of patients with HCC. (A) CDK1. (B) CCNA2. (C) CCNB1. (D) PPP2R1A. (E) CHEK1. (F) KIF23. (G) BIRC5. (H) OIP5. (I) RAD51. (J) RACGAP1. |
Using the TCGA-LIHC database to incorporate these Hub genes into a single-factor, multi-factor COX risk regression model, CCNB1, OIP5, and RACGAP1 showed significant prognostic value for HCC patients. The risk values of these 3 Hub genes were combined to draw the ROC curve, and the AUC value was calculated as 0.771 (Figure 10A). This indicates that CCNB1, OIP5, and RACGAP1 in the established ceRNA network might be potential biomarkers for predicting the prognosis of HCC.
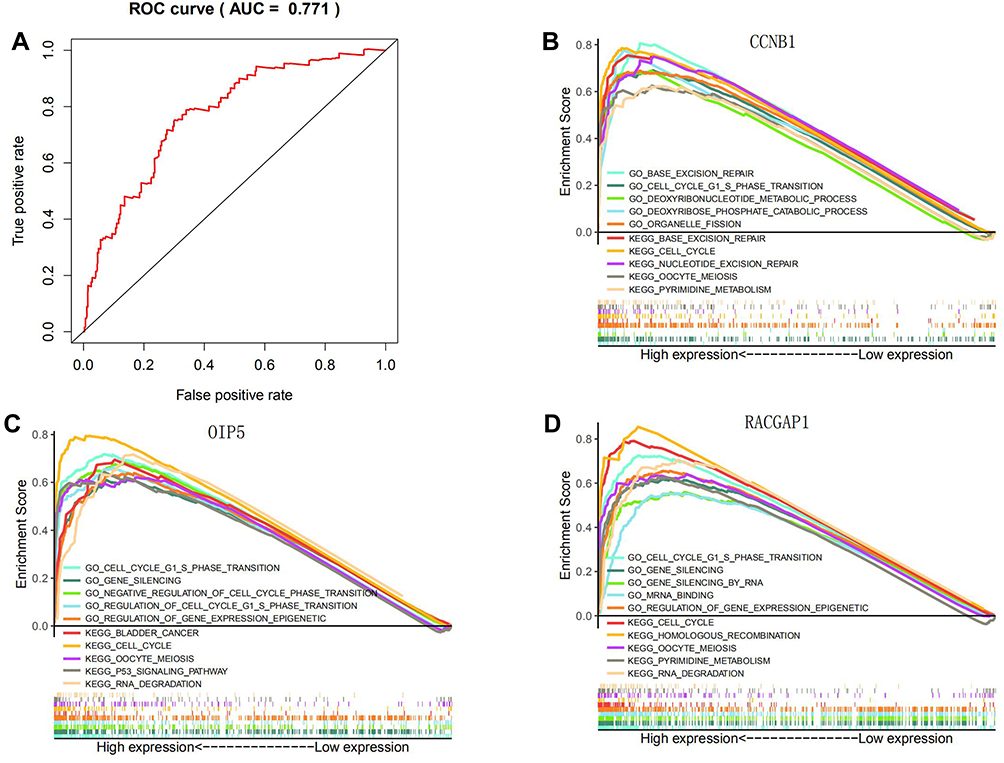 |
Figure 10 (A) ROC analysis of 3 Hub genes in TCGA-LIHC database. The Enrichment plots from GSEA analysis of Hub genes in the TCGA-LIHC dataset. (B) CCNB1. (C) OIP5. (D) RACGAP1. |
GSEA
The TCGA database was used to further determine the possible pathways and mechanisms of these 3 Hub genes in regulating the progress of HCC. Based on their median, the Hub genes were divided into high expression and low expression groups (Figure 10B–D). GSEA was conducted to search for the GO and KEGG pathways enriched in the highly-expressed samples. The results revealed that the high expression of these 3 Hub genes were enriched in the pathways such as “kegg_cell_cycle”, “go_cell_cycle_g1_s_phase_transition” (FDR q<0.05). Therefore, the ceRNA network might promote the development of tumors by regulating the cell cycle and other pathways.
Discussion
Based on the latest data released by the National Cancer Center, hepatocellular carcinoma (HCC) is the 4th leading cancer in China with its 2014 incidence being about 26.67 per 100,000. It causes the cancer-related deaths of approximately 319, 000 people annually, with its death rate only second to lung cancer.23 The existing guidelines recommend that hepatectomy is a vital method to ensure the long-term survival of HCC patients, and significantly improves the prognostic quality of life among patients.24 Nonetheless, the high recurrence rate and short survival time of HCC patients negatively impact the improvement of the overall treatment level of HCC. Also, this is attributed to its biological characteristics including strong invasiveness, high malignancy, and poor prognosis.
Accumulating evidence has shown that circRNA plays an important role in the progression of HCC. As hypothesized by the ceRNA network, circRNA regulates the expression of mRNA through the sponge miRNA of MREs structure.9 As such, circRNA exerts its biological functions through an extensive gene expression regulatory network, and can as well be used as a diagnostic and prognostic biomarker. For instance, Exosomal circPACRGL acts as a sponge for miR-142-3p/miR-506-3p and promotes progression of colorectal cancer via the miR-142-3p/miR-506-3p- TGF-β1 axis. Shang et al established a ceRNA network that might play a role in the early recurrence of HCC, thereby providing new ideas for monitoring the early recurrence of HCC.25
Furthermore, studies conducted by Xu et al have shown that the upregulated expression of circ-CCAC1 in cholangiocarcinoma increases cell progression by sponging miR-514a-5p and up-regulating YY1. Besides, it promotes the occurrence and metastasis of cholangiocarcinoma, and might be an important organism markers for the treatment of cholangiocarcinoma.26
Based on qRT-PCR results, circ_0130911 was relatively and highly expressed in HCC tissues and was closely related to the poor prognosis of HCC. Importantly, the biological function of circ_0130911 in tumors is largely understudied. Therefore, our research is of great significance for further understanding the mechanism of circRNA in HCC.
By predicting the miRNAs bound to circRNA, 7 down-regulated DEmiRNAs were identified, including: hsa-miR-590-3p, hsa-miR-15a-5p, hsa-miR-23c, hsa-miR-29a-3p, hsa- miR-424-5p, hsa-miR-497-5p, and hsa-miR-592. In addition, several lines of evidence have shown that these 7 DEmiRNAs are related to the development and metastasis of cancer and that the expression of miR-590-3p is downregulated in triple-negative breast cancer. Besides, miR-590-3p directly inhibits the invasion and migration ability of triple-negative breast cancer by sponge slugs.27 A research by He et al found that circZNF609 activates the Hedgehog pathway through the sponge miR-15a-5p/15b-5p and promotes the expression of GLI2 protein, thereby enhancing the proliferation and metastatic capability of HCC cells.28 In bladder cancer, lncRNA CASC9 acted as a miR-497-5p sponge to up-regulate the expression of FZD6, activate theWnt/β-cateninsignaling pathway, as well as promote the growth and metastasis of bladder cancer.29 Additional studies have revealed that circFGFR3 participates in the miR-29a-3p/E2F1 axis by inducing the EMT process of ovarian cancer cells and promote the initiation of ovarian cancer.30
In total, we found 113 DEmRNAs in the established ceRNA network. KEGG pathway enrichment analysis suggests that the up-regulated genes were primarily implicated in the following pathways: PI3K-Akt signaling and cell cycle and p53 signaling pathways. Reports indicate that changes in cell cycle rhythm are closely related to the occurrence and development of HCC.31 The imbalance of the p53 signaling pathway promotes the proliferation of HCC cells, thereby contributing to the occurrence of HCC.32
To further determine the key genes in the ceRNA network, we considered the top 10 genes in the PPI network as Hub genes including, CDK1, CCNA2, CCNB1, PPP2R1A, CHEK1, KIF23, BIRC5, OIP5, RAD51, and RACGAP1. Then, the circRNA-miRNA-Hub mRNA network was constructed. The Oncomine and HPA databases were used to verify the true expression of Hub genes in HCC at the transcription and translation levels. The results indicated that the mRNA levels and immunohistochemical staining of these genes in HCC tissues were significantly higher compared to those in normal tissues, suggesting that these Hub genes regulate the development of HCC. To identify the potential prognostic markers in HCC, the GEPIA database was used to analyze the OS and DFS of Hub genes in the prognosis of HCC. We found that the high expression of Hub genes was significantly related to the poor prognosis of HCC patients. Moreover, as demonstrated by the results of the COX regression model, the 3 genes (CCNB1, OIP5, and RACGAP1) were the most significant genes influencing the prognosis of HCC. The ROC curve of these 3 Hub genes was used to analyze the relationship with the prognosis of HCC, and the AUC value was calculated as 0.771. Therefore, these 3 Hub genes were predicted as the potential biomarkers for the prognosis of HCC.
We used the TCGA database to perform GESA enrichment analysis and discovered that when these 10 Hub genes were overexpressed in HCC, they were primarily enriched in Cell cycle, Regulation of gene expression epigenetic, Gene silencing, Cell cycle g1 s phase transition, etc. Reports have confirmed that the dysregulation of cell cycle pathways regulates tumor development and prognosis. For example, TRIM59 regulates the cell cycle by degrading protein phosphatase 1B, thereby promoting the development of HCC.33 Based on various sources of evidence, the high expression of p38 MAPKγ acts as a CDK-like kinase, which can act synergistically with CDK in regulating cell cycle and promotes the occurrence of liver tumors.34 Also, it has been widely reported that the Hub gene discovered in this work can affect tumor development through the cell cycle pathway. Notably, CCNB1 was found to be highly expressed in HCC. LINC00346 regulates the expression of CCNB1 through sponge miR-199a-3p, which influences the p53 signaling pathway and ultimately regulates the cell cycle process of HCC cells.35 He et al found that OIP5 was up-regulated in patients with glioblastoma, and its expression was positively correlated with the poor prognosis of patients. Further studies highlighted that OIP5 promotes cell cycle progression, and knocking down OIP5 will trigger G2/M cell cycle arrest.36 RACGAP1 is highly expressed in HCC, gastric cancer, and other cancers.37,38 For instance, knocking down RACGAP1 in basal-like breast cancer inhibits the growth of cancer cells through the failure of cytokinesis.39
Here, we propose a novel circRNA–miRNA–mRNA network related to the poor prognosis of HCC. This network might promote the progress of HCC through cell cycle and other pathways, hence it is expected to become a novel biomarker for predicting the poor prognosis of HCC. Nevertheless, our findings primarily relied on qRT-PCR and bioinformatics predictions. Thus, additional experiments are necessary to validate our hypothesis.
Conclusion
In conclusion, this work used qRT-PCR to validate that the expression level of circ_0130911 in HCC tissues is significantly different from that in adjacent tissues. As a consequence, we effectively established a complete circRNA-miRNA- Hub genes network by the analysis of bioinformatics research strategies of multiple databases. Moreover, it was found that the CCNB1, OIP5, and RACGAP1 in the network might influence the poor prognosis of HCC patients by regulating the cell cycle and other pathways. Eventually, we uncovered that circ_0130911 acts as a ceRNA in regulating the cell cycle geared towards promoting the progress of HCC and providing novel biomarkers for the prognosis of HCC. Nevertheless, the specific mechanism by which this ceRNA network regulates the development of HCC merits further research.
Abbreviations
BP, biological process; CC, cellular component; CI, confidential interval; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; MF, molecular function; HCC, hepatocellular carcinoma; PPI, protein–protein interaction network; qRT-PCR, quantitative real time PCR; GSEA, Gene Set Enrichment Analysis.
Ethics and Consent Statement
This research was conducted ethically in accordance with the World Medical Association Declaration of Helsinki. Subjects have given their written informed consent and the study protocol regarding human specimen were approved by the Ethics Committee of the Affiliated Tumor Hospital of Guangxi Medical University.
Funding
This study was supported by grants from the National Natural Science Foundation of China (grant number 81960534 and 81860512) and the Innovation Driven Development Project of Guangxi (AA18221001).
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Fitzmaurice C, Allen C, Barber RM, et al.; Global Burden of Disease Cancer C. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 Cancer Groups, 1990 to 2015: a systematic analysis for the Global Burden of Disease Study. JAMA Oncol. 2017;3(4):524–548. doi:10.1001/jamaoncol.2016.5688
2. Yang JD, Hainaut P, Gores GJ, et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589–604.
3. Omata M, Cheng AL, Kokudo N, et al. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: a 2017 update. Hepatol Int. 2017;11(4):317–370.
4. Kristensen LS, Andersen MS, Stagsted LVW, et al. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20(11):675–691. doi:10.1038/s41576-019-0158-7
5. Meng S, Zhou H, Feng Z, et al. CircRNA: functions and properties of a novel potential biomarker for cancer. Mol Cancer. 2017;16(1):94. doi:10.1186/s12943-017-0663-2
6. Vo JN, Cieslik M, Zhang Y, et al. The landscape of circular RNA in cancer. Cell. 2019;176(4):869–81 e13. doi:10.1016/j.cell.2018.12.021
7. Conn SJ, Pillman KA, Toubia J, et al. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160(6):1125–1134. doi:10.1016/j.cell.2015.02.014
8. Zhao Z-J, Shen J. Circular RNA participates in the carcinogenesis and the malignant behavior of cancer. RNA Biol. 2017;14(5):514–521. doi:10.1080/15476286.2015.1122162
9. Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–388. doi:10.1038/nature11993
10. Xie M, Yu T, Jing X, et al. Exosomal circSHKBP1 promotes gastric cancer progression via regulating the miR-582-3p/HUR/VEGF axis and suppressing HSP90 degradation. Mol Cancer. 2020;19(1):112. doi:10.1186/s12943-020-01208-3
11. Song LN, Qiao GL, Yu J, et al. Hsa_circ_0003998 promotes epithelial to mesenchymal transition of hepatocellular carcinoma by sponging miR-143-3p and PCBP1. J Exp Clin Cancer Res. 2020;39(1):114. doi:10.1186/s13046-020-01576-0
12. Yang HB, Li XB, Meng QT, et al. CircPTK2 (hsa_circ_0005273) as a novel therapeutic target for metastatic colorectal cancer. Mol Cancer. 2020;19(1). doi:10.1186/s12943-020-1139-3
13. Zhou J, Sun HC, Wang Z, et al. Guidelines for diagnosis and treatment of primary HCC in China (2017 edition). HCC. 2018;7(3):235–260.
14. Xia SY, Feng J, Chen K, et al. CSCD: a database for cancer-specific circular RNAs. Nucleic Acids Res. 2018;46(D1):D925–D929. doi:10.1093/nar/gkx863
15. Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. Rna. 2014;20(11):1666–1670. doi:10.1261/rna.043687.113
16. Wong NW, Chen YH, Chen S, et al. OncomiR: an online resource for exploring pan-cancer microRNA dysregulation. Bioinformatics. 2018;34(4):713–715. doi:10.1093/bioinformatics/btx627
17. Xie B, Ding Q, Han H, et al. miRCancer: a microRNA-cancer association database constructed by text mining on literature. Bioinformatics. 2013;29(5):638–644. doi:10.1093/bioinformatics/btt014
18. Chou CH, Shrestha S, Yang CD, et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018;46(D1):D296–D302. doi:10.1093/nar/gkx1067
19. Tang Z, Li C, Kang B, et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. doi:10.1093/nar/gkx247
20. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi:10.1093/nar/gky1131
21. Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9(2):166–180. doi:10.1593/neo.07112
22. Thul PJ, Akesson L, Wiking M, et al. A subcellular map of the human proteome. Science. 2017;356(6340):6340. doi:10.1126/science.aal3321
23. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132.
24. Department of Medical Administration N H, Health Commission of the People’s Republic of C. Guidelines for diagnosis and treatment of primary HCC in China (2019 edition). Zhonghua Gan Zang Bing Za Zhi. 2020;28(2):112–128.
25. Shang A, Gu C, Wang W, et al. Exosomal circPACRGL promotes progression of colorectal cancer via the miR-142-3p/miR-506-3p- TGF-beta1 axis. Mol Cancer. 2020;19(1):117. doi:10.1186/s12943-020-01235-0
26. Xu Y, Leng K, Yao Y, et al. A novel circular RNA, circ-CCAC1, contributes to CCA progression, induces angiogenesis, and disrupts vascular endothelial barriers. Hepatology. 2020. doi:10.1002/hep.31493
27. Yan M, Ye L, Feng X, et al. MicroRNA-590-3p inhibits invasion and metastasis in triple-negative breast cancer by targeting Slug. Am J Cancer Res. 2020;10(3):965–974.
28. He YK, Huang H, Jin L, et al. CircZNF609 enhances hepatocellular carcinoma cell proliferation, metastasis, and stemness by activating the Hedgehog pathway through the regulation of miR-15a-5p/15b-5p and GLI2 expressions. Cell Death Dis. 2020;11(5):1–12.
29. Zhan Y, Zhang L, Yu S, et al. Long non-coding RNA CASC9 promotes tumor growth and metastasis via modulating FZD6/Wnt/β-catenin signaling pathway in bladder cancer. J Exp Clin Cancer Res. 2020;39(1):136. doi:10.1186/s13046-020-01624-9
30. Zhou J, Dong ZN, Qiu BQ, et al. CircRNA FGFR3 induces epithelial-mesenchymal transition of ovarian cancer by regulating miR-29a-3p/E2F1 axis. Aging (Albany NY). 2020;12(14):14080–14091. doi:10.18632/aging.103388
31. Liu S, Yang TB, Nan YL, et al. Genetic variants of cell cycle pathway genes predict disease-free survival of hepatocellular carcinoma. Cancer Med. 2017;6(7):1512–1522. doi:10.1002/cam4.1067
32. Liu WL, Li XX, Chu ESH, et al. Paired box gene 5 is a novel tumor suppressor involved in the pathogenesis of hepatocellular carcinoma through interaction with p53 signaling pathway. Gastroenterology. 2011;140(5):S145. doi:10.1016/S0016-5085(11)60589-1
33. Ying H, Ji L, Xu Z, et al. TRIM59 promotes tumor growth in hepatocellular carcinoma and regulates the cell cycle by degradation of protein phosphatase 1B. Cancer Lett. 2020;473:13–24.
34. Tomas-Loba A, Manieri E, Gonzalez-Teran B, et al. p38gamma is essential for cell cycle progression and liver tumorigenesis. Nature. 2019;568(7753):557–560. doi:10.1038/s41586-019-1112-8
35. Jin JL, Xu HQ, Li WY, et al. LINC00346 acts as a competing endogenous RNA regulating development of hepatocellular carcinoma via modulating CDK1/CCNB1 axis. Front Bioeng Biotech. 2020;8. doi:10.3389/fbioe.2020.00054
36. He J, Zhao YZ, Zhao EH, et al. Cancer-testis specific gene OIP5: a downstream gene of E2F1 that promotes tumorigenesis and metastasis in glioblastoma by stabilizing E2F1 signaling. Neuro-Oncology. 2018;20(9):1173–1184. doi:10.1093/neuonc/noy037
37. Wang SM, Ooi LLPJ, Hui KM. Upregulation of rac GTPase-activating protein 1 is significantly associated with the early recurrence of human hepatocellular carcinoma. Clin Cancer Res. 2011;17(18):6040–6051. doi:10.1158/1078-0432.CCR-11-0557
38. Saigusa S, Tanaka K, Mohri Y, et al. Clinical significance of RacGAP1 expression at the invasive front of gastric cancer. Gastric Cancer. 2015;18(1):84–92. doi:10.1007/s10120-014-0355-1
39. Lawson CD, Fan C, Mitin N, et al. Rho GTPase transcriptome analysis reveals oncogenic roles for rho GTPase-activating proteins in basal-like breast cancers. Cancer Res. 2016;76(13):3826–3837. doi:10.1158/0008-5472.CAN-15-2923
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.