")
Back to Journals » Drug Design, Development and Therapy » Volume 17
Identification of a Class of WNK Isoform-Specific Inhibitors Through High-Throughput Screening
Authors Chlebowicz J , Akella R , Humphreys JM , He H , Kannangara AR, Wei S, Posner B, Goldsmith EJ
Received 10 September 2022
Accepted for publication 17 November 2022
Published 20 January 2023 Volume 2023:17 Pages 93—105
DOI https://doi.org/10.2147/DDDT.S389461
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Julita Chlebowicz,1 Radha Akella,1 John M Humphreys,1 Haixia He,1 Ashari R Kannangara,1 Shuguang Wei,2 Bruce Posner,2 Elizabeth J Goldsmith1
1Department of Biophysics, The University of Texas Southwestern Medical Center, Dallas, TX, USA; 2Department of Biochemistry, The University of Texas Southwestern Medical Center, Dallas, TX, USA
Correspondence: Elizabeth J Goldsmith, Department of Biophysics, The University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX, 75390-8816, USA, Tel +1 214 645 6376, Email [email protected]
Introduction: WNK [with no lysine (K)] kinases are serine/threonine kinases associated with familial hyperkalemic hypertension (FHHt). WNKs are therapeutic targets for blood pressure regulation, stroke and several cancers including triple negative breast cancer and glioblastoma. Here, we searched for and characterized novel WNK kinase inhibitors.
Methods: We used a ~210,000-compound library in a high-throughput screen, re-acquisition and assay, commercial specificity screens and crystallography to identify WNK-isoform-selective inhibitors.
Results: We identified five classes of compounds that inhibit the kinase activity of WNK1: quinoline compounds, halo-sulfones, cyclopropane-containing thiazoles, piperazine-containing compounds, and nitrophenol-derived compounds. The compounds are strongly pan-WNK selective, inhibiting all four WNK isoforms. A class of quinoline compounds was identified that further shows selectivity among the WNK isoforms, being more potent toward WNK3 than WNK1. The crystal structure of the quinoline-derived SW120619 bound to the kinase domain of WNK3 reveals active site binding, and comparison to the WNK1 structure reveals the potential origin of isoform specificity.
Discussion: The newly discovered classes of compounds may be starting points for generating pharmacological tools and potential drugs treating hypertension and cancer.
Keywords: WNK, inhibition screen, kinase inhibitor, WNK3-specific, structure–activity relationship, crystallography
Graphical Abstract:

Introduction
WNK [With No lysine (K)] kinases are cytoplasmic serine/threonine protein kinases, named for their unique constellation of catalytic residues.1 WNK kinases are involved in transepithelial ion transport, cell volume control and cell motility,2–5 making them drug targets for several disease indications. WNK1 and WNK4 are associated with familial hyperkalemic hypertension (FHHt) or Gordon Syndrome.6–8 Knockout or knockdown of WNK1 and WNK3 induces low blood pressure9,10 making WNKs established drug targets for hypertension. WNKs are also implicated in breast, lung, ovarian, and brain cancers.11–14 A transposon insertional analysis has identified WNK1 as a proto-oncogenic signature gene for triple negative breast cancer.15 WNK3 knockout mice have reduced edema in a stroke model.16,17 Further, WNK3 is over-expressed in the hippocampus in certain forms of epilepsy.18 Thus, inhibitors of these kinases may be used to determine the biological roles of WNK kinases and can serve as starting points for drug discovery. Indeed, Yamada et al reported a potent pan-WNK inhibitor (WNK463)19,20 as well as a pan-WNK allosteric inhibitor (WNK476).21 WNK463, the pan-WNK inhibitor, is ATP competitive,19,20 whereas WNK476 and other analogs are allosteric, binding in a site adjacent to helix C. Here we present inhibitors of WNK1 and WNK3 in new scaffolds that exhibit discrimination in inhibition constants between the two WNK isoforms. Crystallography of one class of WNK3-specific inhibitors in complex with WNK3 reveals the structural basis for WNK3 specificity.
Materials and Methods
Reagents
The Kinase-Glo® luminescence reagent was purchased from Promega Inc. A plasmid encoding a substrate peptide derived from OSR1 (GST-TEV-GAM314RAKKVRRVPGSSGRLHKTEDGGWEWSDDEF344) was a gift from Melanie Cobb (University of Texas Southwestern Medical Center) (Unpublished). The plasmid was verified by sequencing at UT Southwestern sequencing facility and also the expressed protein was verified by tandem mass spectrometry. WNK1 inhibitors identified by screening were re-acquired from ChemBridge Corp. (San Diego, CA), ChemDiv Inc. (San Diego, CA), ComGenex (Hungary), Prestwick Chemical (San Diego, CA), Collaborative Drug Discovery, Inc. (Newark, DE), and UT Southwestern chemical synthesis laboratories. WNK1 (1–491) and WNK3 (1–431) used in mobility shift assays were obtained from Carna Biosciences USA Inc. Fluorescein-labeled OSR1 peptide (Fluorescein-5-carbonyl-RVPGSSGRLHK-NH2) was obtained from Bachem America Inc, CA.
WNK1, WNK3 and GST-OSR1 Expression and Purification
Cloning, expression, and purification of the pWNK1 (WNK1-KDm/194-483) protocols are described by Min et al22 pWNK1 was expressed in E. coli. Here benzonase nuclease and Protease Inhibitor Cocktail (PIC) (Sigma) were added to cultured cells prior to lysis in a EmulsiFlex-C5 cell disrupter (Avestin). Eight hundred milligrams of pWNK1 was synthesized and purified in 25 preparations. The state of phosphorylation of the pWNK1 on Ser382 was confirmed by mass spectrometry.
An optimized clone for bacterial expression of pWNK3 (118–409) was obtained from GenScript (Piscataway, NJ). pWNK3 was expressed and purified by the same protocol used for pWNK1.23 pWNK3 is phosphorylated on Ser308. The GST-OSR1 (314–344) peptide plasmid was expressed in Rosetta (DE3) pLysS competent E. coli cells. The GST-OSR1 peptide was purified using glutathione beads (GE Healthcare). Three grams of substrate was generated for the screen and used without further purification.
High-Throughput Screening
The high-throughput screening of 210,000 compounds was conducted in 384-well plates using Kinase-Glo ® (Promega Inc.). Seven hundred and forty-one plates were screened, 36 plates per day, in the UT Southwestern High-Throughput Screening Core (HTS). The substrate solution was comprised of 55.3 mM HEPES (pH 7.4), 55.3 mM MgCl2, 4.7 µM pWNK1, and 19.3 µM GST-OSR1. Fifteen microliters of it was added to each well by multiplexed dispensing (Multidrop384, Thermo Scientific). Then, 0.2 µL of pure DMSO (negative control, columns 2 and 23) or 0.5 mM compound (columns 3 to 22) or 0.4 mM control quinazoline inhibitor (Figure S1A) (column 24) were added using a Biomek Liquid handler. Next, 10 µL of 53 µM ATP stock solution was added to each well with a different multiplex dispenser (Biotech Inc.) to a final ATP concentration of 21 μM. The plates were centrifuged for 1 minute at 1000 rpm then incubated for 2 hours at room temperature with shaking. Then, 15 µL of Kinase-Glo (Promega Inc.) diluted 2:1 with solution of 55.33 mM HEPES, 55.33 mM MgCl2, pH 7.4 was added to each well using a Multidrop dispenser. Plates were centrifuged again for 1 minute at 1000 rpm and incubated for 10 minutes under agitation at room temperature. Luminescence was read on an EnVision Multilabel plate reader (PerkinElmer, Inc.).
Chemical Library
The UT southwestern chemical library consists of ~210,000 small-molecule compounds obtained from the following vendors: ChemBridge Corporation (75,000), Chemical Diversity Labs (100,000), ComGenex (22,000), TimTek (1200), Prestwick (1100), and the NIH Clinical Collection (450). Another 2500 compounds were synthesized by UTSW chemistry labs. Compounds in the library approximately satisfy Lipinski’s rules with 99% having a molecular weight less than 550 Da (average 250–300 Da). The library also contains approximately 6500 partially purified natural product fractions isolated from unique marine bacteria by Professor John MacMillan. Each natural product fraction contains 3–10 natural products in DMSO and is suitable for high-throughput screening.
Analysis of High-Throughput Screening Data
All data were analyzed using Genedata Screener® (version 10.1, GeneData, Inc. Basel, Switzerland) as described for a previous screen conducted in our laboratory,24 Supplemental Data.
Confirmation and Refined Screens
The top 1275 hits from the 210,000-compound screen possessing Z-scores >3σ were rescreened for both pWNK1 and pWNK3 in the HTS core facility by the methods described above. Three different inhibitor concentrations, 6 μM, 4 μM, and 0.5 μM were used in the rescreen. The HTS core was also used to further refine IC50’s on 96 of the best compounds using 9 concentrations ranging from 50 nM to 49.5 μM.
Mobility-Shift Assays
WNK activity was measured by phosphorylation-induced mobility shift of fluorescently tagged substrate peptides (FAM-OSXR1).25 For consistency with Yagi et al and studies of WNK463 inhibition of WNKs,19 we used the same Carna Biosciences WNK enzymes (GST tagged WNK1/1-491 (GST WNK1) and GST tagged WNK3/1-434 (GST WNK3)). The assays were conducted in 40 μL reaction volumes and 384-well plates.26 GST-WNK1 or GST-WNK3 was prepared at twice the final (12.5 nM) concentration in 20 μL assay buffer (20 mM HEPES-Na (pH 7.5), 1 mM MnCl2, 0.01% Tween 20, and 2 mM dithiothreitol). 2X enzyme–inhibitor mixtures were pre-incubated for 1 hour with inhibitor varying final concentrations of 0.04, 0.08, 0.16, 0.32, 0.64, 1.25. 2.5, 5 and 10 μM. A solution of 1 μM fluorescently labeled FAM-OXSR1 peptide25 (Bachem Americas, Inc) and 100 μM ATP was mixed 1/1 with WNK1 or WNK3 inhibitor mixtures to final concentrations of 0.5 μM FAM-OXR1 peptide and 50 μM ATP to start the reaction. Reactions were incubated for 3 hours and were stopped by adding 40 μL of quench buffer (100 mM HEPES-Na (pH 7.5), 0.015% Brij-35, 1mM EDTA, 5% DMSO, 20 mM EDTA). Aliquots from each well were sipped onto a 12-sipper chip on PerkinElmer LabChip® EZ reader for the electrophoretic separation. Fluorescence intensity was measured under a pressure of ~2.0 PSI and a voltage of 1650ΔV. Data were analyzed according to a previously published protocol in Liu et al26
Specificity Screen
SW133708, SW120619 and SW182086 were tested for specificity against 50 kinases by Eurofins Inc. (France). Kinases in the Eurofins screen span all five protein kinase classes.27 The activity measurements were conducted in duplicate using myelin basic protein (MBP) as substrate or the native substrate when available. The compound concentration used was 10 μM.
Structure Determination
Crystals were obtained for WNK3-KDm/S308A (118–409, S308A) (WNK3/SA) preincubated with SW120619. The complex crystallized in 50 mM NaCl, 50 mM HEPES (pH 8.0), 1mM EDTA and 1 mM (2-carboxyethyl) phosphine hydrochloride (TCEP). A 1:1 ratio of protein and precipitant solution was mixed (2 μL each). Precipitant solution was 0.1M Na Malonate pH 5.0 and 12% PEG3350. Crystals were grown at 16 °C and cryoprotected in 20% glycerol. Data up to 3.1 Å resolution for the WNK3/SW120619 complex were collected at the APS 19-ID beamline. Integration and scaling were performed with the HKL3000 software suite (HKL Research, Charlottesville VA). The structure was solved using molecular replacement with 5DRB19 as a starting model. The structure was refined using REFMAC in the CCP4 suite. Model-building was performed in Coot using the ligand docking function.28 Data collection and refinement parameters are shown in Table S1. The coordinates and structure factors have been deposited (PDB file 8EDH).
Differential Scanning Fluorimetry
Five micromolar pWNK1 or pWNK3 kinase domains was incubated with 50 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM inhibitor and 5x SYPRO Orange (Invitrogen) to make a 25 μL final reaction volume per well on a 96-well clear bottom plate. The temperature was increased from an initial 4 °C to 80 °C using 0.5 °C increments in a Bio-Rad CFX96 real-time PCR machine. The fluorescence intensity of SYPRO Orange was probed using the fluorescein amidite channel of the RT-PCR machine.29
Results and Discussion
Bootstrap Screens
The phosphorylated kinase domain (pWNK1-KDm (pWNK1)) expressed in bacteria was used in inhibitor screens. A bootstrap procedure was used to identify WNK1 inhibitors in the UTSW compound library. The only WNK1 inhibitor published when the screen was conducted, hypericin (US Patent application US 2008/0286809 A1, Dario Alessi) (Figure S1B), had an IC50 of 200 μM. Therefore, a sub-collection of 3500 representative compounds was screened to identify a better positive control. Myelin basic protein was used as the substrate for the initial 3500 compound screen. This small screen revealed SW133708 as a good hit (Figure S1C). The commercial Eurofins screen revealed significant pan-WNK specificity for SW133708, inhibiting WNKs 1, 2, and 4 (Figure 1A). SW133708 was then used in a slightly larger 8K screen, revealing SW137446 as a major hit (Figure S1A). SW137446 was then used in the 210K screen as the positive control. The 8K and 230K screens for inhibitors were conducted using a GST fusion construct with a peptide encompassing the pan-WNK phosphorylation site of OSR1.30,31 The full screen required 3 grams of the OSR1 fusion peptide, and 800 mg of pWNK1-KDm. Both the 8 K screen and the 210K screen had a Z′-factor of 0.91, reflecting significant differences between the positive and neutral controls on each plate.24 Only seven plates were poor by this measure (Figure 1B). The top 1275 compounds exhibiting a Z-score ≥3σ in the 230K screen were re-screened in triplicate (at 6 μM, 4 μM and 0.5 μM, Figure 1C and D). The Z′-factor for the 36 plates in the confirmation screen was 0.94. The results from the confirmation screen and the HTS screen are well-correlated, as is clear from the diagonal pattern in Figure 1C. Figure 1D shows that the % inhibition is dose dependent, and again shows the correlation between the 210K screen and the confirmation screen. Ninety-six compounds were rescreened at nine concentrations against pWNK1. Selected data are presented in Figures 2–6 as IC50*.
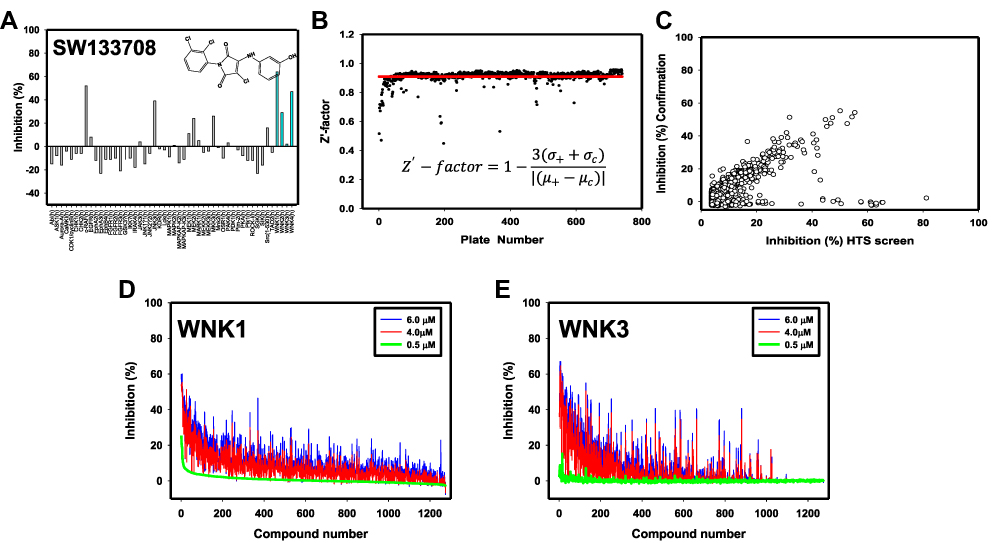 |
Figure 1 High-throughput and specificity screen results. (A) Commercial screen (Eurofins, Inc.) used to test cross-reactivity of SW133708, initial hit in the 3500 compound screen, against 50 diverse kinases. (B) Z′ score for each plate of the 210,000 compound (blue) screen is shown. The average Z′ score across the screen is shown as a red line. (C) % inhibition in the confirmation screen vs % inhibition in the original HTS screen is plotted to show consistency between screens. The % inhibition of each compound is plotted at 6 μM (blue), 4 μM (red) and 0.5 μM (green) toward (D) WNK1 and (E) WNK3. |
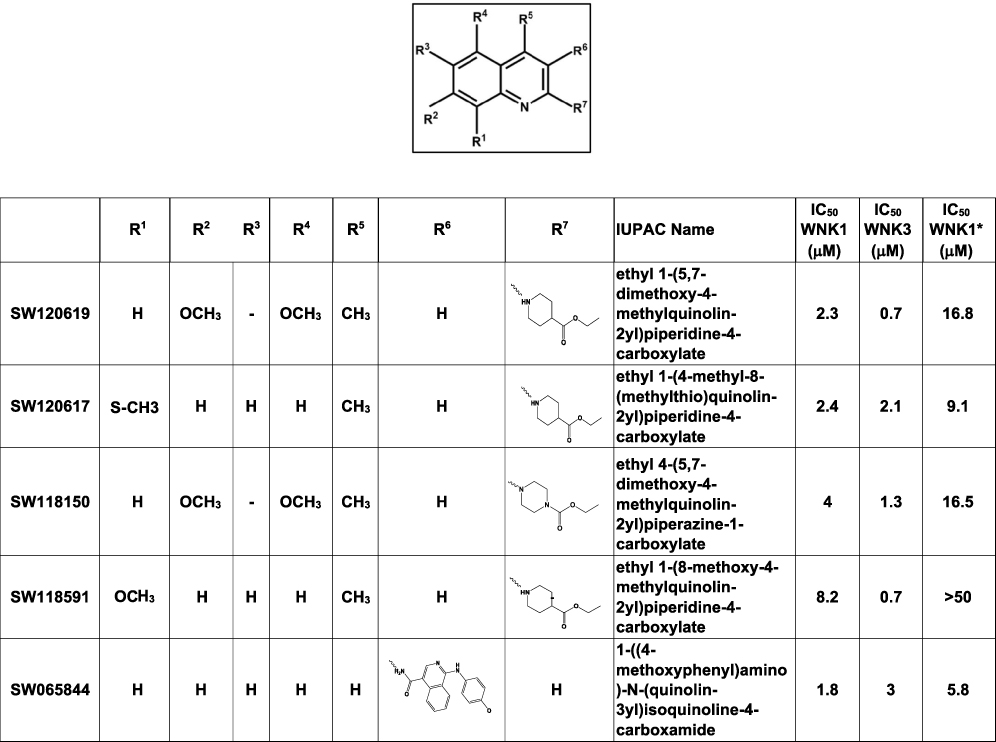 |
Figure 2 Quinoline-derived inhibitors identified from the 210,000 compound screen. IC50 WNK1 and IC50 WNK3 are calculated using non-linear regression method in GraphPad from three inhibitor concentrations (6 μM, 4 μM and 0.5 μM). IC50WNK1* is calculated from the refined HTS screen conducted at nine inhibitor concentrations. |
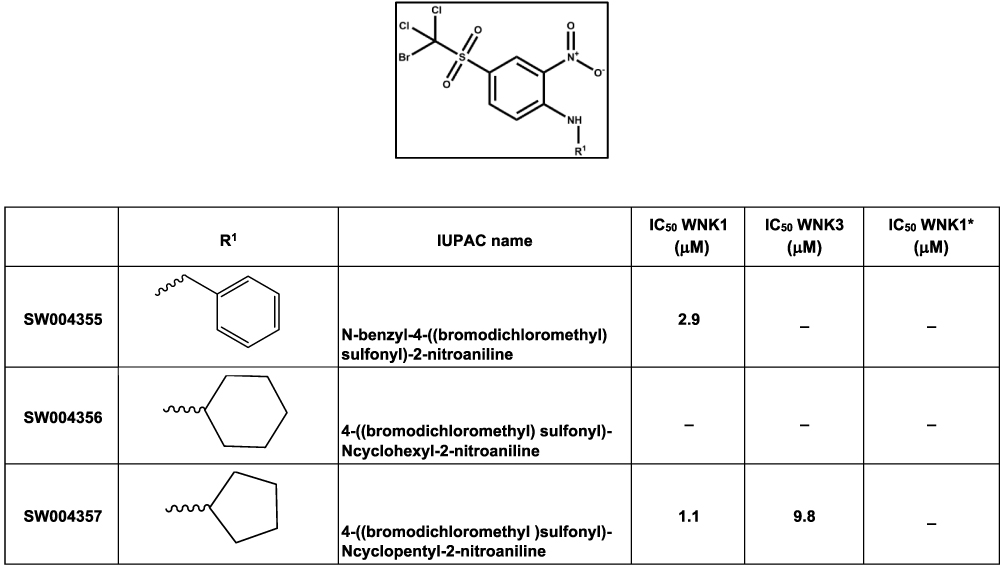 |
Figure 3 Halo-sulfone inhibitors identified from the 210,000 compound screen. IC50 WNK1 and IC50 WNK3 are calculated from three inhibitor concentrations (6 μM, 4 μM and 0.5 μM). - indicates compounds that do not meet a 3 σ criterion for the confirmation screen. |
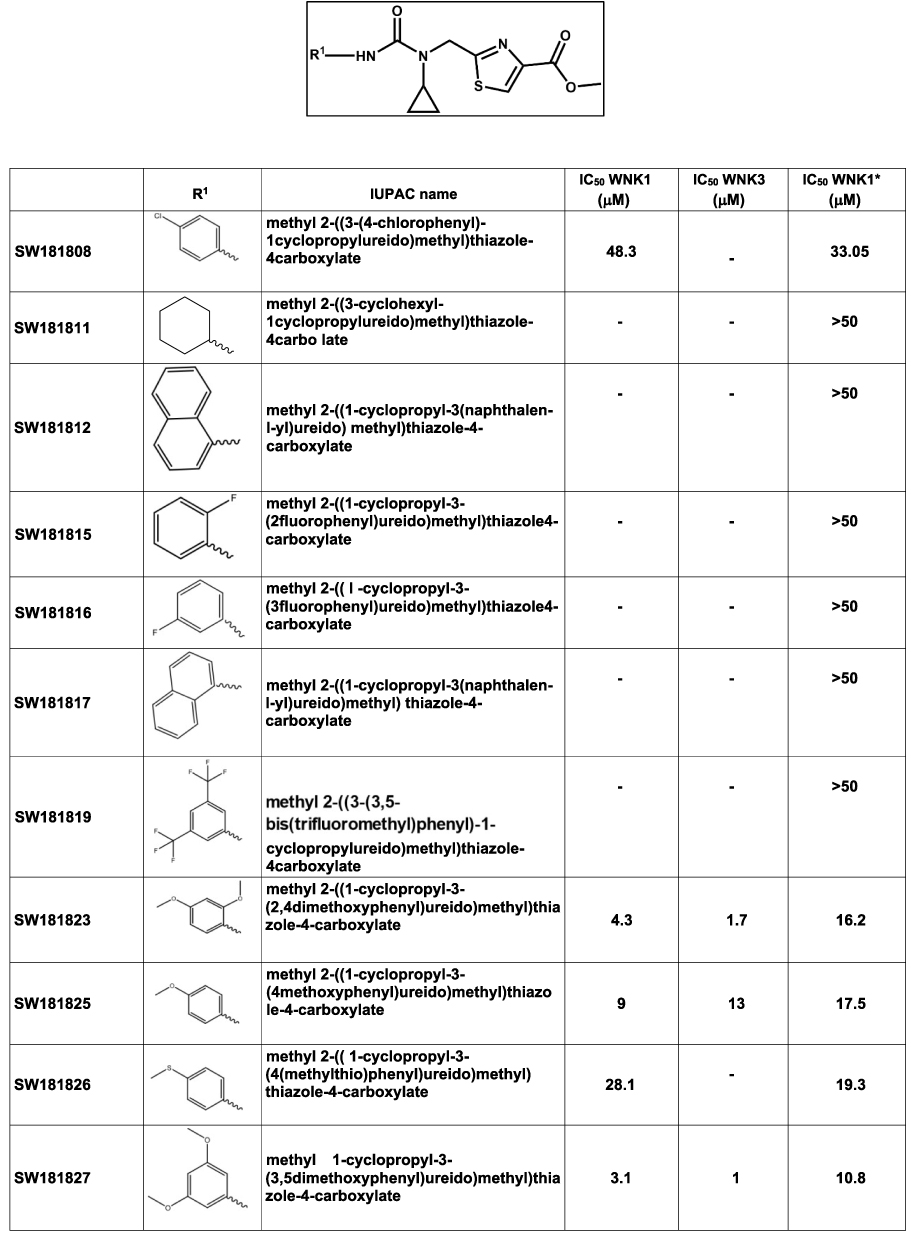 |
Figure 4 Cyclopropane-containing thiazole inhibitors identified from the 210,000 compound screen. IC50 WNK1 and IC50 WNK3 are calculated from three inhibitor concentrations (6 μM, 4 μM and 0.5 μM). IC50 WNK1* is calculated from the refined HTS screen conducted at nine inhibitor concentrations. |
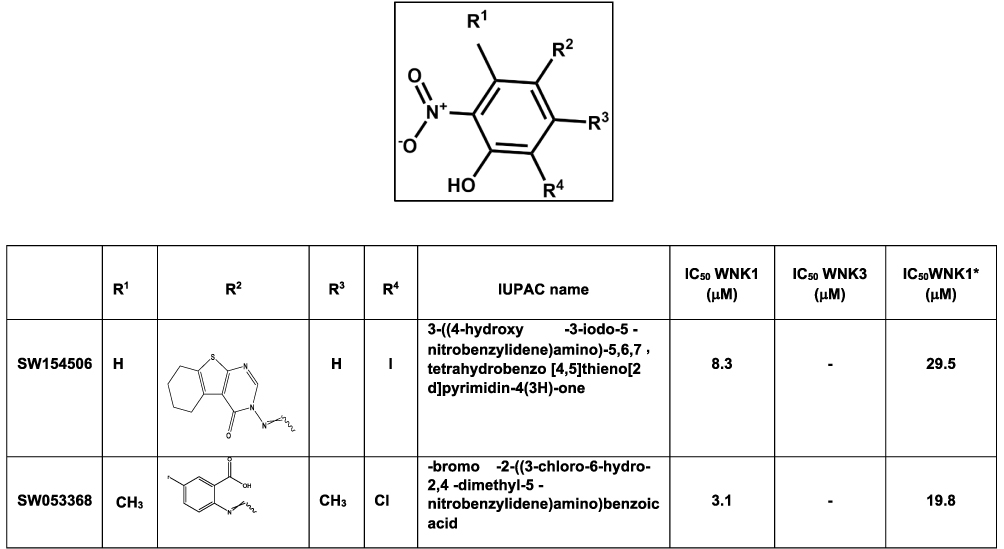 |
Figure 5 Nitrophenol-derived inhibitors identified from the 210,000 compound screen. IC50 WNK1 and IC50 WNK3 are calculated from three inhibitor concentrations (6 μM, 4 μM and 0.5 μM). IC50 WNK1* is calculated from the refined HTS screen conducted at nine inhibitor concentrations. |
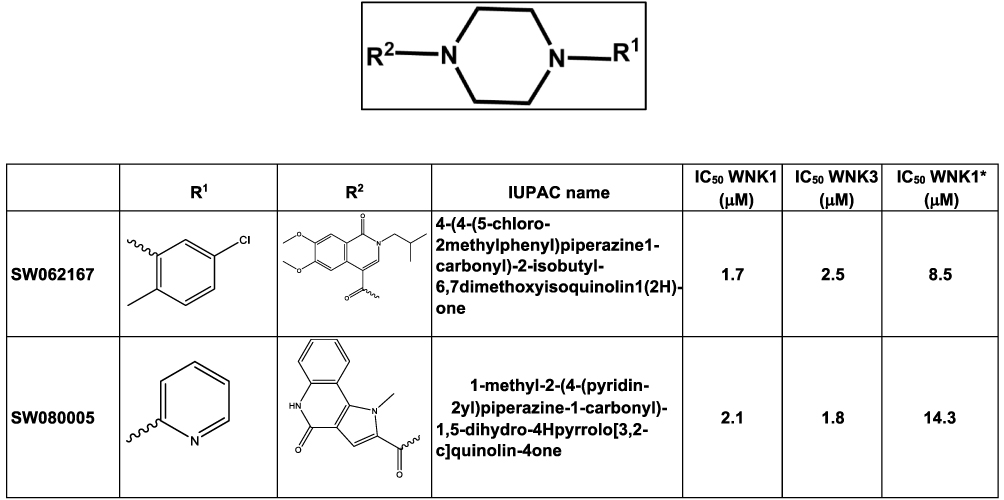 |
Figure 6 Piperazine-derived inhibitors identified from the 210,000 compound screen. IC50 WNK1 and IC50 WNK3 are calculated from three inhibitor concentrations (6 μM, 4 μM and 0.5 μM). IC50. WNK1* is calculated from the refined HTS screen conducted at nine inhibitor concentrations. |
Using the screening facility, the confirmation screen was cross-analyzed with phosphorylated kinase domain of WNK3 (pWNK3-KDm (pWNK3)). This screen gave similar results with respect to quality and correlation as the WNK1 screen.
Classes of Compounds
Five prominent classes have been identified. These five classes are named quinoline-derived (Figure 2), halo-sulfone (Figure 3), cyclopropane-containing thiazoles (Figure 4), piperazine-derived (Figure 5), and nitrophenol-derived (Figure 6). The chemical structures of the best hits from each class are presented in Figures 2–6 together with the inhibition constants against pWNK1 and pWNK3. Figure 2 shows the class of quinoline-derived compounds. Members of the quinoline class were more potent inhibitors of WNK3 than WNK1, discussed more fully below. Another class of inhibitors contains a halo-sulfone group (Figure 3) and is the subject of a separate publication.32 Figure 4 shows the members of the cyclopropane-containing thiazole series. Other classes identified include two piperazine-containing compounds (Figure 5) and two nitrophenol-containing compounds (Figure 6).
Compound Specificity
Two compounds, one from the quinoline class (SW120619) and the other from the cyclopropane-containing thiazole (SW182086), were tested at 10 μM for specificity against 46 different kinases and the four isoforms of WNKs at Eurofins Inc. (France) (Figure 7). Both the inhibitors showed high specificity toward WNK isoforms. Based on a single concentration in the Eurofins screen, SW120619 showed the order of specificity among WNK kinase isoforms as WNK3>WNK1>WNK4>WNK2. SW182086, on the other hand, had nearly equal specificity for WNK1 and WNK3.
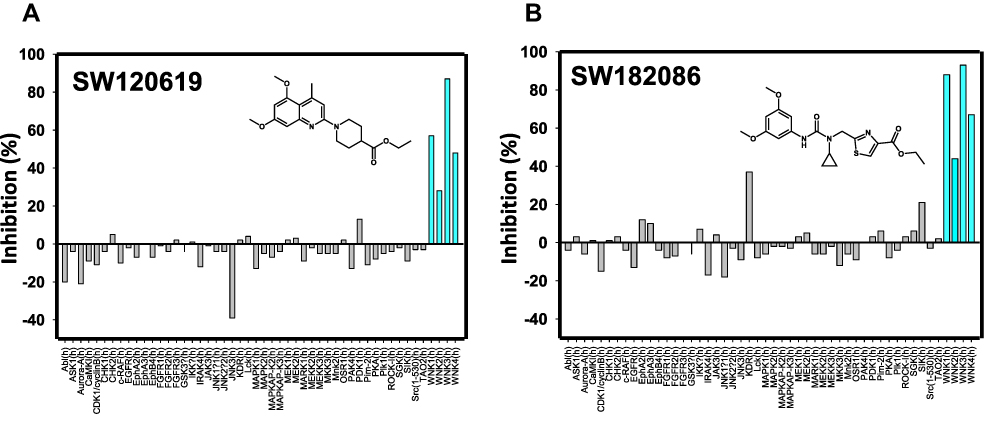 |
Figure 7 Specificity screen. Commercial specificity screen from Eurofins Inc. for the best compounds from (A) the quinoline class and (B) the cyclopropane-containing thiazole class. Compound concentration used in these screens was 10 μM. Blue lines are for the four human WNK isoforms. |
Four quinoline class compounds identified in the HTS screen were acquired from commercial sources. Peptide mobility shift assays showed SW120617, SW120619, and SW118150 were more potent inhibitors of WNK3 than WNK1 (Figure 8A–C). On the other hand, SW118591 showed no inhibition or very weak inhibition toward WNK3 and WNK1, respectively (Figure 8D).
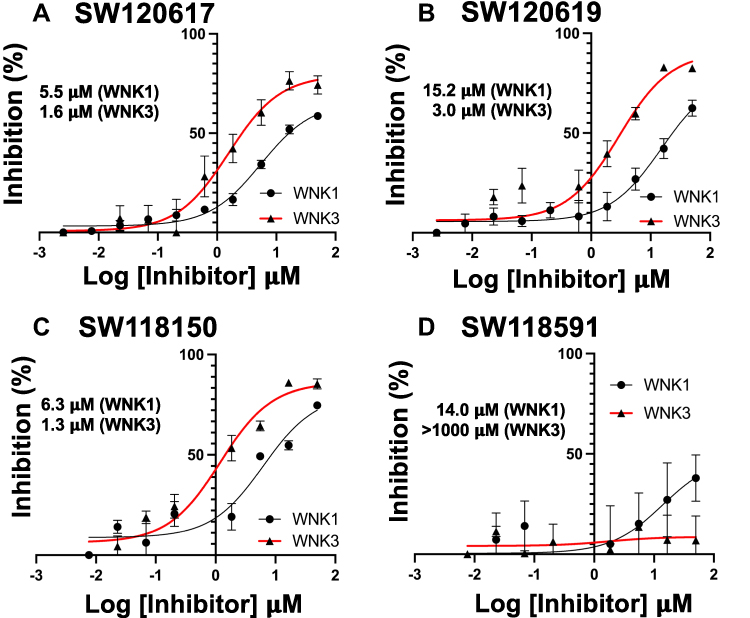 |
Figure 8 Peptide mobility shift assays for quinoline class inhibitors. (A) SW120617, (B) SW120619, (C) SW118150, and (D) SW118591. GST-WNK1 (black line) and GST-WNK3 (red line) using a fluorescently labeled FAM-OXSR1 substrate peptide. |
Differential Scanning Fluorimetry (DSF) measures protein melt temperature (Tm) as a function of inhibitor binding. Inhibitor binding has a stabilizing effect, and this is seen by an increase in melt temperature (Table 1 and Figure S2). All the four quinoline-derived compounds showed positive shifts for pWNK1 and pWNK3. The ∆Tm values ranged from 0°C to 2.5°C for pWNK1 and 6.5°C to 14°C for pWNK3, indicating tighter binding to pWNK3 than pWNK1. These ∆Tm values correlate well with the IC50 values determined by mobility shift assay (Figure 8).
 |
Table 1 Change in Melting Temperature Tm (∆Tm) Upon Binding of Quinoline-Derived Compounds to pWNK1 and pWNK3 Using Differential Scanning Fluorimetry |
Crystal Complex of SW120619 with WNK3/SA
The primary activation loop phosphorylation site of WNK3 is S308. WNK3 S308A does not autophosphorylate. The 3.1 Å X-ray structure of the kinase domain of WNK3/S308A (WNK3/SA) complexed with SW120619 (SW120619-WNK3/SA) was solved by molecular replacement using the WNK1/SA complex (PDB file 6CN9,22).
SW120619 (ethyl 1-(5,7-dimethoxy-4-methylquinolin-2-yl) piperidine-4-carboxylate) is composed of a quinoline moiety fused to piperidine. The inhibitor is well-ordered in the structure (Figure 9A). SW120619 binds approximately perpendicular to the beta-strands of the N-terminal domain, contacting two ridge lines of the beta-sheet. It contacts residues L153, V161, C176, and L225 in one ridge line, and A174 and T227 in a second ridge line (Figure 9B). The inhibitor also contacts the backbone of L229-T231 in the crossover connection between the two domains of the kinase. On the opposite face, SW120619 makes extensive contacts with F282, and interacts with the catalytic lysine (K159) and aspartic acid (D294). The methoxy moieties of SW120619 contact the chloride-binding helix (CBH) at the N-terminus of the Activation Loop (referred to previously as the “3/10 helix”24 through the mainchain of residue L295, and the side-chain of L297 (Figure 9B). SW120619 has similarities with the Novartis pan-WNK inhibitor WNK463 (Figure S1D) both in molecular architecture and binding mode (Figure 9C). Like WNK463, SW120619 is composed of a linear string of heterocycles. SW120619 binds in the same site near the crossover connection but does not penetrate as deeply as WNK463.
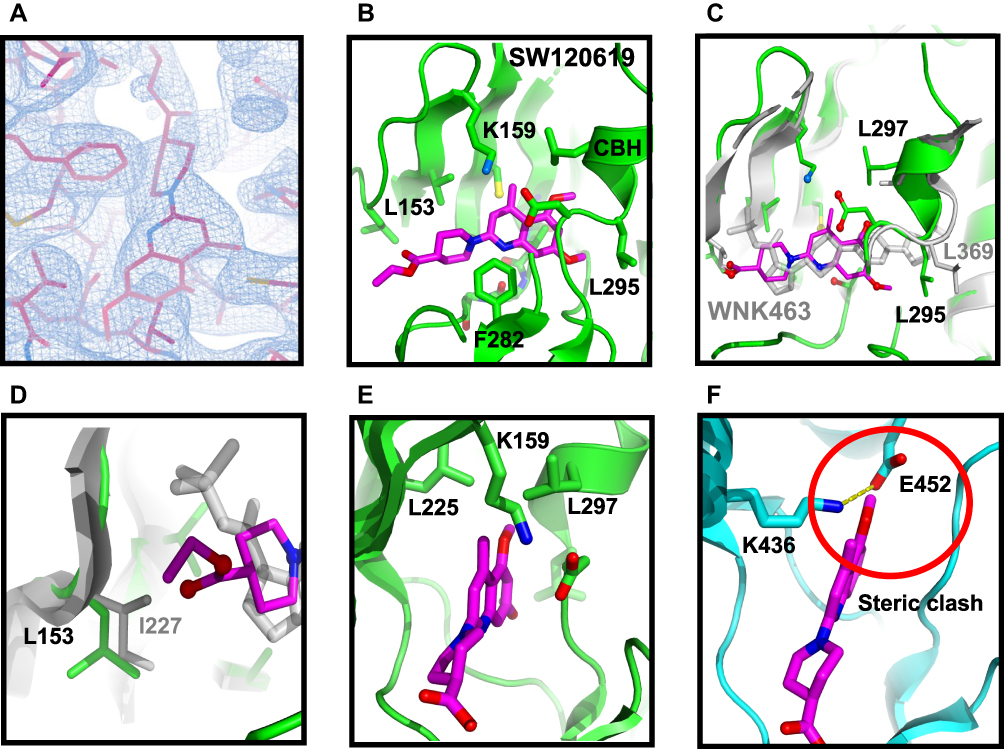 |
Figure 9 Structure of complex between WNK3/SA and SW120619. (A) Electron density of the inhibitor SW120619 contoured at 0.5 σ. (B) Schematic of the inhibitor binding drawn in Pymol (C) Superposition of WNK3/SW120619 with WNK1/6CN9. The superposition encompassed residues forming the binding site, WNK1/225–236, 298–308, 353-375 (in WNK3, WNK3/151-162, 223–233, 279-301) (D) Closeup of SW120619 and WNK463 near L153 in WNK3 (I227 in WNK1). (E) The WNK3/SW120619 complex oriented to highlight the differences between WNKs and more standard protein kinases such as PAK6 (PDB file 2C30). (F) PAK6 oriented in comparison with (E). Red circle indicates steric clash between SW120619 and PAK6. |
Specificity of SW120619 Toward WNK3
There is a single amino acid replacement in residues contacting SW120619 between WNK3 (L153), and WNK1 (I227) (Figure 9D). This amino acid replacement together with the binding mode of SW120619 may contribute to WNK3 selectivity. To compare binding sites, the structure of WNK1 bound to WNK463 was superimposed locally on WNK1 bound to SW120619, using secondary structure elements that form the inhibitor binding site (Figure 9C). The superposition shows that SW120619 contacts WNK3/L153, whereas WNK463 does not (Figure 9D). There are also subtle differences in the CBH that may contribute to the unique specificity of SW120619 for WNK3 (WNK3/L295 and L297, Figure 9C). Since the quinoline class of compounds are more potent WNK3 inhibitors, there may be opportunities to generate isoform-specific inhibitors. The pan-WNK specificity can be understood by comparing our WNK3/SW120619 complex (Figure 9E) with SW120619 docked into a canonical protein kinase structure (Figure 9F). Figure 9E presents the structure of WNK3/SW120619 in an orientation to highlight where SW120619 does not fit well in a canonical kinase PAK6.33 The pocket occupied by the quinoline ring in WNK3/SW10619 is filled with the ion pair between the standard catalytic lysine in β-strand 3 and a conserved glutamate in helix C (Figure 9F), potentially explaining the pan-WNK specificity of these inhibitors.
Crystallography of the quinoline class inhibitors in WNK3-KDm/SA revealed binding to the same pockets as the pan WNK inhibitor, WNK463, but showed unique interactions. SW120619 is smaller than WNK463 and binds only Pockets I and II (as defined by Gray).34 Model-building of other quinoline class analogues points to positions of modifications for further improvement. This suggests that it is possible to identify better WNK isoform-specific inhibitors. We anticipate obtaining potent isoform-specific inhibitors of WNK1 and WNK3 through structure-based drug design.
Conclusions
We have discovered inhibitors of WNK1 and WNK3 through high-throughput screening. The inhibitors obtained from our screen are very selective for WNK kinases when screened in a panel of 46 kinases. Comparison of the molecular structure of WNK kinases with standard Ser/Thr kinases reveals differences in the active site that account for this specificity. Inhibitors derived from quinolines were better inhibitors of WNK3 than WNK1. We anticipate the inhibitors discovered here may be starting points for the development of pharmacological tools to elucidate isoform-specific roles of WNKs in signal transduction and involvement of WNK kinases in hypertension, cancer, and other diseases.
Abbreviations
WNK, With No lysine; OxSR1, oxidative stress responsive kinase 1; ATP, adenosine 5′-triphosphate; IC50, half maximal inhibitory concentration; FHHt, familial hyperkalemic hypertension; HTS, high-throughput screen; CBH, chloride-binding helix.
Data Sharing Statement
X-ray coordinates and structure factors are deposited at RCSB PDB (PDB 8EDH).
Acknowledgments
We thank Clinton Taylor and Melanie Cobb for the gOSR1 peptide. We thank Anwu Zhou for help in analyzing the HTS data. Results shown in this report were derived from work performed at Argonne National Laboratory, Structural Biology Center (SBC) at the Advanced Photon Source. The SBC is operated by the U Chicago Argonne, LLC, for the US Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. Crystallographic studies were coordinated by Diana Tomchick in the UT Southwestern Structural Biology Laboratory. We thank Kenneth Westover for helping with the mobility-shift assays.
Funding
This work was supported by the American Heart Association grants 16SA285300002 and 14GRNT20500035, the Welch Foundation grants I1128 and I-2100-20220331, and CPRIT grant RP190421 to EJG and a pilot grant from Harold C. Simmons Comprehensive Cancer Center to EJG and RA.
Disclosure
All authors report no conflicts of interest in this work.
References
1. Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem. 2000;275(22):16795–16801. doi:10.1074/jbc.275.22.16795
2. Kahle KT, Rinehart J, Lifton RP. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim Biophys Acta. 2010;1802(12):1150–1158. doi:10.1016/j.bbadis.2010.07.009
3. Cruz-Rangel S, Gamba G, Ramos-Mandujano G, Pasantes-Morales H. Influence of WNK3 on intracellular chloride concentration and volume regulation in HEK293 cells. Pflugers Arch. 2012;464(3):317–330. doi:10.1007/s00424-012-1137-4
4. Choe KP, Strange K. Evolutionarily conserved WNK and Ste20 kinases are essential for acute volume recovery and survival after hypertonic shrinkage in Caenorhabditis elegans. Am J Physiol Cell Physiol. 2007;293(3):C915–C927. doi:10.1152/ajpcell.00126.2007
5. Tu SW, Bugde A, Luby-Phelps K, Cobb MH. WNK1 is required for mitosis and abscission. Proc Natl Acad Sci U S A. 2011;108(4):1385–1390. doi:10.1073/pnas.1018567108
6. Wilson FH, Disse-Nicodeme S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293(5532):1107–1112. doi:10.1126/science.1062844
7. Tobin MD, Raleigh SM, Newhouse S, et al. Association of WNK1 gene polymorphisms and haplotypes with ambulatory blood pressure in the general population. Circulation. 2005;112(22):3423–3429. doi:10.1161/CIRCULATIONAHA.105.555474
8. Dbouk HA, Huang CL, Cobb MH. Hypertension: the missing WNKs. Am J Physiol Renal Physiol. 2016;311(1):F16–F27. doi:10.1152/ajprenal.00358.2015
9. Zambrowicz BP, Abuin A, Ramirez-Solis R, et al. Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc Natl Acad Sci U S A. 2003;100(24):14109–14114. doi:10.1073/pnas.2336103100
10. Oi K, Sohara E, Rai T, et al. A minor role of WNK3 in regulating phosphorylation of renal NKCC2 and NCC co-transporters in vivo. Biol Open. 2012;1(2):120–127. doi:10.1242/bio.2011048
11. Davies H, Hunter C, Smith R, et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005;65(17):7591–7595. doi:10.1158/0008-5472.CAN-05-1855
12. Jinawath N, Vasoontara C, Jinawath A, et al. Oncoproteomic analysis reveals co-upregulation of RELA and STAT5 in carboplatin resistant ovarian carcinoma. PLoS One. 2010;5(6):e11198. doi:10.1371/journal.pone.0011198
13. Zhu W, Begum G, Pointer K, et al. WNK1-OSR1 kinase-mediated phospho-activation of Na+-K+-2Cl-cotransporter facilitates glioma migration. Mol Cancer. 2014;13(1):1. doi:10.1186/1476-4598-13-31
14. Xie T, d’Ario G, Lamb JR, et al. A comprehensive characterization of genome-wide copy number aberrations in colorectal cancer reveals novel oncogenes and patterns of alterations. PLoS One. 2012;7(7):e42001. doi:10.1371/journal.pone.0042001
15. Chen L, Jenjaroenpun P, Pillai AM, et al. Transposon insertional mutagenesis in mice identifies human breast cancer susceptibility genes and signatures for stratification. Proc Natl Acad Sci U S A. 2017;114(11):E2215–E2224. doi:10.1073/pnas.1701512114
16. Begum G, Yuan H, Kahle KT, et al. Inhibition of WNK3 kinase signaling reduces brain damage and accelerates neurological recovery after stroke. Stroke. 2015;46(7):1956–1965. doi:10.1161/STROKEAHA.115.008939
17. Zhao H, Nepomuceno R, Gao X, et al. Deletion of the WNK3-SPAK kinase complex in mice improves radiographic and clinical outcomes in malignant cerebral edema after ischemic stroke. J Cereb Blood Flow Metab. 2017;37(2):550–563. doi:10.1177/0271678X16631561
18. Jeong KH, Kim SH, Choi YH, Cho I, Kim WJ. Increased expression of WNK3 in dispersed granule cells in hippocampal sclerosis of mesial temporal lobe epilepsy patients. Epilepsy Res. 2018;147:58–61. doi:10.1016/j.eplepsyres.2018.09.006
19. Yamada K, Park HM, Rigel DF, et al. Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat Chem Biol. 2016;12(11):896–898. doi:10.1038/nchembio.2168
20. Yamada K, Levell J, Yoon T, et al. Optimization of allosteric With-No-Lysine (WNK) kinase inhibitors and efficacy in rodent hypertension models. J Med Chem. 2017;60(16):7099–7107. doi:10.1021/acs.jmedchem.7b00708
21. Yamada K, Zhang JH, Xie X, et al. Discovery and characterization of allosteric WNK kinase inhibitors. ACS Chem Biol. 2016;11(12):3338–3346. doi:10.1021/acschembio.6b00511
22. Min XS, Lee BH, Cobb MH, Goldsmith EJ. Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension. Structure. 2004;12(7):1303–1311. doi:10.1016/j.str.2004.04.014
23. Akella R, Humphreys JM, Sekulski K, et al. Osmosensing by WNK kinases. Mol Biol Cell. 2021;32(18):1614–1623. doi:10.1091/mbc.E20-01-0089
24. Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal. 2014;7(324):ra41. doi:10.1126/scisignal.2005050
25. Yagi YI, Abe K, Ikebukuro K, Sode K. Kinetic mechanism and inhibitor characterization of WNK1 kinase. Biochemistry. 2009;48(43):10255–10266. doi:10.1021/bi900666n
26. Liu Y, Westover KD. Rapid assessment of DCLK1 inhibitors using a peptide substrate mobility shift assay. STAR Protoc. 2021;2(2):100587. doi:10.1016/j.xpro.2021.100587
27. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi:10.1126/science.1075762
28. Emsley P. Tools for ligand validation in coot. Acta Crystallogr D Struct Biol. 2017;73(Pt 3):203–210. doi:10.1107/S2059798317003382
29. Pantoliano MW, Petrella EC, Kwasnoski JD, et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 2001;6(6):429–440. doi:10.1177/108705710100600609
30. Anselmo AN, Earnest S, Chen W, et al. WNK1 and OSR1 regulate the Na+, K+, 2Cl(-) cotransporter in HeLa cells. Proc Natl Acad Sci U S A. 2006;103(29):10883–10888. doi:10.1073/pnas.0604607103
31. Vitari AC, Thastrup J, Rafiqi FH, et al. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J. 2006;397:223–231. doi:10.1042/BJ20060220
32. Rodriguez M, Kannangara A, Chlebowicz J, et al. Synthesis and structural characterization of novel trihalo-sulfone inhibitors of WNK1. ACS Med Chem Lett. 2022;13(10):1678–1684. doi:10.1021/acsmedchemlett.2c00216
33. Eswaran J, Lee WH, Debreczeni JE, et al. Crystal structures of the p21-activated kinases PAK4, PAK5, and PAK6 reveal catalytic domain plasticity of active group II PAKs. Structure. 2007;15(2):201–213. doi:10.1016/j.str.2007.01.001
34. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28–39. doi:10.1038/nrc2559
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.