")
Back to Journals » International Journal of General Medicine » Volume 15
Identification and Verification of Immune-Related Genes Prognostic Signature Based on ssGSEA for Adrenocortical Carcinoma (ACC)
Authors Yuan K , Zeng R, Deng P, Zhang A, Liu H, Wang N, Tang Y, Yin Z, Liu H
Received 29 October 2021
Accepted for publication 26 January 2022
Published 15 February 2022 Volume 2022:15 Pages 1471—1483
DOI https://doi.org/10.2147/IJGM.S345123
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Kaisheng Yuan, Ruiqi Zeng, Pengteng Deng, Aiping Zhang, Huiqian Liu, Ning Wang, Yongxi Tang, Zhikang Yin, Hang Liu
Department of Urology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China
Correspondence: Hang Liu, Department of Urology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China, Tel +86-185-8030-9681, Email [email protected]
Purpose: Adrenocortical carcinoma (ACC) is an endocrine malignant tumor with poor prognosis. The study aimed to construct ACC immune-related gene prognostic signature and verify the efficacy of prognostic signature.
Methods: ACC RNA-seq data and clinical information are downloaded from TCGA databases and GEO databases. We used single sample gene set enrichment analysis (ssGSEA) to assess immune cell infiltration in ACC patients and ACC patients were divided into high- and low-immune cell infiltration clusters. The validity of ssGSEA grouping was verified using the ESTIMATE algorithm. A total of 275 differentially expressed immune-related genes (IRGs) were obtained from the intersection of IRGs and differentially expressed genes (DEGs) in high and low immune cell infiltration clusters. LASSO analysis was used to identify 13 IRGs that regulate the prognosis of ACC patients through immune infiltration. Kaplan–Meier analysis, ROC curve, univariate and multivariate Cox regression further confirmed that these 13 immune-related gene signatures were innovative and significant prognostic factors, which were independent of clinical features. Finally, ACC prognostic nomogram was constructed, ROC curve and calibration curve were drawn to evaluate the accuracy of the prognostic nomogram.
Results: LASSO regression analysis was used to screen out ACC survival-related genes. Univariate and multivariate Cox proportional risk regression models were used to analyze and construct the ACC prognosis nomogram. The AUC for predicting 1-, 3- and 5-year overall survival rate of ACC patients was 0.799, 0.966 and 0.969, suggesting good prediction accuracy. The calibration curve shows that the predicted results of the prognostic nomogram are in good agreement with the actual situation.
Conclusion: ssGSEA technique plays an important role in the construction of ACC prognostic model. Based on IRGs associated with survival independently predicted ACC prognosis, we identified thirteen immune-related genes as prognostic signature for ACC.
Keywords: immune, prognostic, adrenocortical carcinoma, ACC, single sample gene set enrichment analysis, ssGSEA, risk score
Introduction
Adrenocortical carcinoma (ACC) is an invasive endocrine malignant tumor originating in the adrenal cortex with poor prognosis. Statistically, the incidence worldwide of ACC is between 0.7 and 2 cases per million population.1 Clinically, patients with adrenal cortical carcinoma are more likely to be diagnosed at an advanced stage with a 5-year survival rate of less than 15%, the rate of metastasis and recurrence is very high even after radical resection.2 However, globally, there are relatively few cases of ACC available for study, and the potential molecular mechanism and prognostic factors of ACC are not particularly clear. Therefore, robust and trustworthy prognostic biomarkers associated with biological heterogeneity of ACC, except for the usual clinical characteristics, are necessary to improve prognosis.
Today, immunotherapy is a new treatment that has revolutionized cancer treatment. Based on observations of adrenalitis in patients receiving immune checkpoint blockade, the presence of programmed death-ligand 1 (PD-L1) expression in the tumor cell membrane and tumor-infiltrating mononuclear cells in surgically treated ACC, we think that evaluation of immunotherapy in ACC was convincing.3,4 Furthermore, Bagante et al constructed a multi-centre study and found that upregulation of immunologic markers neutrophil–lymphocyte ratio (NLR) and platelet–lymphocyte ratio (PLR) was significantly associated with poor prognosis after resection of ACC patients.5 Although there is a lot of evidence that immunotherapy may be a breakthrough treatment for ACC, the related molecular mechanisms, especially the regulation of immune-related genes (IRGs), are still unclear. Therefore, systematic analysis of the relationship between immune-related genes and prognosis may provide motivation and target for further study on immunotherapy of ACC.
Therefore, in this study, ACC patients were divided into high- and low-immune cell infiltration clusters based on single sample gene enrichment analysis (ssGSEA), which was verified by ESTIMATE and CIBERSORT analysis. Gene expression and clinical data of ACC patients were downloaded from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO). The prognostic genes of ACC were identified by survival analysis. The prognostic model was constructed by using prognostic genes and the accuracy of the model would be verified. Finally, we constructed the nomogram to predict the survival rate of ACC patients, which will provide reference for the prognosis of ACC patients.
Materials and Methods
Collect and Cluster of ACC Data
ACC (n = 92) RNA sequencing data can be downloaded from TCGA (The Cancer Genome Atlas) (https://portal.gdc.cancer.gov/). Seventy-nine of the samples had relevant clinical information. Supplement Table S1A shows all the patients’ data from TCGA dataset. All IRGs can be obtained from IMMPORT (https://www.immport.org/shared/home).6 We performed a systematic search on the GEO database (https://www.ncbi.nlm.nih.gov/geo/) to find ACC gene expression datasets. GSE76201 dataset from the GEO database was used for external validation in this study. GSE76201 is a microarray dataset that includes 29 adrenal carcinoma tissues with available clinical information. The platform of the dataset is GPL96.7 Supplement Table S1B shows all the patients’ data from GEO dataset. We downloaded ACC mutation data from TCGA and used “perl” to calculate tumor mutation burden (TMB).8 SsGSEA was used to score IRGs and immune-related functions using the R package “GSVA”.9 Based on the results of ssGSEA score, immune cells in ACC expression profile in TCGA database were divided into high- and low-immune cell infiltration cluster using the R package “sparcl”.10 Supplement Figure S1 shows a flow diagram of the design of the work in this study.
Validation of the Effectuality of Immune Clustering
According to the expression levels of immune cells and stromal cells, the scores of stromal cells infiltration levels, the scores of immune cells infiltration levels and tumor purity in ACC tumor microenvironment were calculated using the R package “ESTIMATE”.11
First, we use ESTIMATE algorithm to calculate Stromal Score, Immune Score, ESTIMATE Score and Tumor Purity of 79 ACC samples from two clusters in TCGA database. Then, R Package “Pheatmap” was used to draw the heatmap.12 The R package “ggpubr” was used to draw violin plots (vioplot) of tumor purity, estimated score, immunity score and matrix score of two clusters. Then, we used the R package “CIBERSORT” to count the contents of 22 immune cells in all ACC samples, and plotted the boxplots of differently expressed immune cells in different cluster groups using the R package “GGplot2”.13 Furthermore, we reconfirmed differences in human leukocyte antigen (HLA)-related gene expression between the high- and low-immune cell infiltration cluster.14
GSEA Enrichment Analysis
GSEA software was used for gene enrichment analysis of ACC data. R Package “GGplot2” was applied to transform the results of GSEA enrichment analysis into annotated visualized bar graph and bubble graph.15 P < 0.05 was considered statistically significant.
Identification of Differentially Expressed IRGs
To identify the DEGs between two clusters in the TCGA-ACC cohort, the R package “limma” was applied in the standard comparison mode.16 The significance criteria of DEGs were set as | logFC | >0.585, FDR < 0.05. The volcano map was also plotted to identify DEGs with “ggplot2” package in R software. Common genes of IRGs and DEGs were obtained by Venn analysis. Then, R package “pheatmap” was used to draw the heatmap of DEGs and differentially expressed IRGs. Finally, we used R package “sva” to calculate the differentially expressed IRGs and their expression levels in TCGA and GEO, respectively.
Identification of Immune-Related Prognostic Genes for ACC
Univariate Cox regression analysis was used to distinguish immune prognosis genes among ACC samples (n = 79) with complete clinical information from TCGA database and draw the corresponding forest map.17 The significance criteria for determining ACC immune prognostic genes was set as p < 0.05. Hazard radio >1 is the high-risk gene, <1 is the low-risk gene.
Establishment of PPI Network
Then, a PPI network of correlated nodes was established by the STRING (v11.5; https://string-db.org/).18 Nodes represent genes or proteins, and the lines represent protein–protein associations.
Construction of IRGs Prognostic Signature for TCGA-ACC
In the next study, the data extracted from the TCGA dataset were used as the experimental group, and the GEO dataset was set as the verification group. First, we screened survival-related genes from TCGA dataset using LASSO regression analysis to obtain significant results from univariate Cox regression by using R “glmnet” package.17 One thousand rounds of cross validation for tuning parameter selection are used to prevent overfitting and partial likelihood bias meets minimum criteria. Second, we constructed prognostic markers of ACC based on the expression of IRGs and correlation coefficients of LASSO regression analysis. These genes were selected to further calculate the risk score of each patient.19 The median value of the risk score was defined by using the R package “survminer”, where ACC patients were divided into high-risk and low-risk groups. Time-dependent receiver-operating characteristic (ROC) and Kaplan–Meier (K-M) curves were used to evaluate the clinical prognostic ability of risk scores using the R “timeROC”, “survival”, and “survminer” packages.20,21 Third, to evaluate whether risk score can be regarded as an independent predictor of overall survival in ACC patients, univariate Cox and multivariate Cox regression analysis were performed with risk score, gender and tumor grade as variables using the R “survival” package. Finally, we used R package “ggplot2” to analyze the co-expression of 22 immune cells and 13 ACC prognosis genes and draw relevant heat maps.
Verification of IRGs Prognostic Signature Used by GSE76201
In the verification group, ACC patients were also divided into high-risk and low-risk groups according to LASSO regression analysis. Then, ROC and K-M curves were used to verify the clinical prognostic ability of risk scores using the R “timeROC”, “survival”, and “survminer” packages.
The Establishment and Evaluation of the Nomogram
The nomogram was constructed to predict the survival status of ACC patients. Gender, N classification, risk, clinical stage, and T classification were used to construct the nomogram using R “rms” and “survival” packages.22 ROC curves and calibration curves were used to assess the accuracy of the nomogram using R “regplot”, “survminer” and “timeROC” packages.
Statistical Analysis
All statistical analyses were conducted using the R software version 4.1.0 and perl version 5.32.1. p < 0.05 was regarded as statistically significant.
Results
Validation of the Effectuality of Immune Clustering
We obtained 79 ACC samples with relevant clinical information from the GDC database. All IRGs were obtained from IMMPORT. We used ssGSEA to quantify ACC RNA sequencing data for immune cell infiltration. Finally, 29 infiltrating immune cells were obtained: aDCs, Mast cells, Type II IFN Response, Type I IFN Response, DCs, MHC class I, B cells, Neutrophils, pDCs, APC co-stimulation, HLA, T helper cells, iDCs, NK cells, CD8+ T cells, Inflammation promoting, Th1 cells, Check point, T cell co-inhibition, T cell co-stimulation, Cytolytic-activity, TIL, Tfh, APC co-inhibition, Parainflammation, CCR, Treg, Macrophages, Th2 cells. By applying an unsupervised hierarchical clustering algorithm (cutoff = 1.0), ACC samples were divided into two groups according to immune infiltration: high immune cell infiltration cluster (n = 17) and low immune cell infiltration cluster (n = 62) (Figure 1A). To verify the effectiveness of ssGSEA grouping, we used ESTIMATE algorithm to calculate the stromal score, immune score, ESTIMATE score and tumor purity of two clusters, respectively. The results show that stromal score, immune score, and ESTIMATE score in high immune cell infiltration cluster were higher than those in low immune cell infiltration cluster. However, tumor purity was the opposite (Figure 1B). The violin plot also shows that stromal score, immune score and ESTIMATE score in the two clusters have significant differences (P < 0.001) (Figure 1C). In addition, the box plot also showed that the expression of high immune cell infiltration cluster was significantly higher than that of low immune cell infiltration cluster in 24 HLA. (“***”, “**”, “*”, “ns” are “P<0.001” “P<0.01” “P<0.05” “no significance”) (Figure 1D).
 |
Figure 1 Validation of the effectuality of immune clustering. (A) 79 ACC samples were divided into high immune cell infiltration group (Red) and low immune cell infiltration group (Blue). (B) The enrichment levels of 29 immune-related cells and types in the high immune cell infiltration group (Immunity_H) and the low immune cell infiltration group (Immunity_L). The tumor purity, ESTIMATE score, immune score and stromal score of every patient’s gene were showed combine with the clustering information. (C) The violin plot showed the difference in ESTIMATE score, immune score and stromal score between two clusters. (D) The expression of most HLAs was a significant difference in high- (red) and low- (blue) immune cell infiltration cluster. *p < 0.05, **p < 0.01, ***p < 0.001. Abbreviation: ns, no statistically significant. |
GSEA Enrichment Analysis
KEGG analysis of genes in high and low immune cell infiltration clusters showed that these genes were related to KEGG HEMATOPOIETIC CELL LINEAGE, TYPE I DIABETES MELLITUS, AUTOIMMUNE THYROID DISEASE, ALLOGRAFT REJECTION, CYTOKINE CYTOKINE RECEPTOR INTERACTION, INTESTINAL IMMUNE NETWORK FOR IGA PRODUCTION, CELL ADHESION MOLECULES CAMS (Figure 2A and B).
 |
Figure 2 GSEA enrichment analysis. KEGG analysis of differentially expressed IRGs was performed and visualized via a dot plot (A) and bar plot (B). |
Identification of Differentially Expressed IRGs Between High and Low Immune Cell Infiltration Clusters
By using the threshold value of |lgFC| > 0.585 and FDR < 0.05, we identified 2890 DEGs with high and low immune cell infiltration in the 79 ACC samples of TCGA. It included 769 upregulated genes and 2111 downregulated genes (Figure 3A). A total of 1793 IRGs were downloaded from IMMPORT and Venn analysis was used to yield 275 differentially expressed IRGs by making intersection with DEGs (Figure 3B). At the same time, we plot the heat map (Figure 3C and D) for DEGs and differentially expressed IRGs.
 |
Figure 3 Identification of differentially expressed IRGs between high and low immune cell infiltration clusters. (A) The volcano plot showed that 769 up-regulated genes (red) and 2111 down-regulated genes (green) between high and low immune cell infiltration cluster in TCGA dataset. Black dots mean meaningless (P>0.05). (B) Using venn diagram to pick up the intersection, 275 differentially expressed IRGs were obtained. (C) The heat map showed the expression levels of 2890 DEGs with high and low immune cell infiltration in 79 ACC samples from TCGA. (D) The heat map showed the expression levels of 275 differentially expressed IRGs. |
Identification of IRGs Prognostic Signature for ACC
We used univariate Cox regression analysis to analyze data from 79 ACC patients with complete clinical data. Finally, 57 differentially expressed IRGs were significantly associated with ACC overall survival rate (P < 0.05), with 38 high-risk genes and 19 low-risk genes (Figure 4). Supplement Table S2 shows differentially expressed IRGs.
 |
Figure 4 Identification and verification of immune-related gene prognostic signature for ACC. The p-value and HR of selected genes in univariate Cox regression analysis (p < 0.05). Hazard radio>1 are the high-risk genes, shown in red. Hazard radio<1 are the low-risk genes, shown in green. |
Establishment of PPI Network
A protein interaction network analysis was performed on 109 co-expressed nodes and these nodes representing genes or proteins. If there was a connection between nodes, there was a protein interaction relationship between nodes (Figure 5). The lines of different colors represent different meanings. The lines represent protein–protein associations. The light blue line means that both proteins are from curated databases. The pink line means that both proteins are experimentally determined. The green line means that both proteins are in a gene neighborhood. The red line means that both proteins are gene fusions. The dark blue line means that both proteins are gene co-occurrence. The yellow line means that both proteins are text mining. The black line means that both proteins are co-expression and the purple line means that both proteins are protein homology.
 |
Figure 5 Establishment of Sankey diagram and PPI network. The protein interaction network analysis shows the relationship of protein interaction between nodes, and different color lines represent different meanings. |
Construction of IRGs Prognostic Signature
The data extracted from the TCGA dataset was used as the experimental group, and the GEO dataset was set as the verification group. We used the GSE76201 dataset to verify the validity of immune-related gene prognostic signature. LASSO Regression analysis was performed on 57 differentially expressed IRGs. Eventually, 13 genes including RAC2, KRAS, IL11RA, C3, GHR, VGF, TOR2A, PTGFR, TGFBR2, TNFAIP3, PAK4, IRF7, SDC4 were validated (Figure 6A and B). Risk scores were calculated based on the expression levels and their coefficients of these 13 genes. Risk score = (−0.0722415*expression of RAC2) + (0.6064765* expression of KRAS) + (0.6911548*expression of IL11RA) + (−0.0375132 * expression of C3) + (−0.1608705*expression of GHR) + (0.0646402 *expression of VGF) + (0.4313901*expression of TOR2A) + (−0.0413595 * expression of PTGFR) + (−0.0223629*expression of TGFBR2) + (0.3557257 *expression of TNFAIP3) + (0.1153306*expression of PAK4) + (0.5941446 *expression of IRF7) + (−0.0655002*expression of SDC4).
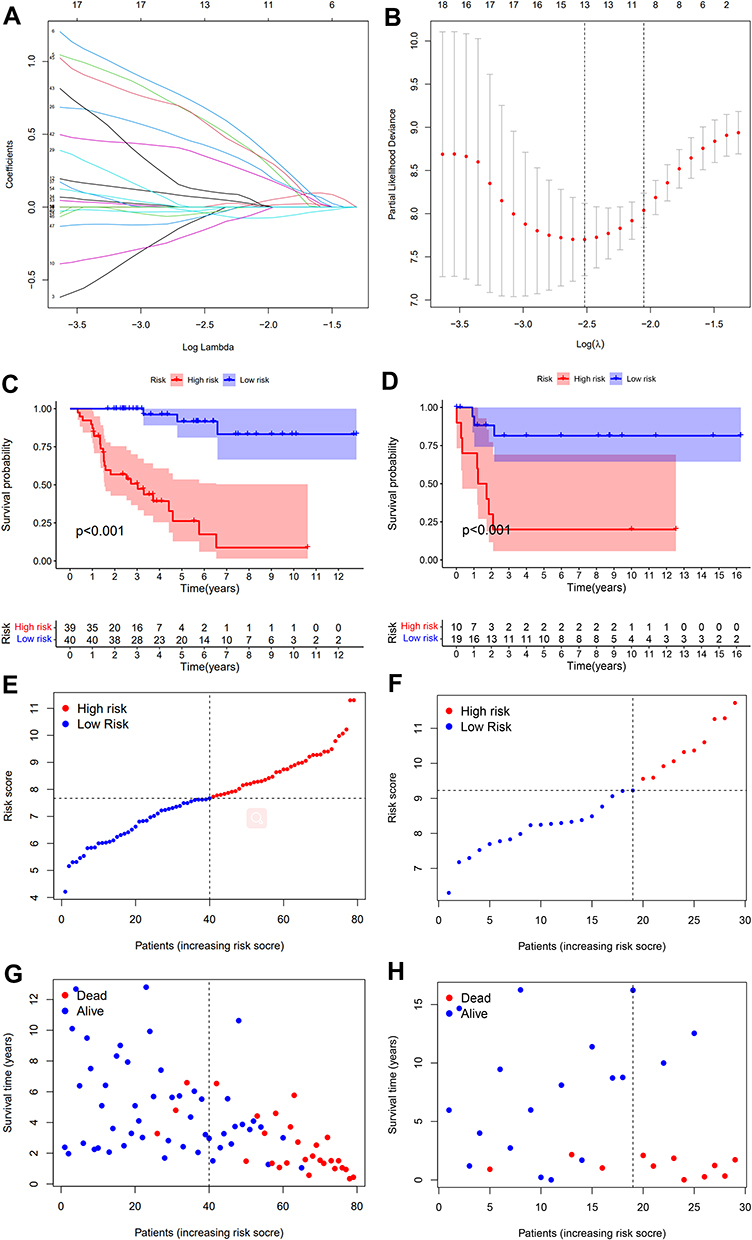 |
Figure 6 Construction of IRGs prognostic signature (A and B). The LASSO Cox analysis identified 13 genes associated with prognosis. (C) Train group, (D). Test group). The results of Kaplan–Meier survival analysis revealed that the survival rate of the low-risk group was significantly higher than that of the high-risk group (P<0.001). ((E) Train group, (F) Test group). The risk curve of every sample arranged by risk score. (G) Train group, (H) Test group). The scatter plot of ACC samples survival overview. The red and green dots stand for death and survival, respectively. |
Patients were divided into high-risk group and low-risk group by using the median of the risk score as the critical value in both groups. The results of Kaplan–Meier survival analyses revealed that the survival rate of the low-risk group was significantly better than that of the high-risk group (P < 0.001), indicating that the risk score predicted the prognosis of ACC patients effectively (Figure 6C and D). Risk curves (Figure 6E and F) and scatter plots (Figure 6G and H) show the risk scores and survival status for all ACC patients.
Verification of IRGs Prognostic Signature
The ROC curve was used to assess the accuracy of the 13 IRGs in predicting the 1-, 3-, and 5-year overall survival of ACC patients. The AUC of 1, 3 and 5 years were 0.973, 0.970 and 0.936, respectively (Figure 7A) and the AUC of 1, 3 and 5 years were 0.722, 0.822 and 0.817, respectively (Figure 7B). It showed that the risk score has a strong predictive power. The larger the area under the curve, the better the prediction effect. These results suggest that 13 IRGs signature can be used as an important indicator for the assessment of ACC prognosis. The predictive power of the nomogram was evaluated and quantified by measuring the degree of fit between the C index in the standard curve and the baseline time predicted by the model. From the 1-, 3- and 5-year relapse calibration curves shown in Figure 7C and D, the nomogram has a good predictor on the outcome of ACC patients.
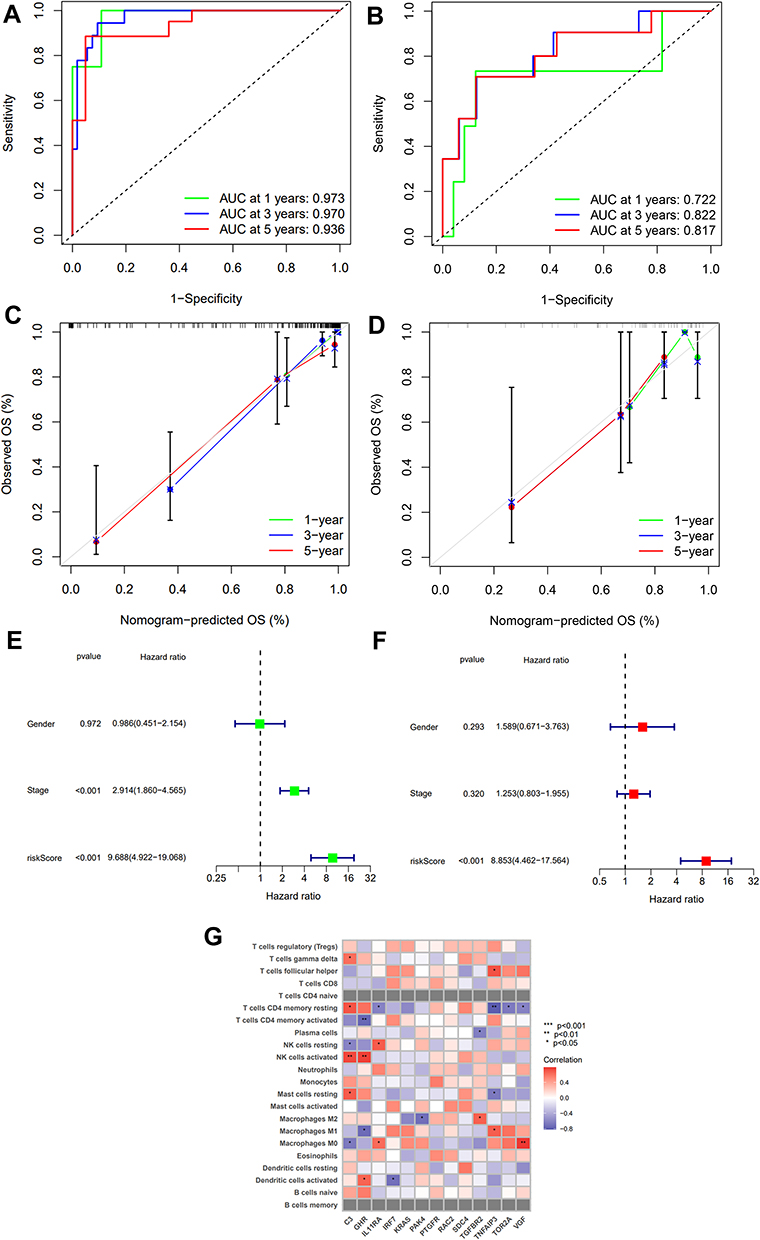 |
Figure 7 Verification of IRGs prognostic signature. ((A) Train group, (B) Test group). The ROC curve was used to evaluate the precision of risk score in predicting overall survival of ACC patients at 1-, 3-, and 5- year. ((C) Train group, (D) Test group). Calibration curve for 1-, 3-, and 5-year recurrence rate of ACC in TCGA datasets. (E) Univariate Cox regression demonstrated that clinical stage and the risk score could predict Overall Survival (OS). (F) Multivariate Cox regression showed that the risk score was independent prognostic factors. (G) Heatmap of correlation matrix between the immune cell and 13 IRGs signature. *p < 0.05, **p < 0.01, ***p < 0.001. |
We applied univariate (Figure 7E) and multivariate regression analysis (Figure 7F) to verify whether the 13 IRGs signature is an independent predictor of other features, eg, gender, tumor, stage, and risk score. The results showed that the risk score is an independent predictor (P < 0.001). Finally, the heatmap was used to show the relationship between 22 immune cells and 13 IRGs signature (Figure 7G).
The Establishment and Evaluation of the Nomogram
The nomogram was constructed to predict survival status in ACC patients. Gender, N classification, risk, clinical stage, and T classification were treated as variables. Each factor in this nomogram has a specific score. Based on the reality of each sample, the score of each prognostic factor was added to get a total score (Figure 8A). We can obtain patients’ survival status of 1, 3 and 5 years and plot the corresponding ROC curves (Figure 8B). The ROC curve predicted that the AUC of ACC patients 1-, 3- and 5-year overall survival rate was 0.799, 0.969 and 0.969, indicating good prediction accuracy. The calibration curve obtained compared well with the ideal model (Figure 8C).
 |
Figure 8 The establishment and evaluation of the nomogram (A). The nomogram used to predict the survival time (B). The ROC curve was used to evaluate the precision of nomogram in predicting overall survival of ACC patients at 1-, 3-, and 5- year. (C) Calibration maps used to predict the 1-, 3- and 5-year survival. |
Discussion
ACC is a rare heterogeneous malignancy with unclear mechanisms of tumorigenesis and poor prognosis.23 In recent years, the findings of the tumor microenvironment have increased a deep understanding of tumor progression and immunotherapy. Habra et al conducted a single-center Phase II clinical trial on 16 ACC patients with a previous history of treatment failure in the past 6 months.24 They found that single-agent pembrolizumab has modest efficacy as a salvage therapy in ACC regardless of the tumor’s hormonal function, MSI status, or PD-L1 status. There was a low rate of severe adverse events in most study participants. Many studies have shown that immunotherapy plays a critical role in many tumors and is likely to be a key part of the clinical management of tumors.25 Therefore, the identification of ACC IRGs will be critical to predict the prognosis of ACC and the development of immunotherapy. In the study, we identified 13 IRGs regulating ACC prognosis by using the TCGA ACC dataset combined with ssGSEA and LASSO analysis. Then, we used the GSE76201 dataset to verify the validity of 13 IRGs. And we screened independent risk factors influencing ACC prognosis and constructed an ACC prognostic nomogram for clinical reference.
In this study, we applied an unsupervised hierarchical clustering algorithm to classify 79 samples based on the enrichment of 29 immune cell types into high- (n = 17) and low immune cell infiltration (n = 62). The stromal score, immune score, ESTIMATE score and tumor purity varied significantly between the two clusters (P < 0.05). Then, the variability in the ACC immune microenvironment was verified by the expression levels of HLA. Finally, LASSO regression analysis of differentially expressed IRGs was performed to construct 13 IRGs signature to predict the prognosis of ACC patients. The samples were divided into a low-risk group and a high-risk group based on the median risk score, with a lower survival rate for ACC patients in the high-risk group. K-M curve and ROC curve showed that 13 IRGs had good predictive ability.
KRAS, IL11RA, C3, TOR2A, TNFAIP3, PAK4, IRF7 were highly expressed in ACC and the rest of the IRGs (RAC2 GHR, VGF PTGFR, TGFBR2, SDC4) were low. Most of the 13 IRGs have been found to be associated with prognosis in different types of cancer. KRAS G12Dresulted in the transition of macrophages into M2-like tumor-promoting phenotype by STAT3-dependent fatty acid oxidation. Thus, disruption of KRAS G12D release and uptake eliminated macrophage-mediated pancreatic cancer stimulation in mouse models. Some studies showed that KRAS in macrophages expression levels were correlated with poor survival in pancreatic cancer patients.26 IL11RA may play a role in the progression of endometrial cancer, and IL11 can be used as a diagnostic marker for early endometrial cancer.27 Local C3 deposition in the tumor microenvironment is a relevant immune feature for predicting gastric cancer prognosis. It may abnormally activate the JAK2/STAT3 pathway, thereby allowing for progression of gastric cancer.28 The inhibition of enzyme topoisomerase 2 alpha (TOP2a) during replication and transcription is one of the main mechanisms of anthracyclines, it perturbs DNA structure and function, leading to cell death. Elevated TOP2a activity and elevated TOP2a gene expression levels caused by amplification of the TOP2a site (17q21.2) in tumor cells were correlated with anthracycline chemosensitivity.29 TNFAIP3, is a potent regulator of ubiquitin (UB)-dependent signaling. Deletion of the TNFAIP3 gene is associated with the NFκB pathway in lymphoma patients and can slow down recurrence or progression in diagnosed patients.30 It was shown that PAK4 as a promoter of breast cancer, can overcome the caducity of oncogene-induced and reveal a selective vulnerability of cancer to PAK4 inhibition.31 A large number of genes suppressed in bone metastasis in breast cancer are targets of the interferon regulator Irf7. Recovery of Irf7 or interferon administration in tumor cells resulted in reduced bone metastasis and prolonged survival.32
We then verified whether gender, tumor, stage, and risk score were independent predictors of other features by applying both univariate and multivariate regression analyses, showing that the risk score was an independent predictor. Finally, we constructed a nomogram predicting the survival status of ACC patients based on gender, N classification, risk, clinical stage, T classification. From this nomogram, we were able to predict survival at 1, 3, and 5 years for ACC patients, with 79 samples having 100% survival for less than 5 years. The calibration curves indicate the nomogram could better predict prognosis in ACC patients.
Nevertheless, there are still some limitations of our study. First, larger samples are needed to further strengthen our findings. Second, all data downloaded online come from public datasets and require further validation in prospective clinical trials, but ACC cases are clinically rare, so the feasibility of prospective studies needs to be further explored.
In conclusion, this study identified 13 IRGs regulating ACC prognosis and constructed a nomogram predicting survival status in ACC patients. This may provide a reference for immunotherapy in ACC patients.
Statement of Ethics
The study was submitted for review by the Ethics Committee of The First Affiliated Hospital of Chongqing Medical and a waiver for ethics was granted.
Consent for Publication
All authors have read and approved the content, and they agree to submit it for consideration for publication in the journal.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jasim S, Habra MA. Management of adrenocortical carcinoma. Curr Oncol Rep. 2019;21(3):20. doi:10.1007/s11912-019-0773-7
2. Pittaway JFH, Guasti L. Pathobiology and genetics of adrenocortical carcinoma. J Mol Endocrinol. 2019;62(2):R105–r19. doi:10.1530/JME-18-0122
3. Ryder M, Callahan M, Postow MA, et al. Endocrine-related adverse events following ipilimumab in patients with advanced melanoma: a comprehensive retrospective review from a single institution. Endocr Relat Cancer. 2014;21(2):371–381. doi:10.1530/ERC-13-0499
4. Fay AP, Signoretti S, Callea M, et al. Programmed death ligand-1 expression in adrenocortical carcinoma: an exploratory biomarker study. J Immunother Cancer. 2015;3:3.
5. Bagante F, Tran TB, Postlewait LM, et al. Neutrophil-lymphocyte and platelet-lymphocyte ratio as predictors of disease specific survival after resection of adrenocortical carcinoma. J Surg Oncol. 2015;112(2):164–172. doi:10.1002/jso.23982
6. Rubio AJ, Porter T, Zhong X. Duality of B cell-CXCL13 axis in tumor immunology. Front Immunol. 2020;11:521110.
7. Legendre CR, Demeure MJ, Whitsett TG, et al. Pathway implications of aberrant global methylation in adrenocortical cancer. PLoS One. 2016;11(3):e0150629. doi:10.1371/journal.pone.0150629
8. Jang BS, Han W, Kim IA. Tumor mutation burden, immune checkpoint crosstalk and radiosensitivity in single-cell RNA sequencing data of breast cancer. Radiother Oncol. 2020;142:202–209.
9. Finotello F, Trajanoski Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol Immunother. 2018;67(7):1031–1040. doi:10.1007/s00262-018-2150-z
10. Rui X, Shao S, Wang L, et al. Identification of recurrence marker associated with immune infiltration in prostate cancer with radical resection and build prognostic nomogram. BMC Cancer. 2019;19(1):1179. doi:10.1186/s12885-019-6391-9
11. Ren N, Liang B, Li Y. Identification of prognosis-related genes in the tumor microenvironment of stomach adenocarcinoma by TCGA and GEO datasets. Biosci Rep. 2020;40(10). doi:10.1042/BSR20200980
12. Cheng Q, Wang L. LncRNA XIST serves as a ceRNA to regulate the expression of ASF1A, BRWD1M, and PFKFB2 in kidney transplant acute kidney injury via sponging hsa-miR-212-3p and hsa-miR-122-5p. Cell Cycle. 2020;19(3):290–299. doi:10.1080/15384101.2019.1707454
13. Kawada JI, Takeuchi S, Imai H, et al. Immune cell infiltration landscapes in pediatric acute myocarditis analyzed by CIBERSORT. J Cardiol. 2021;77(2):174–178. doi:10.1016/j.jjcc.2020.08.004
14. Yu X, Guo J, Zhou Q, et al. A novel immune-related prognostic index for predicting breast cancer overall survival. Breast Cancer. 2021;28(2):434–447. doi:10.1007/s12282-020-01175-z
15. Wu M, Li X, Zhang T, et al. Identification of a nine-gene signature and establishment of a prognostic nomogram predicting overall survival of pancreatic cancer. Front Oncol. 2019;9(996). doi:10.3389/fonc.2019.00996
16. Shen Y, Liu J, Zhang L, et al. Identification of potential biomarkers and survival analysis for head and neck squamous cell carcinoma using bioinformatics strategy: a study based on TCGA and GEO datasets. Biomed Res Int. 2019;2019(7376034):1–14. doi:10.1155/2019/7376034
17. Liang JY, Wang DS, Lin HC, et al. A novel ferroptosis-related gene signature for overall survival prediction in patients with hepatocellular carcinoma. Int J Biol Sci. 2020;16(13):2430–2441. doi:10.7150/ijbs.45050
18. Nangraj AS, Selvaraj G, Kaliamurthi S, et al. Integrated PPI- and WGCNA-retrieval of hub gene signatures shared between barrett’s esophagus and esophageal adenocarcinoma. Front Pharmacol. 2020;11(881). doi:10.3389/fphar.2020.00881
19. Chen X, Jin Y, Gong L, et al. Bioinformatics analysis finds immune gene markers related to the prognosis of bladder cancer. Front Genet. 2020;11:607.
20. Chen L, Lu D, Sun K, et al. Identification of biomarkers associated with diagnosis and prognosis of colorectal cancer patients based on integrated bioinformatics analysis. Gene. 2019;692(119):119–125. doi:10.1016/j.gene.2019.01.001
21. Ni J, Liu S, Qi F, et al. Screening TCGA database for prognostic genes in lower grade glioma microenvironment. Ann Transl Med. 2020;8(5):209. doi:10.21037/atm.2020.01.73
22. Wang Z, Gao L, Guo X, et al. Development and validation of a nomogram with an autophagy-related gene signature for predicting survival in patients with glioblastoma. Aging. 2019;11(24):12246–12269. doi:10.18632/aging.102566
23. Fassnacht M, Libé R, Kroiss M, et al. Adrenocortical carcinoma: a clinician’s update. Nat Rev Endocrinol. 2011;7(6):323–335. doi:10.1038/nrendo.2010.235
24. Habra MA, Stephen B, Campbell M, et al. Phase II clinical trial of pembrolizumab efficacy and safety in advanced adrenocortical carcinoma. J Immunother Cancer. 2019;7(1):253. doi:10.1186/s40425-019-0722-x
25. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi:10.1016/j.immuni.2013.07.012
26. Dai E, Han L, Liu J, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16(11):2069–2083. doi:10.1080/15548627.2020.1714209
27. Yap J, Salamonsen LA, Jobling T, et al. Interleukin 11 is upregulated in uterine lavage and endometrial cancer cells in women with endometrial carcinoma. Reprod Biol Endocrinol. 2010;8(63):1–11.
28. Yuan K, Ye J, Liu Z, et al. Complement C3 overexpression activates JAK2/STAT3 pathway and correlates with gastric cancer progression. J Exp Clin Cancer Res. 2020;39(1):9. doi:10.1186/s13046-019-1514-3
29. Litviakov N, Tsyganov M, Larionova I, et al. Expression of M2 macrophage markers YKL-39 and CCL18 in breast cancer is associated with the effect of neoadjuvant chemotherapy. Cancer Chemother Pharmacol. 2018;82(1):99–109. doi:10.1007/s00280-018-3594-8
30. Marguet F, Fontanilles M, Bohers E, et al. [Low-grade dural extranodal marginal zone lymphoma]. Ann Pathol. 2020;40(3):243–247. French. doi:10.1016/j.annpat.2019.12.006
31. Costa TDF, Zhuang T, Lorent J, et al. PAK4 suppresses RELB to prevent senescence-like growth arrest in breast cancer. Nat Commun. 2019;10(1):3589. doi:10.1038/s41467-019-11510-4
32. Bidwell BN, Slaney CY, Withana NP, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med. 2012;18(8):1224–1231. doi:10.1038/nm.2830
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.