Back to Archived Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 7
Identification and analysis of potential targets in Streptococcus sanguinis using computer aided protein data analysis
Authors Chowdhury Md, Bhuiyan Md, Saha A, Mosleh IM, Mondol S, Ahmed CM
Received 7 May 2014
Accepted for publication 4 August 2014
Published 25 November 2014 Volume 2014:7 Pages 45—54
DOI https://doi.org/10.2147/AABC.S67336
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Juan Fernandez-Recio
Video abstract presented by Rabiul Hossain Chowdhury.
Views: 1471
Md Rabiul Hossain Chowdhury,1 Md IqbalKaiser Bhuiyan,2 Ayan Saha,2 Ivan MHAI Mosleh,2 Sobuj Mondol,2 C M Sabbir Ahmed3
1Department of Pharmacy, University of Science and Technology Chittagong, Chittagong, Bangladesh; 2Department of Genetic Engineering and Biotechnology, University of Chittagong, Chittagong, Bangladesh; 3Biotechnology and Genetic Engineering Discipline, Khulna University, Khulna, Bangladesh
Purpose: Streptococcus sanguinis is a Gram-positive, facultative aerobic bacterium that is a member of the viridans streptococcus group. It is found in human mouths in dental plaque, which accounts for both dental cavities and bacterial endocarditis, and which entails a mortality rate of 25%. Although a range of remedial mediators have been found to control this organism, the effectiveness of agents such as penicillin, amoxicillin, trimethoprim–sulfamethoxazole, and erythromycin, was observed. The emphasis of this investigation was on finding substitute and efficient remedial approaches for the total destruction of this bacterium.
Materials and methods: In this computational study, various databases and online software were used to ascertain some specific targets of S. sanguinis. Particularly, the Kyoto Encyclopedia of Genes and Genomes databases were applied to determine human nonhomologous proteins, as well as the metabolic pathways involved with those proteins. Different software such as Phyre2, CastP, DoGSiteScorer, the Protein Function Predictor server, and STRING were utilized to evaluate the probable active drug binding site with its known function and protein–protein interaction.
Results: In this study, among 218 essential proteins of this pathogenic bacterium, 81 nonhomologous proteins were accrued, and 15 proteins that are unique in several metabolic pathways of S. sanguinis were isolated through metabolic pathway analysis. Furthermore, four essentially membrane-bound unique proteins that are involved in distinct metabolic pathways were revealed by this research. Active sites and druggable pockets of these selected proteins were investigated with bioinformatic techniques. In addition, this study also mentions the activity of those proteins, as well as their interactions with the other proteins.
Conclusion: Our findings helped to identify the type of protein to be considered as an efficient drug target. This study will pave the way for researchers to develop and discover more effective and specific therapeutic agents against S. sanguinis.
Keywords: Streptococcus sanguinis, essential proteins, unique metabolic pathways, therapeutic targets
Introduction
Streptococcus sanguinis, known earlier as Streptococcus sanguis, is a facultative, aerobic, Gram-positive, coccoid bacterium.1 It is one of the microorganisms that pioneers the colonizing process because it binds to specific receptors on the obtained pellicle of the enamel surface.2 Many species of viridans streptococcus cause endocarditis, although these bacteria rarely cause meningitis.3 An intimate association between S. sanguinis and infection has been stated, and it has been seen that the clinical presentation and outcome are very much influenced by this bacterium.4–8 S. sanguinis, moreover, is considered one of the most widespread agents of infective endocarditis among the viridans streptococci.9–11 It is a typical occupant of the human mouth and is predominantly found in dental plaque. It makes its surroundings favorable for other strains of Streptococcus, such as S. mutans, that are capable of causing cavities. An advantageous role within the oral cavity is normally played by the oral streptococci. Because of their habitation, the oral streptococci can be injected into the bloodstream through oral surgery or daily actions such as chewing and brushing.12,13 Blood-borne bacteria may perhaps colonize in the endocardium or the cardiac valves that have been weakened by congenital circumstances or degenerative developments; this could result in transferable endocarditis.14,15
Oral streptococci are continuously subjected to environmental stressors. Saliva flow and intake of food cause vacillations in nutrient supply, pH, temperature, and other environmental conditions. S. sanguinis has the aptitude to use and integrate free extracellular DNA from the environments, an attribute also known as genetic competence.16 This characteristic is shared by certain other bacteria, including more than a few oral streptococcal species.17
This study investigated the essential nonhuman homologue proteins of the unique pathways of S. sanguinis to determine their functions, their druggable pockets, the details of their metabolic pathways, and their associated proteins in other organisms.
Materials and methods
The methodical designation and delineation of the purported drug target of S. sanguinis was done strategically by the following methods.
Recognition of probable target proteins
Reclamation of indispensable proteins of S. sanguinis
In the beginning, with the help of the database of essential genes (DEG), 218 crucial genes of S. sanguinis were retrieved from the National Center for Biotechnology Information in FASTA format.18,19
Identifying human nonhomologous essential proteins in S. sanguinis
While the default constraints were kept unchanged, the retrieved 218 crucial proteins were put through BlastP at the National Center for Biotechnology Information server against Homo sapiens. To distinguish human nonhomologous essential proteins of S. sanguinis, proteins that have dissimilarities in threshold expectation values greater than 104 were considered. To do this, a protein database named Refseq was utilized.20 Thus, the human nonhomologous essential proteins of S. sanguinis were screened, as they form the bacterium’s 218 crucial proteins.
Metabolic pathway analysis of human nonhomologous essential proteins in S. sanguinis
The screened human nonhomologous proteins of S. sanguinis were then subjected to a metabolic pathway investigation; this investigation was carried out by the KEGG Automatic Annotation Server (KAAS) at the Kyoto Encyclopedia of Genes and Genomes. The functional footnotes of the genes were made available by KAAS, along with the physically pastured KEGG GENES database. The functional annotation was also carried out by this server through a BlastP comparison of the genes.21 Genes were separated according to their metabolic functions.
Exclusive metabolic pathway identification for S. sanguinis
Then, we identified those pathways of S. sanguinis that were not present in H. sapiens. Confrontation and comparisons of metabolic pathways of both S. sanguinis and H. sapiens were done by the use of the KEGG Genome Database, and incomparable metabolic pathways of S sanguinis were identified.22 Among the distinct metabolic pathways of S. sanguinis, human nonhomologue essential proteins were identified.
Target protein identification of S. sanguinis
Subcellular localization of exclusive metabolic proteins (essential nonhuman homologues) of S. sanguinis was done by PSORTb to spot the proteins that are or might be available on the surface membrane.23 Such an approach can be helpful to discover putative therapeutic targets.
Uncharacterized membrane-bound protein characterization
SVM-PORT was used for the depiction of uncharacterized membrane-bound hypothetical proteins.24
Characterization of target proteins
Three-dimensional structure development of target proteins
The three-dimensional (3D) structure for each of those four proteins was developed using Phyre2.25 Phyre2 generates homology models by detecting homologues of known 3D structures. This is called template-based homology modeling, or protein recognition. Protein Data Bank files of those proteins were also downloaded from here.
Active site prediction of target proteins
CastP, a Web-based tool, was utilized in forecasting the active sites of the sequences of interest.26 For the weighted Delaunay triangulation and the contour measurements of the alpha complex, CastP was utilized to identify and measure the surface attainable pockets, as well as accessible interior cavities of proteins and other molecules. Methodologically, it determines the volume, as well as the area of each cavity and pocket. Both the molecular surface (Connolly’s surface) and the solvent accessible surface (Richards’ surface) are calculated. For each pocket, the number of mouth openings, the circumferences of the mouth lips, and the areas of the openings – both the solvent accessible surface and the molecular surface – can be calculated with this tool.27
Druggable pocket identification
DoGSiteScorer is a Web-based tool that can provide qualitatively and quantitatively valuable data for druggability assessment.28 By the use of this tool, the druggability of a pocket can be automatically predicted on the basis of size, shape, and chemical features. Moreover, DoGSiteScorer is helpful to assess amino acid composition, functional groups, and elements that can be present on the targeted pocket. Considering all descriptors, DoGSiteScorer provides a drug score value (0–1) for a selected pocket. The higher the score, the more druggable the pocket is estimated to be.
Functional properties analysis of target proteins
A specially designed tool, the Protein Function Prediction server from Kihara Bioinformatics Laboratory (Purdue University, West Lafayette, IN, USA), was utilized for the functional analysis of these proteins.29 The amino acid sequences that were used as the input data were taken in FASTA format.
Protein–protein interaction predictions for target proteins
For conceding and prognosticating protein–protein interactions, an infallible database named STRING (Search Tools for the Retrieval of Interacting Genes/Proteins) was used.30 The association of both physical and functional traits was assembled from four sources: Genomic Context; High-throughput Experiments; (Conserved) Co-expression; and Previous Knowledge. STRING quantitatively consolidates data interaction from these sources for a large number of organisms and sends information between these organisms when it is pertinent. This database covers 5,214,234 proteins from 1,133 organisms.31
Results and discussion
The present study revealed 81 essential nonhomologous metabolic proteins, of which 17 are membrane-bound proteins and 15 are metabolic proteins in unique pathways. Of these 15 proteins, only four proteins are essential nonhuman homologous membrane-bound proteins and, therefore, can be potential drug targets. For more specification, we have also examined the active sites of these four proteins, as well as their interactions with other proteins. On the basis of these results, drugs can be synthesized to more specifically target and more effectively kill S. sanguinis without harming the host.
This study was conducted with the intention of establishing possible therapeutic targets for alternative treatments of MRSA (methicillin-resistant S. aureus) and also to classify and investigate the targets. Utilization of the subtractive genomic approach was carried out in this study. The fundamental principle of the subtractive genomic approach is “a good therapeutic target is a gene/protein essential for the bacterial survival, [but] which cannot be found in host”.32 At the beginning, indispensable proteins of S. sanguinis were recovered from the DEG. The definition of essential genes is a minimal set of genes that is sufficient for the continued existence of a cellular life form under complementary conditions.33
With an intention to avoid any cross-reactivity of the recognized drug with the human host, BlastP was performed alongside the 218 essential proteins and then the 81 nonhuman homologue proteins having a threshold expectation value greater than 104. To find out additional characteristics of the nonhuman homologous vital proteins of S. sanguinis, a metabolic pathway investigation was carried out by the KAAS server at the KEGG. The outcome was a set of 65 proteins that were involved in 37 metabolic pathways that were particularly classified into 12 classes (Table 1). With an aim to determine which of the 65 metabolic proteins were possible drug targets, a comparative study was carried out in the metabolic pathways of human host and S. sanguinis in the KEGG Genome database. This analysis revealed 15 exclusive pathways; this finding indicated that these metabolic pathways are in S. sanguinis, but not in the human host. Also, 15 proteins were found that are involved in these exclusive pathways. Four of the 15 proteins in these pathways were found in cytoplasmic membranes. We examined these four proteins for more details. Those 15 proteins were present on four dissimilar, distinctive pathways. Eight out of the 15 proteins are in the peptidoglycan biosynthesis pathway, four of them are in the photosynthesis pathway, two of them are in the D-alanine metabolism pathway, and the remaining one is in the tetracycline biosynthesis pathway. For additional categorization and forecasting, the subcellular localization of pathogenic proteins and computational characterization of hypothetical proteins were carried out, and this provided a faster way to recognize cell surface drug targets (Table 2).34
 | Table 1 Metabolic pathways of human nonhomologous proteins |
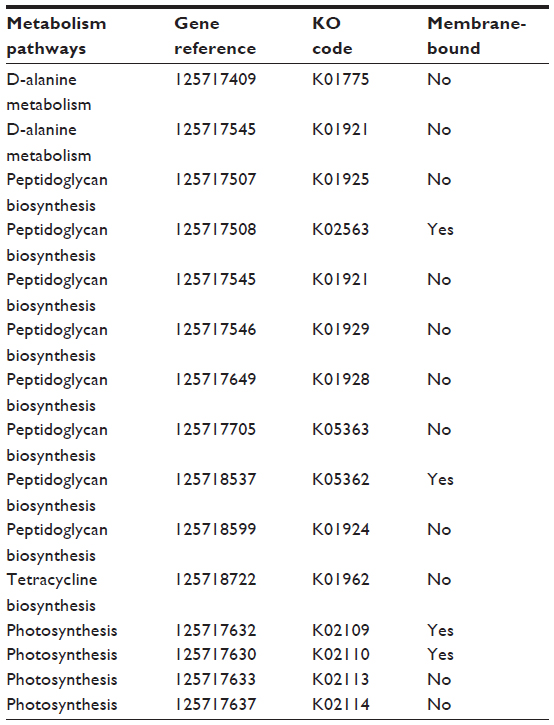 | Table 2 Unique metabolism pathways and related proteins |
Our investigation uncovered 15 essential nonhuman homologue proteins, of which four are membranous proteins. The four proteins are undecaprenyldiphospho-muramoylpentapeptide beta-N-acetylglucosaminyltransferase (gene reference 125717508), polysaccharide biosynthesis protein (gene reference 125718537), adenosine triphosphate (ATP) synthase F0F1 subunit C (gene reference 125717630), and ATP synthase F0F1 subunit B (gene reference 125717632). These four proteins can be potential targets against S. sanguinis.
We have discovered indispensable metabolic nonhuman homologue proteins in the exclusive metabolism pathways. Both in vitro and in vivo studies indicate that inhibitors that are designed for targeting these proteins have the potential to efficiently hold back bacterial growth by passing the bacterial mutation site for drug confrontation. The reason behind such activity is that almost all existing antibiotics are able to target just four pathways (cell wall synthesis, protein synthesis, nucleic acid synthesis, and folate synthesis).35 Thus, such frequent contact with the same site amplifies the possibility of mutation to the site; this results in mutant bacteria.35,36 Targets among these pathways have revealed their potential for antibacterial agents (for example, oligomycin, antimycin A), but are not utilized in human cases because of the unsafe outcomes for both bacteria and humans. However, the proteins that are revealed in this study are human nonhomologues and, therefore, can be used as drug targets to reduce the baneful consequences of the drug for the human host.36
Membrane proteins received particular consideration because among all the drug targets, 60% are membrane proteins. Membrane proteins are also easier to explore by means of computational techniques rather than experimental methods because of their standardized interactions and structures. Proteins have a high tendency to assemble secondary structures consecutively, which lowers the difficulty of predicting the protein structure by computational means. If an elevated resolution structure is not established, computer-based structure forecasting can be done easily with membrane proteins, and the prediction of a structure-based drug can be carried out.37
The 3D structure of undecaprenyldiphospho-muramoylpentapeptide beta-N- acetylglucosaminyltransferase was developed based on the d1f0ka template. Here, 96% of the sequence was covered with 100% confidence (Figure 1A). In the case of the polysaccharide biosynthesis protein, coverage was 80%, but the confidence level was 100%. Here, the c41z9A template was used (Figure 1B). However, the confidence level was much lower in the case of ATP synthase F0F1 subunit B. The c1b9uA template was used here as a base (Figure 1C). The last target protein developed was based on c1wu0A, and the confidence level and coverage were 99.9% and 98%, respectively (Figure 1D).
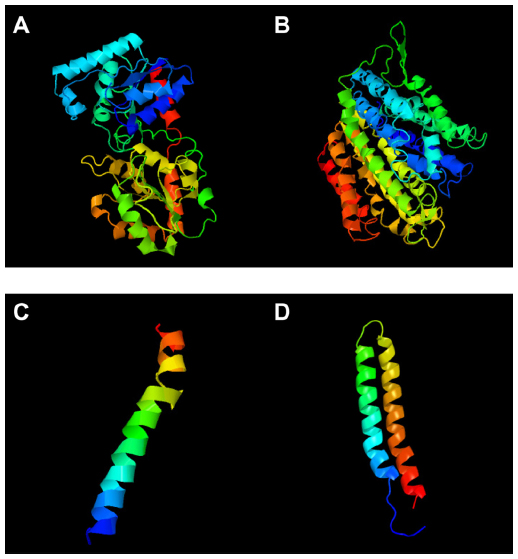 | Figure 1 3D structures. |
In this study, we have also evaluated the best active site area of the aforementioned four experimental proteins, along with the number of amino acids involved with that site.
Undecaprenyldiphospho-muramoylpentapeptide beta-N- acetylglucosaminyltransferase is a human nonhomologous essential protein involved in a unique metabolism pathway named peptidoglycan biosynthesis. This can be found on the inner membrane, the organelle membrane, or the cell membrane. This enzyme is responsible for some biological functions, such as apocarotenoid metabolism, cell wall synthesis, regulation of gene expression via a specific molecular function such as transferase activity, binding, and undecaprenyldiphospho-muramoylpentapeptide beta-N-acetylglucosaminyltransferase activity (Table 3 and Figure 2A).8
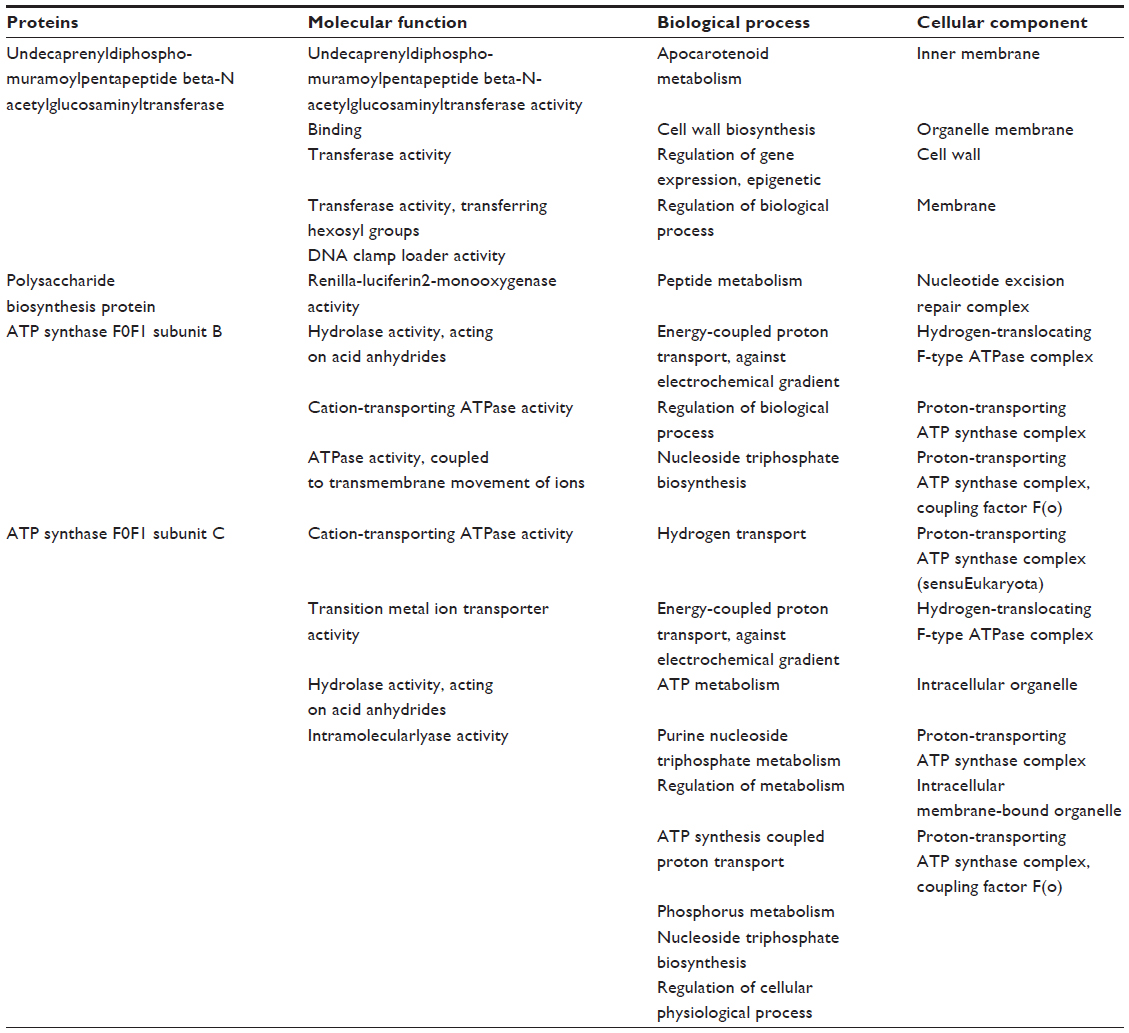 | Table 3 Molecular function, biological process, and cellular components of selected proteins |
 | Figure 2 Active site information by CASTp. |
In S. sanguinis SK36, the polysaccharide biosynthesis protein is also a human nonhomologous protein that plays an identical role in peptidoglycan biosynthesis. Its cellular components include the nucleotide excision repair complex. Besides the biological function of peptidoglycan, the protein shows renilla-luciferin 2-monooxygenase activity as a molecular function (Table 3 and Figure 2B).
ATP synthase F0F1 subunit B is a common enzyme found in various species. This enzyme also shows unique activity in S. sanguinis SK36. Similar to ATP synthase F0F1 subunit C, ATP synthase F0F1 subunit B is also involved in photosynthesis. The hydrogen-translocating F-type ATPase complex and proton-transporting ATP synthase complex are some examples of its cellular components. Nucleoside triphosphate biosynthesis, regulation of biological processes, and energy-coupled proton transport against electrochemical gradients are the biological activities of this subunit. The ATP synthase F0F1 subunit B also performs some molecular functions, such as hydrolase activity, by acting on acid anhydridescation-transporting ATPase (Table 3 and Figure 2C).
ATP synthase F0F1 subunit C is also a membranous protein responsible for photosynthesis. This enzyme is a component of the proton-transporting ATP synthase complex (sensu Eukaryota), and intracellular organelle proton-transporting ATP synthase complex. ATP synthase F0F1 subunit C is responsible for hydrogen transport, ATP metabolism, phosphorus metabolism, and purine nucleoside triphosphate metabolism. ATP synthase F0F1 subunit C is responsible for specific molecular functions such as cation-transporting ATPase activity, transition metal ion transporter activity, intramolecular lyase activity, hydrolase activity, and acting on acid anhydrides (Table 3 and Figure 2D).
Pockets are the surface concavities of protein where a substrate might bind. The prediction of protein druggability has been a widely discussed topic in the pharmaceutical community over the past few decades. The pharmaceutical industry has an urgent need to prioritize suitable drug targets because of the high attrition rate and failure rates in drug discovery and development. In our study, we have already discussed four targeted proteins for S. sanguinis drug targets. These proteins have been further investigated to determine the specific pockets that are best able to bind with a drug. Using DoGSiteScorer, we uncovered 13 pockets from the undecaprenyldiphospho-muramoylpentapeptide beta-N-acetylglucosaminyltransferase protein. Furthermore, the polysaccharide biosynthesis protein has 12 pockets, and ATP synthase F0F1 subunit B and ATP synthase F0F1 subunit C have one pocket each. Among these pockets, we have selected four pockets that have the highest druggability (Figure 3). To determine druggability, we have taken pocket volume, surface area, liposurface area, and depth into consideration (Table 4). According to our inspection, undecaprenyldiphospho-muramoylpentapeptide beta-N-acetylglucosaminyltransferase protein has two pockets with druggability scores of 0.87 each. Similarly, the polysaccharide biosynthesis protein has two pockets whose druggability scores are 0.86 and 0.85.
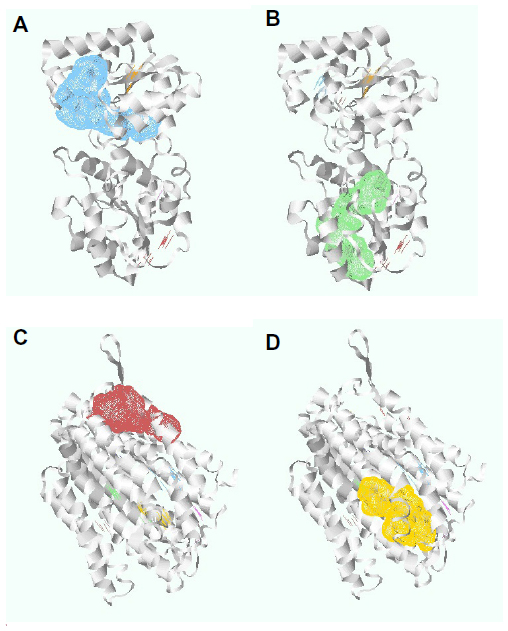 | Figure 3 Undecaprenyldiphospho-muramoylpentapeptide beta-N-acetylglucosaminyltransferase protein pockets 1 and 2, and polysaccharide biosynthesis protein pockets 6 and 7. |
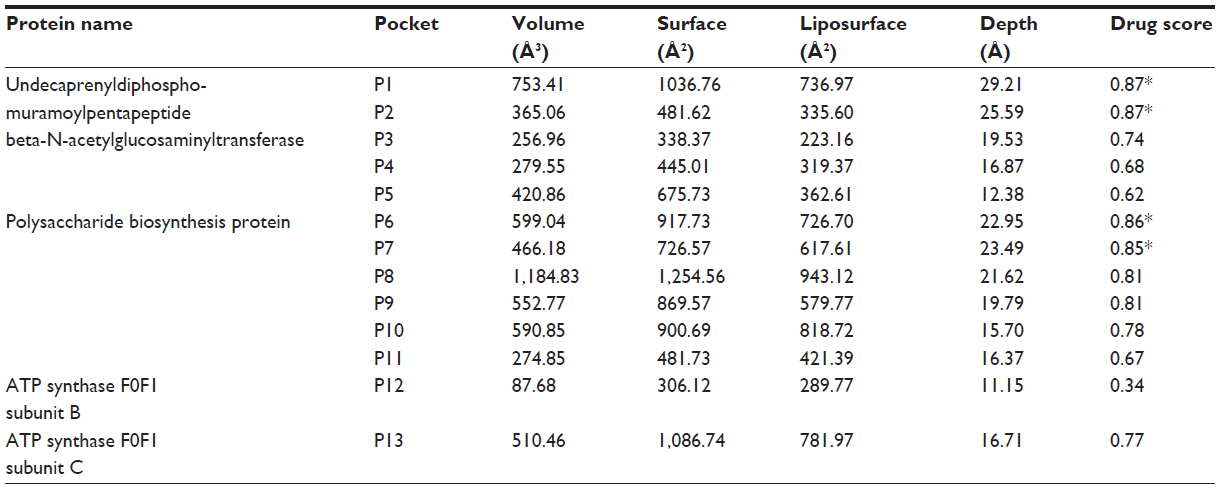 | Table 4 Identified drug targetable pockets of selected proteins |
There are still no gold standards to individualize binding pocket properties, although many studies have showed different parameters. For instance, Volkamer et al38 have shown that large pocket volume, high depth, and a high apolar amino acid ratio are effective parameters to determine druggability. Pocket volumes of selected pockets, namely P1, P2, P6, and P7, are 753.41, 365.06, 599.04, and 466.18, respectively, and the depths of the pockets are more than 20 Å (Table 4). Again, a high apolar amino acid ratio also suggests that those pockets could be potential drug targets (Table 5).
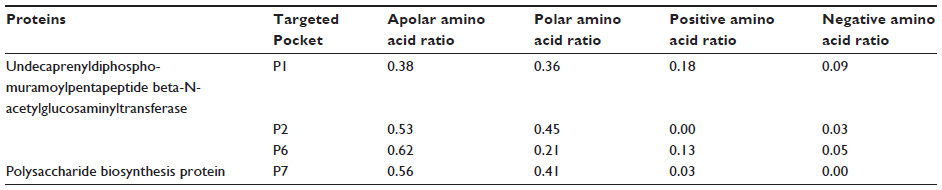 | Table 5 Amino acid composition of selected pockets |
Today, in the postgenomic period, computational analysis of the complex networks formed by interactions between proteins are one of the major challenges. Protein–protein interaction databases are useful to explore biological pathways and networks in cells. We also studied the protein–protein interactions of the selected proteins by STRING. The protein undecaprenyldiphospho-muramoylpentapeptide beta-N-acetylglucosaminyltransferase (ID gi |125717508) was found to interact with cell division protein FtsA; putative, phospho-N-acetylmuramoyl-pentapeptide-transferase, cell division protein FtsW; and putative, undecaprenyl diphospho (UDP)-N-acetylmuramoyl-L-alanyl-D-glutamate synthetase. Another protein of peptidoglycan biosynthesis, polysaccharide biosynthesis protein (ID gi |125718537), was seen to interact with UDP-N-acetylmuramoyl-L-alanyl-D-glutamate–L-lysine ligase, cation efflux family protein, L-cysteine desulfhydrase, and cystathionine gamma-synthase. ATP synthase F0F1 subunit B, (ID gi |125717632) can interact with F0F1 ATP synthase subunit A, F0F1 ATP synthase subunit epsilon, F0F1 ATP synthase subunit gamma, and proton-translocating ATPase. The remaining protein, ATP synthase F0F1 subunit C (ID gi |125717630), was also found to interact with F0F1 ATP synthase subunit B, adenylate kinase, F0F1 ATP synthase subunit A, and putative manganese-dependent inorganic pyrophosphatase (Table 6). Thus, these target proteins have associations and functions in different and important metabolic processes of S. sanguinis. Therefore, the outcomes of this study will expose a new horizon in the treatment against S. sanguinis.
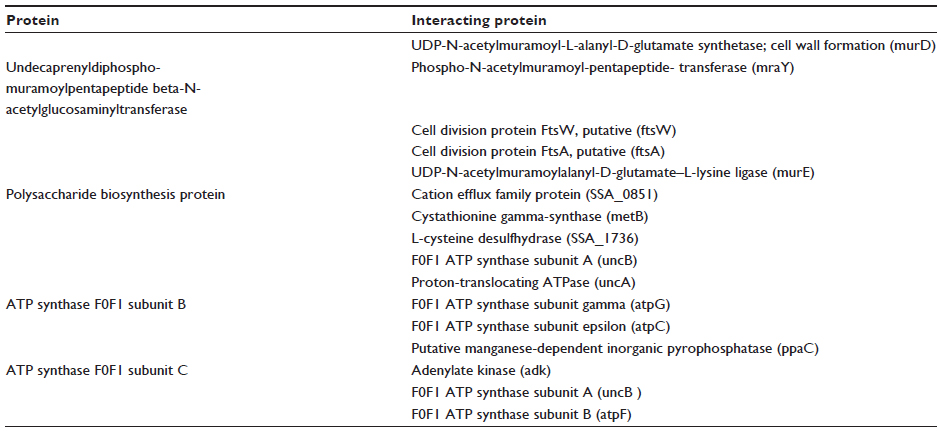 | Table 6 Interacting proteins with target proteins |
Conclusion
This study focuses on several sections of the S. sanguinis genome to reveal essential nonhomologous metabolic membrane-bound proteins as potential drug targets. Molecular modeling and analysis of the targets helped to uncover the best possible active sites that can be targeted for drug design. This pathogen-specific drug will not be lethal to the host because a subtractive genomic approach was applied in this study. Computational screening against these novel targets will be beneficial in the discovery of potential therapeutic agents against S. sanguinis.
Acknowledgments
Mr Adnan Mannan, Assistant Professor, Department of Genetic Engineering and Biotechnology, University of Chittagong, Chittagong-4331, Bangladesh.
Author contributions
Md Rabiul Hossain Chowdhury: data collection; data analysis; literature review. Md IqbalKaiser Bhuiyan: data collection; data interpretation. Ayan Saha: research design; data analysis; literature review; manuscript correction. Ivan MHAI Mosleh: table and figure preparation; manuscript writing. Sobuj Mondol: manuscript writing. C M Sabbir Ahmed: software selection; data collection; literature review. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Paik S, Senty L, Das S, Noe JC, Munro CL, Kitten T. Identification of virulence determinants for endocarditis in Streptococcus sanguinis by signature-tagged mutagenesis. Infect Immun. 2005;73(9):6064–6074. | |
Kolenbrander PE, Palmer RJ Jr, Rickard AH, Jakubovics NS, Chalmers NI, Diaz PI. Bacterial interactions and successions during plaque development. Periodontol 2000. 2006;42:47–79. | |
Koneman EW, Allen SD, Janda WM, Schreckenberger PC, Winn WC. Color Atlas and Textbook of Diagnostic Microbiology. 5th ed. New York, NY: Lippincott; 1997:577–649. | |
Cabellos C, Viladrich PF, Corredoira J, Verdaguer R, Ariza J, Gudiol F. Streptococcal meningitis in adult patients: current epidemiology and clinical spectrum. Clin Infect Dis. 1999;28(5):1104–1108. | |
Kuramitsu HK, He X, Lux R, Anderson MH, Shi W. Interspecies interactions within oral microbial communities. Microbiol Mol Biol Rev. 2007;71(4):653–670. | |
Nyvad B, Kilian M. Microbiology of the early colonization of human enamel and root surfaces in vivo. Scand J Dent Res. 1987;95(5):369–380. | |
Rosan B, Lamont RJ. Dental plaque formation. Microbes Infect. 2000; 2(13):1599–1607. | |
Xu P, Alves JM, Kitten T, et al. Genome of the opportunistic pathogen Streptococcus sanguinis. J Bacteriol. 2007;189(8):3166–3175. | |
Douglas CW, Heath J, Hampton KK, Preston FE. Identity of viridans streptococci isolated from cases of infective endocarditis. J Med Microbiol. 1993;39(3):179–182. | |
Mylonakis E, Calderwood SB. Infective endocarditis in adults. N Engl J Med. 2001;345(18):1318–1330. | |
Tleyjeh IM, Steckelberg JM, Murad HS, et al. Temporal trends in infective endocarditis: a population-based study in Olmsted County, Minnesota. JAMA. 2005;293(24):3022–3028. | |
Forner L, Larsen T, Kilian M, Holmstrup P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J Clin Periodontol. 2006;33(6):401–407. | |
Guntheroth WG. How important are dental procedures as a cause of infective endocarditis? Am J Cardiol. 1984;54(7):797–801. | |
Moreillon P, Que YA. Infective endocarditis. Lancet. 2004;363(9403):139–149. | |
Bashore TM, Cabell C, Fowler JV. Update on infective endocarditis. Curr Probl Cardiol. 2006;31(4):274–352. | |
Håvarstein LS, Hakenbeck R, Gaustad P. Natural competence in the genus Streptococcus: evidence that streptococci can change pherotype by interspecies recombinational exchanges. J Bacteriol. 1997;179(21):6589–6594. | |
Martin B, Quentin Y, Fichant G, Claverys JP. Independent evolution of competence regulatory cascades in streptococci? Trends Microbiol. 2006;14(8):339–345. | |
Zhang R, Lin Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res. 2009;37(Database issue):D455–D458. | |
National Center for Biotechnology Information [webpage on the Internet]. Welcome to NCBI. Bethesda, MD: National Center for Biotechnology Information, US National Library of Medicine; 2013. Available from: http://www.ncbi.nlm.nih.gov. Accessed August 10, 2013. | |
Kerfeld CA, Scott KM. Using BLAST to teach “E-value-tionary” concepts. PLoS Biol. 2011;9(2):e1001014. | |
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35(Web Server issue):W182–W185. | |
Hossain M, Chowdhury DU, Farhana J, et al. Identification of potential targets in Staphylococcus aureus N315 using computer aided protein data analysis. Bioinformation. 2013;9(4):187–192. | |
Nancy YY, Wagner JR, Laird MR, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26(13):1608–1615. | |
Cai CZ, Han LY, Ji ZL, Chen X, Chen YZ. SVM-Prot: Web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 2003;31(13):3692–3697. | |
Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009; 4(3):363–371. | |
Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Web Server Issue):W116–W118. | |
Panda S, Chandra G. Physicochemical characterization and functional analysis of some snake venom toxin proteins and related non-toxin proteins of other chordates. Bioinformation. 2012;8(18):891–896. | |
Volkamer A, Kuhn D, Grombacher T, Rippmann F, Rarey M. Combining global and local measures for structure-based druggability predictions. J Chem Inf Model. 2012;52(2):360–372. | |
Hawkins T, Luban S, Kihara D. Enhanced automated function prediction using distantly related sequences and contextual association by PFP. Protein Sci. 2006;15(6):1550–1556. | |
Franceschini A, Szklarczyk D, Frankild S, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(Database issue):D808–D815. | |
Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–D568. | |
Allsop AE, Brooks G, Bruton G, et al. Penem inhibitors of bacterial signal peptidase. Bioorg Med Chem Lett. 1995;5(2):443–448. | |
Gardy JL, Brinkman FS. Methods for predicting bacterial protein subcellular localization. Nat Rev Microbiol. 2006;4(10):741–751. | |
Freiberg C, Pohlmann J, Nell PG, et al. Novel bacterial acetyl coenzyme A carboxylase inhibitors with antibiotic efficacy in vivo. Antimicrob Agents Chemother. 2006;50(8):2707–2712. | |
Stermitz FR, Lorenz P, Tawara JN, Zenewicz LA, Lewis K. Synergy in a medicinal plant: antimicrobial action of berberine potentiated by 5′-methoxyhydnocarpin, a multidrug pump inhibitor. Proc Natl Acad Sci U S A. 2000;97(4):1433–1437. | |
Kagawa Y, Racker E. Partial resolution of the enzymes catalyzing oxidative phosphorylation. 8. Properties of a factor conferring oligomycin sensitivity on mitochondrial adenosine triphosphatase. J Biol Chem. 1966;241(10):2461–2466. | |
Arinaminpathy Y, Khurana E, Engelman DM, Gerstein MB. Computational analysis of membrane proteins: the largest class of drug targets. Drug Discov Today. 2009;14(23–24):1130–1135. | |
Volkamer A, Kuhn D, Grombacher T, Rippmann F, Rarey M. Combining global and local measures for structure-based druggability predictions. J Chem Inf Model. 2012;52(2):360–372. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.