")
Back to Journals » Drug Design, Development and Therapy » Volume 17
Icariin Induces Triple-Negative Breast Cancer Cell Apoptosis and Suppresses Invasion by Inhibiting the JNK/c-Jun Signaling Pathway
Authors Gao S, Zhang X, Liu J, Ji F, Zhang Z, Meng Q, Zhang Q, Han X, Wu H, Yin Y, Lv Y, Shi W
Received 5 December 2022
Accepted for publication 4 March 2023
Published 20 March 2023 Volume 2023:17 Pages 821—836
DOI https://doi.org/10.2147/DDDT.S398887
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Shenghan Gao,1,2 Xinyu Zhang,1,2 Jie Liu,3 Fuqing Ji,2 Zhihao Zhang,2 Qingjie Meng,2 Qi Zhang,1 Xiaogang Han,2 He Wu,2 Yulong Yin,2 Yonggang Lv,2 Wenzhen Shi3
1The College of Life Sciences, Northwest University, Xi’an, 710069 People’s Republic of China; 2Department of Thyroid Breast Surgery, Xi’an NO.3 Hospital, the Affiliated Hospital of Northwest University, Xi’an, People’s Republic of China; 3Clinical Medical Center, Xi’an NO.3 Hospital, the Affiliated Hospital of Northwest University, Xi’an, People’s Republic of China
Correspondence: Yonggang Lv, Department of Thyroid Breast Surgery, Xi’an NO.3 Hospital, the Affiliated Hospital of Northwest University, Xi’an, People’s Republic of China, Email [email protected] Wenzhen Shi, Xi’an Key Laboratory of Cardiovascular and Cerebrovascular Diseases, Xi’an No.3 Hospital, The Affiliated Hospital of Northwest University, School of Life Sciences and Medicine, Northwest University, Xi’an, People’s Republic of China, Tel +8615037916770, Email [email protected]
Background: Breast cancer is a common cancer worldwide. Triple-negative breast cancer (TNBC) is an aggressive form of breast cancer characterized by a poor prognosis. Icariin (ICA) is a flavonoid glycoside purified from the natural product Epimedium, which is reported to exert an inhibitory effect on a variety of cancers. However, molecular mechanisms behind ICA suppressed TNBC remain elusive.
Methods: The curative effects of ICA on TNBC cells and potential targets were predicted by network pharmacology and molecular biology methods screening, and the mechanism of inhibition was explained through in vitro experiments such as cell function determination, Western blot analysis, molecular docking verification, etc.
Results: This study showed that ICA inhibits TNBC cell functions such as proliferation, migration, and invasion in a dose-dependent manner. ICA could induce redox‐induced apoptosis in TNBC cell, as shown by ROS upregulation. As a result of network pharmacology, ICA was predicted to be able to inhibit the MAPK signaling pathway. ICA treatment inhibited the expression of JNK and c-Jun and downregulated the antiapoptotic gene cIAP-2. Our results suggested that ICA could induce apoptosis by inducing an excessive accumulation of ROS in cells and suppress TNBC cell invasion via the JNK/c-Jun signaling pathway.
Conclusion: We demonstrated that ICA can effectively inhibit cell proliferation and induced apoptosis of TNBC cells. In addition, ICA could inhibit TNBC cell invasion through the JNK/c-Jun signaling pathway. The above suggests that ICA may become a potential drug for TNBC.
Keywords: triple-negative breast cancer, icariin, JNK/c-Jun, apoptosis, ROS, invasion
A Letter to the Editor has been published for this article.
A Response to Letter by Dr Mariya has been published for this article.
Introduction
Female breast cancer is the most common cancer and the leading cause of cancer death in women in 2020.1 Triple-negative breast cancer (TNBC) refers to a collection of diverse and heterogeneous diseases, which lack estrogen receptor (ER), progesterone receptor (PR) and HER2 expression in comparison to other breast cancer subtypes.2 TNBC is an aggressive cancer characterized by a poor prognosis.3 TNBC is not sensitive to endocrine therapy or molecular targeted therapy.4 Relapsed or refractory TNBC patients cannot receive standard chemotherapy, the treatment responses usually last a short time and then cancer relapses rapidly.5 Many side effects brought about by systemic chemotherapy make patients miserable. Therefore, natural medicine with fewer side effects is essential for alternative or complementary chemotherapies for TNBC.
Natural medicine extracts induce apoptosis of tumor cells and have a powerful cancer-fighting effect.6 In the past, several natural medicine molecules have been used successfully as anticancer agents, such as paclitaxel, camptothecin, and vincristine.7 However, further discovery of new cancer treatment molecules of natural origin is necessary.
Icariin (ICA), a flavonoid derived from Epimedium, exerts anti-inflammatory,8 antioxidant, and neuroprotective effects.9,10 ICA inhibited the growth of cancer cells, such as ovarian cancer,11 cervical cancer,12 gastric cancer,13 lung cancer,14 and breast cancer.15 ICA has been used clinically as an anticancer agent against a broad range of cancers.7 For instance, ICA induces mouse melanoma B16 cell cycle arrest via Erk1/2-p38-JNK-dependent signaling pathway.16 Moreover, ICA induces apoptosis via ROS/JNK pathway in hepatoma cells.17 However, this mechanism is rarely reported in breast cancer.
Network pharmacology can comprehensively and systematically study the action mechanism of drugs that can be developed in natural products. In recent years, this method has been used to help scientists predict the mechanisms underlying actions of drugs and visualize the relationships among drugs, diseases, and targets.18 Molecular docking tool can verify the accuracy of drug molecules and predicted targets binding.19 This method has a high computational power and maybe useful for characterizing multi-target drugs in diseases and can also promote the experimental design and verify the role of ICA in TNBC in a more effective way. Therefore, this study first investigates the potential targets of ICA in treating TNBC through this method, and the flow chart is shown in Figure 1.
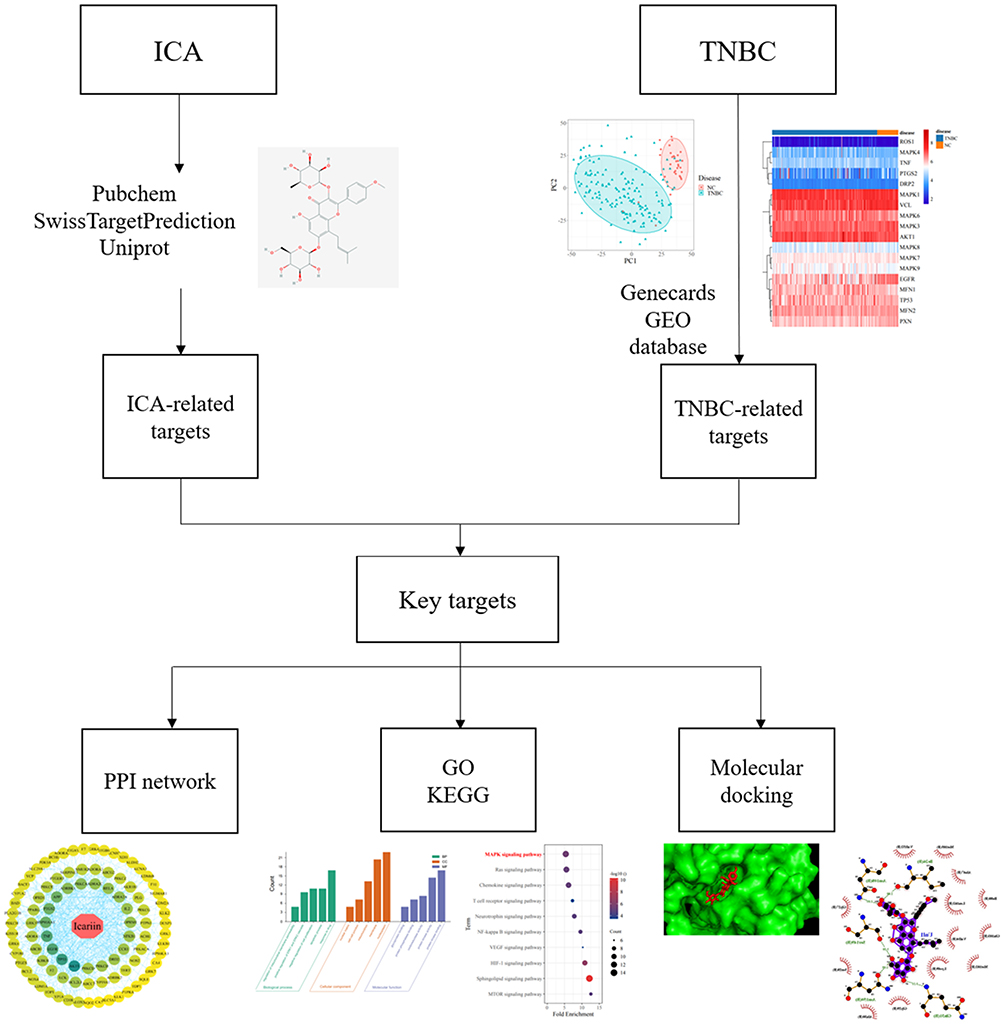 |
Figure 1 The flowchart of Network pharmacology in this study. |
As a result of network pharmacology, ICA was predicted to be able to inhibit the MAPK signaling pathway. JNK as a subfamily of MAPK regulates cell proliferation, apoptosis, and tumor invasion.20 Reactive oxygen species (ROS) relate to multiple signaling pathways affecting cell growth and death.21 It has been reported that ROS accumulation induced colorectal cancer cell apoptosis by activating the JNK/c-Jun signaling pathway.22 Han et al demonstrated that STC-1 induced TNBC cell invasion through the JNK/c-Jun pathway.23 Therefore, excessive accumulation of ROS in cells could be an available treatment strategy for cancer cell, including TNBC cell. Moreover, JNK is also the downstream effector of ROS, which can induce cell apoptosis by affecting mitochondria.24,25 Therefore, whether ICA can treat TNBC through the above mechanisms has aroused our interest.
We first investigated the potential treated targets and mechanisms of ICA acts on TNBC using network pharmacology analysis, and then we conducted in vitro experiments to study the inhibitory activities of ICA on various functions of TNBC cells. In terms of the mechanism, by activating the JNK pathway, we found that ICA inhibited TNBC cell invasion through the JNK/c-Jun pathway. Together, our studies indicate that ICA may be a potential drug for effective treatment of TNBC, and regulating targeted JNK/c-Jun pathway might be therapeutic against TNBC.
Methods and Materials
Screening of the Structure and Potential Targets of ICA
The compound structure of ICA compounds was retrieved from the PubChem database using the keyword “icariin.” For the purpose of obtaining icariin-related predictive target information, the chemical structure of SIN compound was uploaded into the SwissTargetPrediction database. Predictive target information was entered into the UniProt database for retrieval and screening to eliminate duplicate and nonstandard targets. After screening and transformation, the potential therapeutic targets were finally obtained.
Determination of TNBC-Related Targets
The TNBC-associated targets were obtained from the GeneCards database (https://www.genecards.org/). Due to this database and its content being public and users can download data for free for publishing articles, this study would have the need for ethics approval waived by the ethics committee of Xi’an NO.3 Hospital, the Affiliated Hospital of Northwest University. Search for relevant targets in the database by using the keyword “triple negative breast cancer.” The online intersection analysis Venn diagram webtool intersected drug treatment targets with disease-related targets. In the treatment of TNBC, common targets are considered potential targets of ICA.
Constructing the Protein–Protein Interaction Network
The interaction database STRING platform studied the interaction between potential ICA-related target in the treatment of TNBC. The information was uploaded into the STRING database and a protein–protein interaction (PPI) network was built. The relevant targets in TNBC-related proteins were analyzed by Cytoscape software 3.7.1 to visualize and analyze PPI networks.
Gene Ontology Functional Analysis
Gene Ontology (GO) is a powerful bioinformatics resource cataloguing the functions of genes including biological process (BP), cellular component (CC), and molecular function (MF).26 The DAVID database has a comprehensive biological annotation function for large-scale gene and protein lists to help researchers collect biological information from them. The GO enrichment analysis was performed by entering the gene name of the intersection gene to be enriched into the DAVID database. The “Homo” species option was selected after submission.
The KEGG Enrichment Analysis
The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis can provide more information about biological functions related to genes.27 The gene name of the intersection gene to be enriched was added into the DAVID database and the “Homo” species option was selected after submission to perform KEGG enrichment analysis. Using P < 0.05 as the screening criterion, the potential pathway of ICA in the treatment of TNBC was screened.
The GEO Analysis
Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo) is a public resource involving extensive high-throughput gene expression and other functional genomic datasets submitted by global research institutions.28 This study from public resources, the content of which is open and can be reused without restriction, this study would have the need for ethics approval waived (Ethics Committee of Xi’an NO.3 Hospital, the Affiliated Hospital of Northwest University). The TNBC dataset was searched in the GEO database for “triple-negative breast cancer” as a keyword, and the direct expression of core targets in the PPI network in the normal and disease groups was verified.
Molecular Docking Approach
The structure of proteins was downloaded from the Protein Data Bank database (PDB), and protein structure files and drug structure files were preprocessed using PyMOL 2.2.0. After treatment, AutoDock tool was used to dock Jun protein with ICA and SP600125 molecules, and then drew the binding diagram to visually verify the interaction between protein and molecules.
Drugs and Reagents
Icariin (purity >98%) was purchased from Meilunbio (Dalian, PRC), a stocked solution of 75 mM in 100% dimethyl sulfoxide (DMSO) was prepared, stored at −80°C and diluted with medium for later use. A medium containing 0.1% DMSO was used as control. Cell Counting Kit-8 (CCK-8) was bought from Mishushengwu (Xian, PRC). DCFH-DA was purchased from Njjcbio (Nanjing, PRC). A Count & Viability Kit (MCH100102) and Annexin V & Dead Cell Kit (MCH100105) were purchased from Luminex (Austin, US). Anisomycin was purchased from Beyotime (Shanghai, PRC). Anti-SAPK/JNK antibodies (#9252), anti-phospho-SAPK/JNK antibodies (#4668), and anti-c-IAP2 antibodies (#3110) were obtained from Cell Signaling Technology Co., Ltd. (Danvers, US). Anti-E-cadherin antibodies, anti-p-c-Jun antibodies, and anti-β-actin antibodies were acquired from ABclonal (Wuhan, PRC).
Cell Culture
The Hs 578T, MDA-MB-468, and MCF-10A cell lines were obtained from Procell Life Science & Technology Co., Ltd. (Lot No. CL-0290, CL-0114, CL −0525, Wuhan, PRC) and cultured in cell exclusive culture medium, including DMEM+0.01mg/mL Insulin+10% FBS+1% P/S (Lot No. CM-0114), DMEM+10% FBS+1% P/S (Lot No. CM-0290B), DMEM+5% HS+20ng/mL EGF+0.5μg/mL Hydrocortisone+10μg/mL Insulin+1% NEAA+1% P/S (Lot No. CM-0525) in 5% CO2 at 37°C.
Cell Proliferation Assay
The CCK-8 assay evaluated cell proliferation. Hs 578T, MDA-MB-468, and MCF-10A cells were initially plated at a density of 5×103 cells in 96-well culture plates and cultured for more than 4 h for cell adhesion. Then, cells were treated with ICA of low, medium, and high concentrations (12.5, 25, and 50 μM). CCK-8 solution (10 μL) was added in each well after removing the drug (24–48 h after treatment) and the cells were incubated for 3 h at 37°C. The optical density (OD) was then measured at 450 nm by a microplate reader (Bio-Rad) to evaluate the change of cell proliferation. The test was repeated 3 times.
The action of ICA treatment on TNBC cell proliferation was evaluated by the 5-ethynyl-2′-deoxyuridine (EdU) staining assay. Hs 578T and MDA-MB-468 cells were plated at a density of at 3×104 cells/mL in 24-well culture plates. After starvation at 37°C overnight, ICA (12.5, 25, or 50 μM) was administered to cells for 24 h. Then, cells were incubated with EdU solution (10 μM) for another 2 h. Paraformaldehyde 4% fixed the samples for 15 min. After washing three times with PBS containing 3% BSA, samples were treated with PBS having 0.3% Triton X-100 for 15 min and further washed two times with PBS. Each sample was incubated with Click Additive Solution for 30 min in a dark area, washed three times with PBS, and then stained with Hoechst for 10 min, followed by further three washing with PBS washes in a dark area. Finally, EdU-labelled cells were visualized under a fluorescence microscope. The ImageJ software was used to quantify EdU-labelled cells.
Transwell Migration and Invasion Assay
The upper and lower chambers of 24-well culture plates were separated with 8-mm pore size inserts to evaluate cell migration. 100 μL suspension containing 2×104 cells was seeded into the upper chamber, and DMEM (600 μL) with 30% FBS was added to each chamber. After 4 h for cell adhesion, different concentrations of ICA were added to the upper chamber. After 24 h at 37°C, paraformaldehyde 4% was added into each well for 30 min to fix the invasive cells at the lower part of the filter, then non-migrated and noninvasive cells were removed from the upper surface of the filter. 0.1% crystal violet was used to stain cells that were then imaged with a microscope (Leica).
Wound Healing Assay
Hs 5787T and MDA-MB-468 cells were seeded in 24-well plates and treated with ICA for 24 h when they had reached 90% confluence. A 20 μL pipette tip was used to scratch cells that were then cultured in a medium. The motility of cells was observed using the Cell Imaging Multi-Mode Reader (Cytation 1) at baseline, 12, 36, 48, and 72 h.
Cell Apoptosis Assay
Low, medium, and high concentrations of ICA were administered to Hs 578T and MDA-MB-468 cells for 24 h. Then, cells were stained with Annexin V-FITC/PI and quantified with Guava Muse Cell Analyzer. Apoptotic cells contained Annexin V/PI.
Cell Viability Assay
A density of 5×105 cells per well was used to seed Hs 578T and MDA-MB-468 cells into 6-well plates. After starvation at 37°C overnight, cells were treated with ICA (12.5, 25, or 50 μM) for 24 h at 37°C, and then Count & Viability reagent (Luminex) and cell suspension were added to each tube and measured by a Muse Cell Analyzer.
Detection of Total ROS
Hs 578T and MDA-MB-468 cells were seeded in 24-well plates and treated with ICA for 24 h when they had reached 80% confluence. To prepare a working solution, dilute the 10 mM DCFH-DA solution with PBS immediately before adding it to the wells. Cells were incubated at 37°C for 30 min followed the removal of the drug-containing, followed by the addition to each well of 100 μL DCFH-DA working solution. For each well, 500 μL PBS was added after washing. Representative fluorescence images were taken using the green fluorescent protein (GFP) channel on Cell Imaging Multi-Mode Reader (Cytation 1).
Western Blot Analysis
MDA-MB-468 and Hs 578T cells from each group were lysed in RIPA buffer. The proteins were loaded onto a 10% SDS-PAGE gel and transferred to membranes after gel electrophoresis, with the membranes blocked with 5% nonfat dried milk. Then, proteins were incubated with primary antibodies overnight at 4°C and later with secondary antibodies for 2 h at room temperature.
Statistical Analysis
The GraphPad Prism software (version 5.0) was used for statistical analysis. The statistical significance was assessed by one-way ANOVA followed by Tukey’s post-hoc test. A p-value <0.01 was considered significant.
Results
Potential ICA-Related Targets in TNBC Treatment
The 2D structure of ICA (Figure 2A) was retrieved from the PubChem database, which proved that it is similar to the drugs that can be used as drug molecule for further analysis. 4913 TNBC-related targets were acquired from the GeneCards database, and 100 ICA drug treatment targets were predicted from the SwissTargetPrediction database. The key targets were obtained by crossing the ICA-related targets with the TNBC genes and the Venn diagram was drawn (Figure 2B). Potential targets were submitted to the STRING database and the PPI network was constructed. The relevant findings were imported in the Cytoscape software. The result showed that the top four potential targets were found to be AKT 1, TP 53, EGFR, and TNF in the network (Figure 2C).
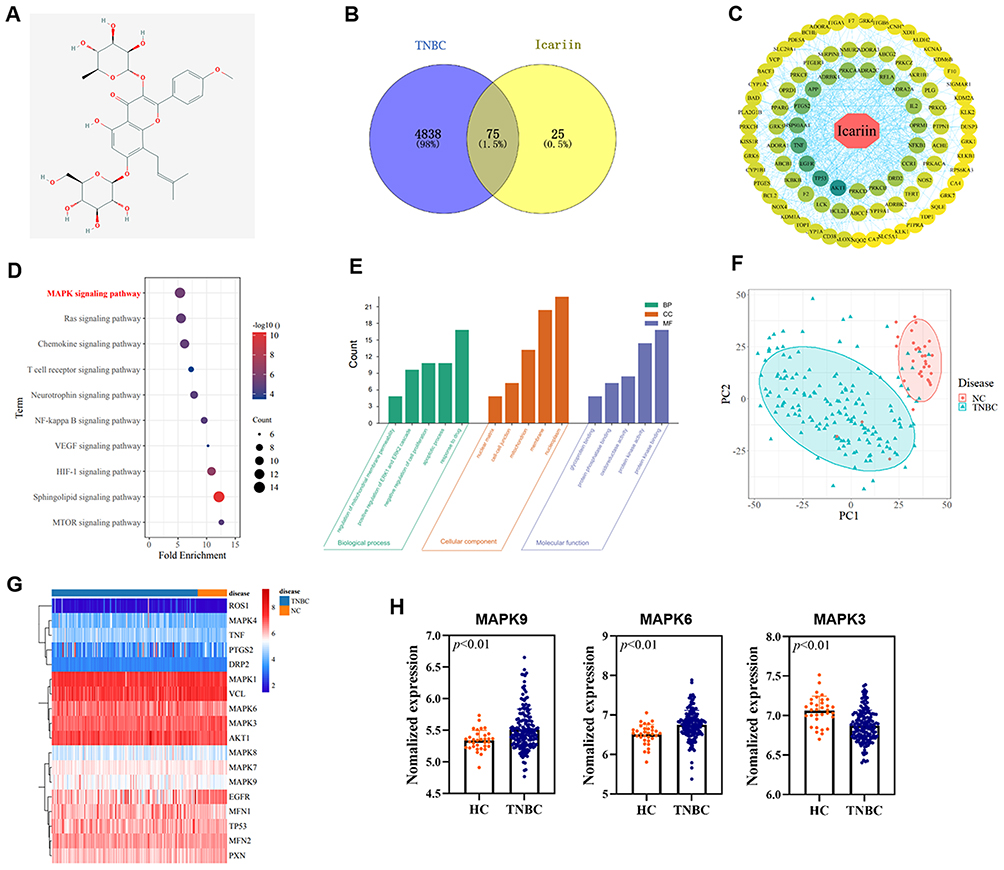 |
Figure 2 (A) The 2D structure of ICA. (B) The Venn diagram reports the intersection of targets in TNBC and ICA. (C) The PPI network with total nodes of ICA. Circular nodes represent targets and rhombic nodes represent ICA. Edges represent interactions between ICA and targets. (D) The KEGG enrichment pathway analysis. (E) The GO enrichment analysis of key targets. (F) Principal component analysis (PCA) plot of stromal molecular signature of TNBC patients tissue samples and normal breast tissue samples. Shapes and Ellipses show clustering of the samples. (G) Heatmap of stromal molecular signatures of TNBC and normal samples. Clustering of the samples showed by the annotations on top of the heatmap. The gene expression is displayed in (H), with significant statistical difference (p < 0.01). Abbreviations: PPI, protein–protein interactions; KEGG, Kyoto Encyclopedia of Genes and Genomes. |
Functional Enrichment Pathway Analysis of Core Targets
The KEGG pathway analysis of potential core targets revealed that genes were mainly enriched in the MAPK pathway (Figure 2D). The top five BP terms for GO analysis including regulation of mitochondrial membrane permeability, regulation of ERK1 and ERK2 cascade, negative regulation of cell proliferation, apoptotic process, and response to drug (Figure 2E). The top five CC terms for GO analysis included nuclear matrix, cell‒cell junction, mitochondrion, membrane, and nucleoplasm. The top five MF terms for GO analysis include glycoprotein binding, protein phosphatase binding, oxidoreductase activity, protein kinase activity, and protein kinase binding. These results indicate that ICA may act on TNBC-related genes, and the MAPK pathway may be a potential pathway for treatment.
Gene Expression Omnibus Analysis
Take “triple-negative breast cancer” as the keyword to retrieve the dataset GSE76250 in GEO databases, including breast tissue samples from 165 clinical TNBC patients and 33 normal individuals. All samples were examined using principal component analysis (PCA), a way to identify strong patterns in large, complex datasets. The normal group was significantly different from the TNBC group (Figure 2F). We combined the key targets from PPI network and the pathway-related targets enriched by KEGG to find that the expression of some genes in the normal group and TNBC group showed significant differences, especially the targets on MAPK pathway (Figure 2G and H). Therefore, these targets can be used as therapeutic targets for our next research, and we hypothesize that ICA may inhibit TNBC via the MAPK pathway. The above results still need to be verified experimentally.
ICA Effectively Inhibits the Proliferation of TNBC Cell
We first determined the concentration conditions and time conditions of ICA treatment in TNBC through preliminary experiments, and we performed cell proliferation experiments on the above two TNBC cell lines with the CCK-8 assay, and human mammary epithelium cell line MCF-10A was used as negative control. ICA was administered to cells at 12.5, 25, and 50 μM for 24 h or 48 h. As shown in Figure 3A–C, ICA had time‐dependent and concentration‐dependent cytotoxicity to both Hs 578T and MDA-MB-468 cells, but it has little cytotoxicity on MCF-10A cells. Due to overtreatment for 48 h and 50μM ICA, we subsequently selected 24 h and 25μM as the treatment time and concentration to measure other cellular functions. After 24 h of ICA treatment toward MDA-MB-468 and Hs 578T at low, medium, and high concentrations, the survival rate of MDA-MB-468 gradually decreased in pace with the increase in ICA concentration, and when the ICA concentration reached 50 μM, the survival rate of cells was only half (Figure 3D), and Hs 578T showed the same trend (Figure 3E). In addition, the 5‐ethynyl‐2′‐deoxyuridine (EdU) staining assay verified that the number of EdU positive MDA-MD-468 and Hs 578T cells (proliferative cells) decreased with the increase in ICA treatment concentration compared with controls (Figure 3F and G). The above results show that the growth of TNBC cells was strongly inhibited by ICA treatment and that ICA induced anti-proliferation in TNBC cells is concentration-dependent.
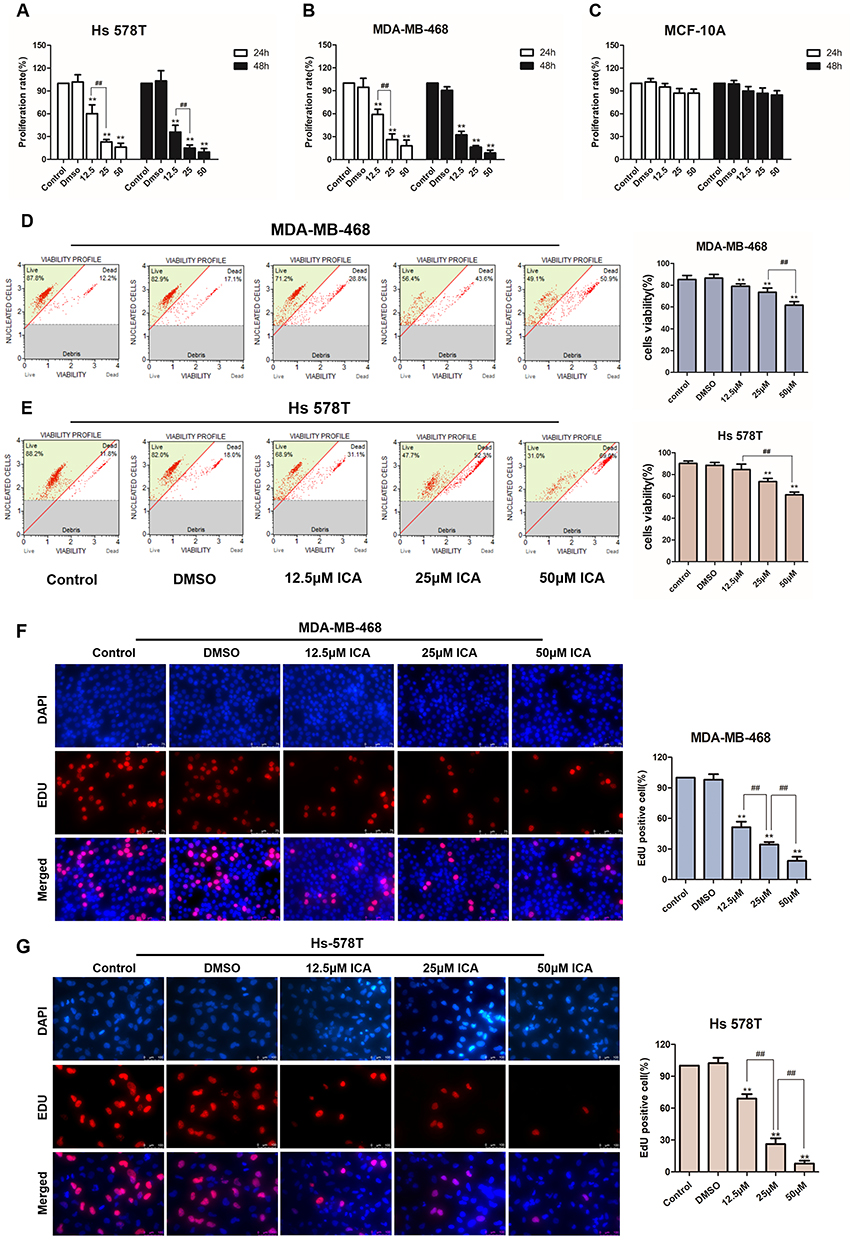 |
Figure 3 TNBC, Hs 578T and MDA-MB-468 cells, or normal human breast epithelial cells, MCF-10A in culture were treated with 0.1% DMSO or ICA (12.5, 25, and 50 μM). (A–C) CCK-8 assays determined the viability of Hs 578T, MDA-MB-468, or MCF-10A cells after 24 h or 48 h; n = 3. (D and E) The viability of MDA-MB-468 and Hs 578T cells were measured by Muse cell analyzer after 24 h treatment; n = 3. (F and G) EdU staining assays were conducted to verify MDA-MB-468 and Hs 578T cell proliferation; n = 3. Data are presented in triplicate as mean ± SD. **p < 0.01, compared with the control group; ##p < 0.01, compared with the experimental group. Bar: 75 μm. |
ICA Suppresses the Migration and Invasion in TNBC Cell
The wound healing assay evaluated the anti-migration effect of ICA in TNBC cells. After 24 h of ICA treatment, the mobilities of Hs 578T and MDA-MB-468 cells were observed under microscope at several time points (Figure 4A and B) and the cell migration of both cells was inhibited to varying degrees with the increase in ICA treatment concentration. Furthermore, the Transwell assay investigated the anti-invasion effect of ICA. Compared with controls and the solvent group (Figure 5C and D), MDA-MB-468 and Hs 578T cells exhibited a concentration-dependent inhibition of invasion after ICA administration for 24 h.
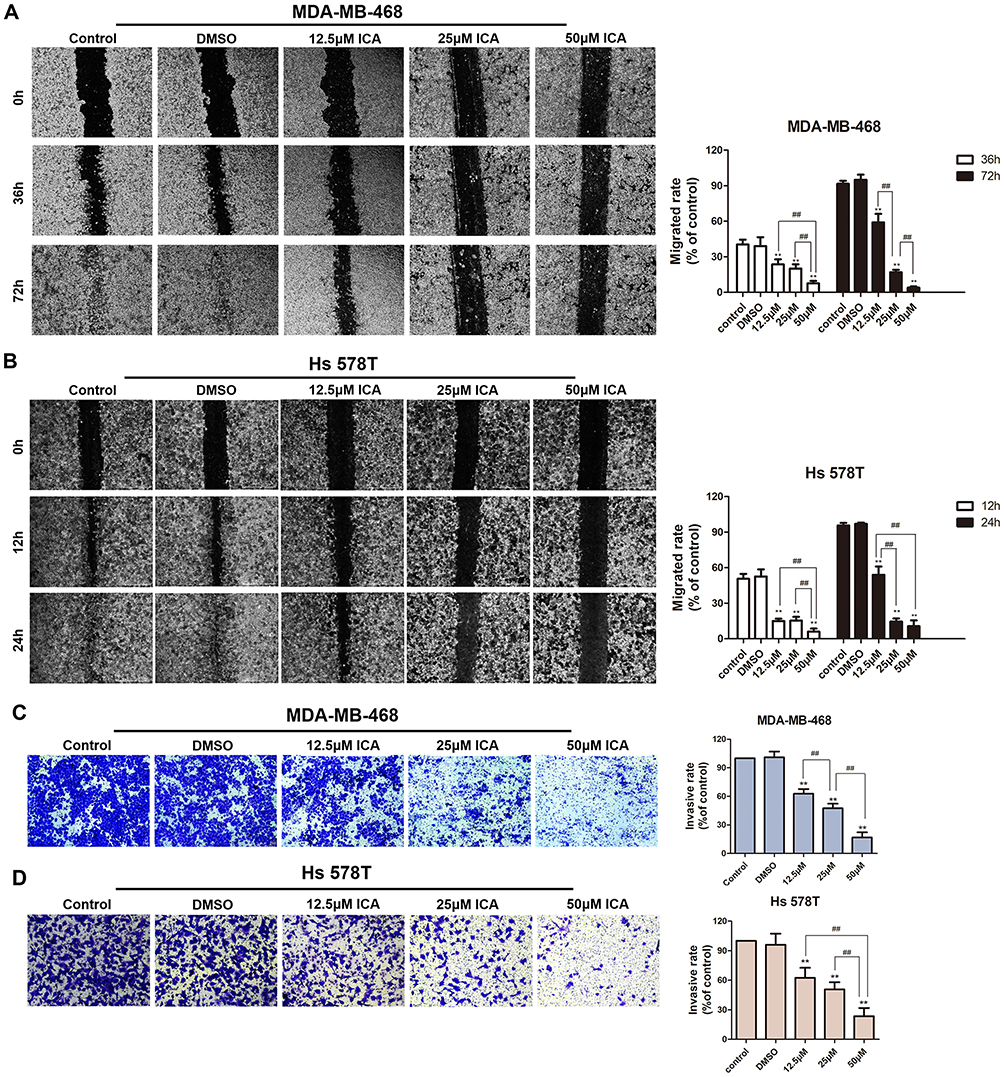 |
Figure 4 MDA-MB-468 and Hs 578T lines in culture were treated with 12.5, 25, 50μM ICA or 0.1% DMSO for 24 h. (A and B) The wound healing assay examined the cell migration ability. Images were obtained at 0, 12, 24, 36 or 72 h; n = 3. (C and D) The transwell assay examined the effects of ICA treatment on MDA-MB-468 and Hs 578T cells invasion ability; n = 3. Mean ± S.D. Data are representative of 3 independent determinations. **p < 0.01, compared with the control group; ##p < 0.01, compared with the experimental group. Bar: 500 μm. |
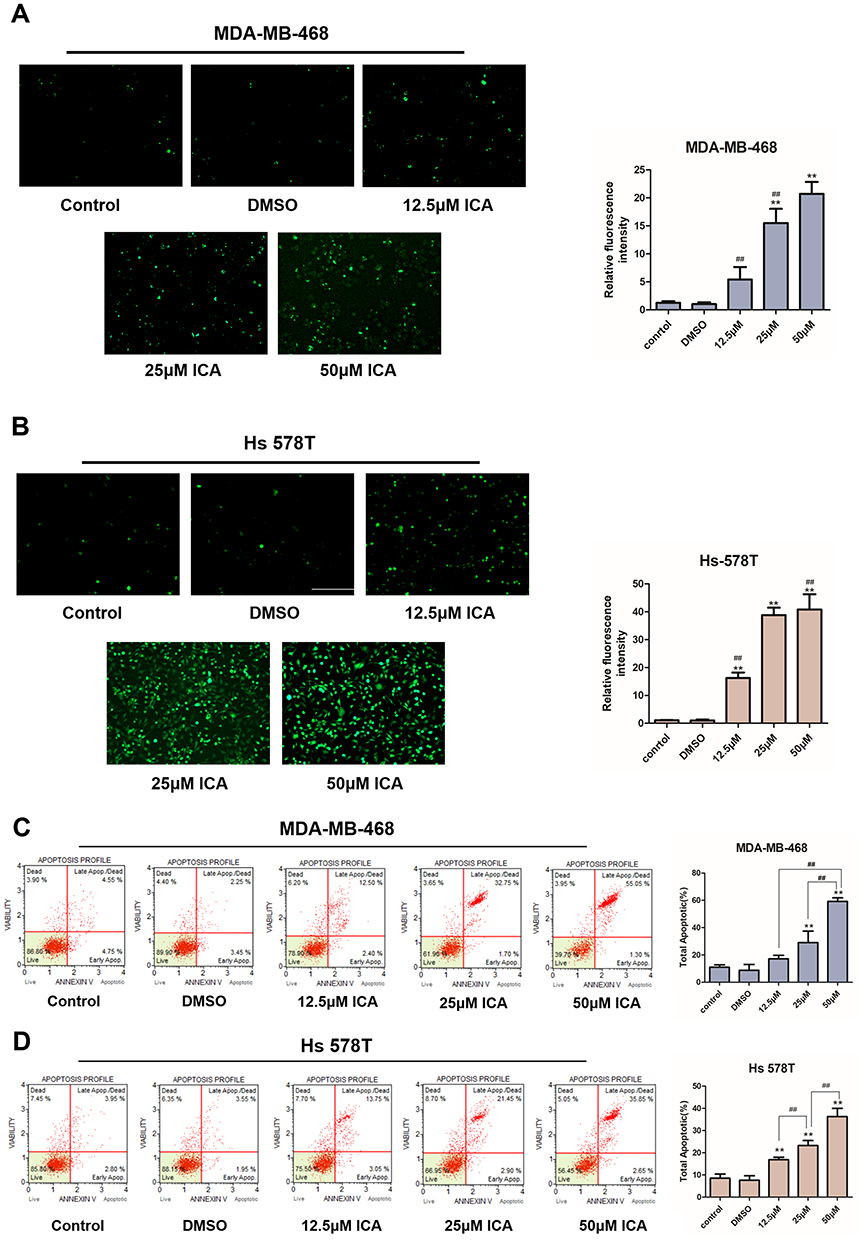 |
Figure 5 MDA-MB-468 and Hs 578T lines in culture were treated with 12.5, 25, 50μM ICA or 0.1% DMSO for 24 h. (A and B) Representative fluorescent images taken by microscope for DCFH-DA staining in Hs 578T and MDA-MB-468 cells and quantified for the relative fluorescence intensity and plotted; n =3. (Bar: 100 μm). (C and D) The population of apoptotic (early + late apoptosis) cells were measured by Muse cell analyzer; n = 3. Mean ± S.D. Data are representative of 3 independent determinations. **p < 0.01, compared with the control group; ##p < 0.01, compared with the experimental group. |
ICA Induces Oxidative Stress and Apoptosis in TNBC Cell
We analyzed the effects of ICA concentrations described above on cell apoptosis. Changes in ROS production after ICA treatment in TNBC cells were measured using fluorescent probe DCFH/DA. ICA was administered in MDA-MB-468 and Hs 578T cells to induce oxidative stress. As shown in Figure 5A and B, green fluorescence increased in a concentration‐dependent manner in MDA-MB-468 and Hs 578T cells. The quantified fluorescence intensity was significantly increased by ICA in TNBC cells. Then, we explore the apoptotic effect of ICA on cells after 24 h treatment of ICA by annexin V-FITC/PI double staining. Following ICA administration, apoptotic cells composed of apoptotic (Annexin V+/PI -) and late apoptotic (Annexin V+/PI +) cells increased in a dose-dependent manner (Figure 4C and D). Similar to the above results, the antiapoptotic protein, c-IAP2, was also decreased in Hs 578T and MBA-MB-468 cells (Figure 6C and D). Altogether, the data suggest that ICA can induce ROS production and promote apoptosis in TNBC cells in a concentration-dependent manner.
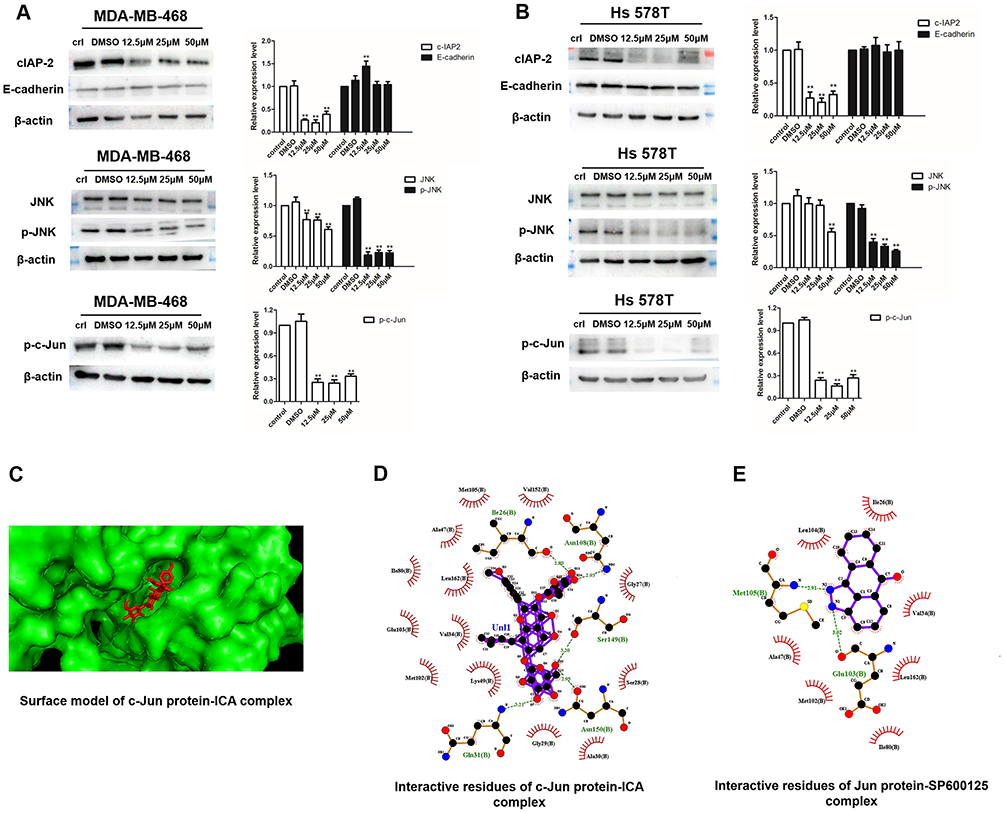 |
Figure 6 (A and B) The levels of c-IAP2, E-cadherin, JNK and c-Jun after 0.1% DMSO or ICA treatment for 24 h in MDA-MB-468 and Hs 578T cells were analyzed by Western blot and quantification of c-IAP2, E-cadherin, JNK and c-Jun protein expression. Results are displayed as mean ± SD (**p < 0.01). (C) The molecular docking simulation diagram of ICA molecular combined with c-Jun protein. The ICA is represented by red stick models while proteins are represented by green models. ICA (D) and JNK inhibitor - SP600125 (E) and Jun protein were verified to establish hydrogen bonds. Green dotted lines: carbon hydrogen bond interactions. |
ICA Decreases the Expression Levels of JNK/c-Jun Signaling Molecules in TNBC Cells
JNK is upregulated in basal-like TNBC.29 Our previous network pharmacological results also confirmed the difference in JNK2 expression between TNBC and non-TNBC patients (Figure 2H). Next, we assessed whether ICA regulates TNBC cell function using the JNK pathway, and Western blot analysis showed significant inhibition of the protein expression in JNK after ICA treatment as expects. JNK phosphorylation and c-Jun phosphorylation after ICA treatment in both cell lines (Figure 6A and B), suggesting JNK/c-Jun pathway inhibition. Similarly, Xie et al indicated that JNK favored c-Jun activation in TNBC cell.30 At the same time, c-IAP2 was also suppressed as an anti-apoptotic gene, and there was no significant change in E-cadherin protein. Molecular docking approach was applied to verify the binding action mode of c-Jun with ICA, according to the network analysis. As shown in Figure 6C, the structure of ICA was wrapped in the c-Jun protein pocket instead of floating on the surface, which conforms to the key structure. In Figure 6D, the structure of ICA could form hydrogen bonds with Ile26, Asn108, Ser149 and Asn150 in c-Jun, with the best binding affinity of −11 kcal mol−1. In Figure 6E, the JNK inhibitor (SP600125) molecule and Jun protein were interacted as a positive control, with the best binding ability of −9 kcal mol−1, docking results of JUN protein showed good binding affinity and modes of interaction with both the ICA molecules and the JNK inhibitor molecule. In conclusion, these results indicate that treatment with ICA inhibits cellular invasion by inhibiting the JNK/c-Jun pathway, and our conclusions need to be further verified.
Anisomycin Activates JNK/c-Jun and Abolishes the Effects of ICA
We activated the JNK pathway in Hs 578T cells by treatment with a nontoxic dose of anisomycin. As Figure 7A shows, anisomycin, an activator of JNK, increased phospho-JNK, phospho-c-Jun, and c-IAP2, as expected. It is worth noting that there was a very significant increase in c-Jun, a cell invasion facilitator, this is consistent with what has been found in previous Western blots (Figure 7B–E). Furthermore, as shown in Figure 7F and G the inhibitory effect of ICA on cell invasion was significantly reduced after anisomycin treatment. Our results further confirm that ICA inhibits the cellular function of TNBC by inhibiting the JNK/c-Jun pathway.
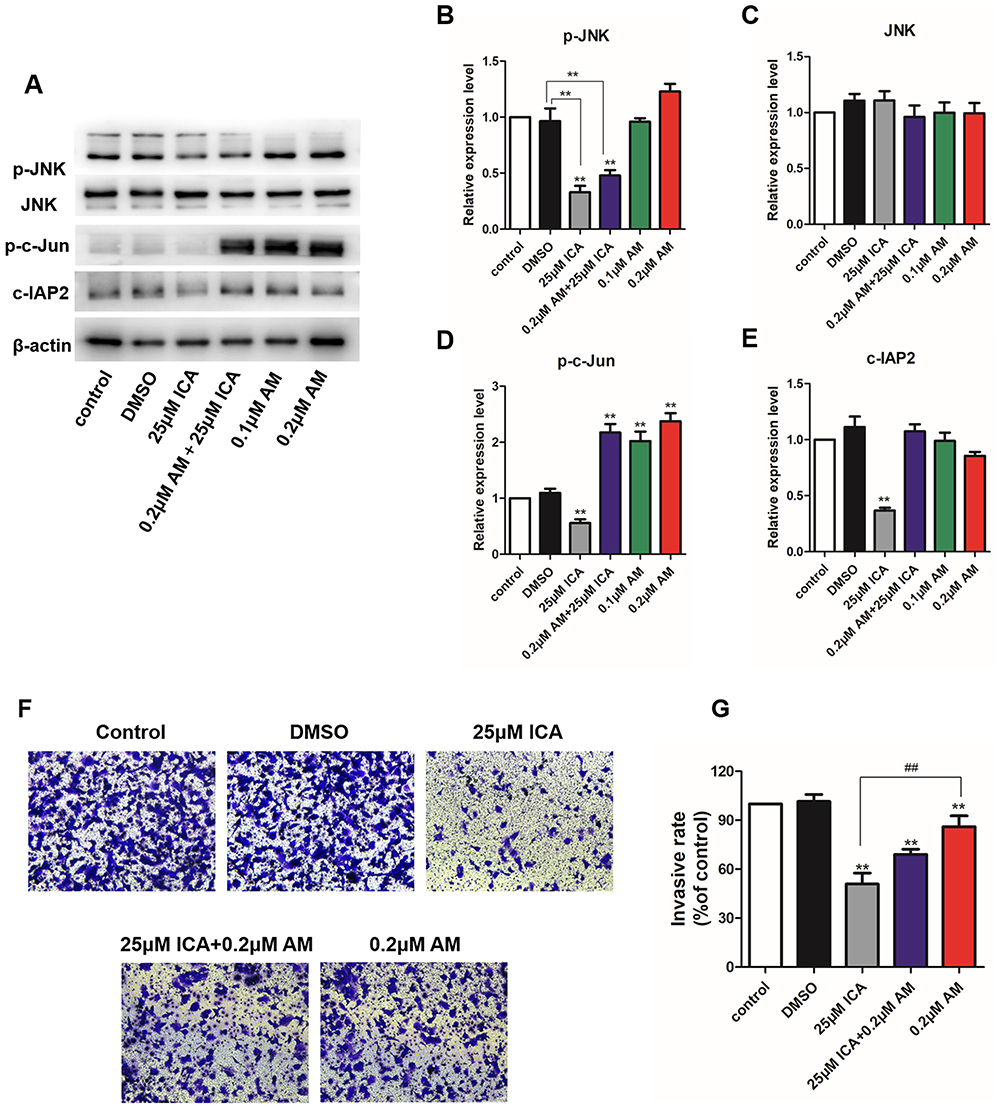 |
Figure 7 (A) Hs 578T cells were administered with 25 μM ICA for 24 h with or without anisomycin for the Western blot analysis. (B–E) The protein expression of Phosphor-JNK, JNK, phosphor-c-Jun and c-IAP2. (F and G) Hs 578T cells invasion ability was detected by the transwell assay; n = 3. Mean ± S.D. Data are representative of 3 independent determinations. **p < 0.01, compared with the control group; ##p < 0.01, compared with the experimental group. Abbreviation: AM, anisomycin. |
Discussion
Current treatment strategies for TNBC are chemotherapy and radiotherapy.31 However, TNBC treatment remains a significant challenge due to its high invasiveness and adverse reaction to therapeutics. Among various treatments for TNBC, natural products have prominent anti-cancer effects with higher efficiency and fewer side effects. Effective drug candidates are needed for TNBC treatment, and natural products are an important source of these candidates.
In this study, we reported ICA, a major active compound from the plant Epimedium, a traditional herb in Asian countries. Scientific studies have reported anticancer abilities of ICA,32 including anti-apoptosis in breast cancer through the SIRT6/NF‐κB pathway.15 Recently, Li et al observed that ICA can efficiently inhibit cell proliferation and invasion, promoting apoptosis through the TLR4/MyD88/NF-κB and Wnt/β-catenin pathways.12 In addition, ICA inhibited proliferation of HeLa cervical cancer cells through the mTOR/PI3K/AKT signaling.33 Additionally, Zhang et al indicated that gastric cancer cells are inhibited by ICA through the hsa_circ_0003159/miR-223-3p/NLRP3 pathway.13
As a result of network pharmacology, ICA was predicted to be able to inhibit the MAPK signaling pathway. The MAPK pathway plays a more significant role in cell proliferation, apoptosis, and invasion compared with other pathways,34 and tumor invasion was promoted by activating the MAPK signaling.35 This evidence suggests that ICA has the potential to act on TNBC, but our hypothesis needs to be further confirmed experimentally.
Our results from CCK-8, EdU, and cell viability assays confirmed that ICA inhibited the proliferation of TNBC cells in a time- and concentration-dependent manner. ICA also induced apoptosis in a concentration-dependent manner. However, further investigation is needed to understand the detailed mechanisms of ICA inhibiting TNBC cell.
Reactive oxygen species (ROS) are oxygen-containing substances produced during cell metabolism.36 As second messengers, ROS amends a variety of cell-signaling proteins, such as MAPKs, PI3K-AKT, and NF‐κB.37 The level of cancer cells is higher than normal cells. Overproduction of ROS occurs in cancer cells’ mitochondria, promoting cancer cell growth,38 but excessive ROS directly insults proteins, DNA, and lipids and can kill cancer cells.39 Due to the increased ROS level and relatively low ROS lack of efficacy, tumor cells are more easily controlled by changes in ROS levels; therefore, the excessive accumulation of ROS in cells should be an effective strategy for cancer cell therapy, including TNBC. Our results indicated that ICA increased ROS level and induced apoptosis in TNBC cells, and ICA also reduced the protein expression of cellular inhibitor of apoptosis-2 (c-IAP2). Similarly, Gu et al reported that ICA efficiently inhibited the proliferation of human esophageal carcinoma cells through ROS-mediated alterations,40 and ICA could induce human hepatoma cells apoptosis by activating the ROS/JNK pathway.17 Therefore, we suggested that ICA exerted inhibitory effects induces TNBC cell apoptosis by favoring an excessive accumulation of ROS in cells.
We used wound healing assay and transwell assay to investigate different antitumor abilities of ICA in TNBC cells. We found that ICA inhibits cell migration and invasion in a concentration-dependent manner. The mechanism of ICA inhibits the invasion of TNBC cells was further investigated. Cai et al confirmed that pancreatic cancer cell invasion and migration could be enhanced via JNK/c-Jun signaling activation, and JNK activity is related to elevated cell migration and invasion in prostate cancer.41
JNK, as a subfamily of MAPK, regulates cell proliferation, tumor invasion, and apoptosis.20 The activation of JNK and its downstream target c-Jun can promote lung cancer cell invasion and migration42 and human glioma cells.43 The JNK pathway also plays a role in ROS-induced apoptosis through mitochondrial damage.24,25 It has been reported that ROS activates JNK causing increased mitochondrial membrane permeability and ultimately apoptosis in lung cancer cells.44 In this study, ICA inhibited the migration and invasion of TNBC cells, and decreased the protein expression level of phosphorylated JNK and phosphorylated c-Jun. Moreover, anisomycin diminished the effects of ICA on TNBC cell invasion. Similarly, Zhou et al proved that JNK activation could abolish the suppressive effect on the invasiveness of cell migration and invasion.43 Together, the above results indicate that ICA inhibits cell invasion of TNBC cell by suppressing the JNK/c-Jun signaling pathway.
However, JNK could also inhibit the apoptosis of cancer cells,45 and researchers have indicated that JNK activation that is sustained promotes apoptosis, whereas activation that is acute or transient promotes cell survival.46,47 Although activation of JNK can increase the expression of antiapoptotic c-IAP2, anisomycin could not fully rescue TNBC cells from apoptosis, which was obtained in this study, suggesting that the different functions of JNK may be related to temporal and dosage aspects. In order to fully reveal the detailed pro- or antiapoptotic mechanisms of JNK in TNBC cells, further studies are needed in the future.
Conclusion
In conclusion, our study showed that ICA could produce cytotoxicity towards TNBC cells in a time- and concentration-dependent manner, and inhibit cellular functions including proliferation, migration, and invasion. ICA could induce apoptosis by inducing an excessive accumulation of ROS in cells and suppress TNBC cell invasion via the JNK/c-Jun signaling pathway. These results suggest that ICA could be a potential drug for TNBC therapeutic. However, the present findings will require additional in vivo evaluation and clinical trials to determine its reliability.
Acknowledgments
This work was supported by the Foundation of Xi’an Municipal Bureau of Science and Technology (Y20201001); the Foundation of Health Commission of Shaanxi Province (2022B003); the Foundation of Xi’an Municipal Bureau of Science and Technology (22YXYJ0018).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Foulkes WD, Smith IE, Reis-Filho, JS. Triple-Negative Breast Cancer. N Engl J Med. 2010;363(20):1938–1948. doi:10.1056/NEJMra1001389
3. Wu N, Zhang J, Zhao J, et al. Precision medicine based on tumorigenic signaling pathways for triple-negative breast cancer. Oncol Lett. 2018;16(4):4984–4996. doi:10.3892/ol.2018.9290
4. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948. doi:10.1056/NEJMra1001389
5. Won KA, Spruck C. Triple‑negative breast cancer therapy: current and future perspectives (Review). Int J Oncol. 2020;57(6):1245–1261. doi:10.3892/ijo.2020.5135
6. Fontana F, Raimondi M, Marzagalli M, Di Domizio A, Limonta P. The emerging role of paraptosis in tumor cell biology: perspectives for cancer prevention and therapy with natural compounds. Biochim Biophys Acta Rev Cancer. 2020;1873(2):188338. doi:10.1016/j.bbcan.2020.188338
7. Chen M, Wu J, Luo Q, et al. The anticancer properties of herba epimedii and its main bioactive components icariin and icariside II. Nutrients. 2016;8:9. doi:10.3390/nu8090563
8. Wei Y, Liu B, Sun J, et al. Regulation of Th17/Treg function contributes to the attenuation of chronic airway inflammation by icariin in ovalbumin-induced murine asthma model. Immunobiology. 2015;220(6):789–797. doi:10.1016/j.imbio.2014.12.015
9. Xiong W, Chen Y, Wang Y, Liu J. Roles of the antioxidant properties of icariin and its phosphorylated derivative in the protection against duck virus hepatitis. BMC Vet Res. 2014;10:226. doi:10.1186/s12917-014-0226-3
10. Liu B, Zhang H, Xu C, et al. Neuroprotective effects of icariin on corticosterone-induced apoptosis in primary cultured rat hippocampal neurons. Brain Res. 2011;1375:59–67. doi:10.1016/j.brainres.2010.12.053
11. Fu Y, Liu H, Long M, et al. Icariin attenuates the tumor growth by targeting miR-1-3p/TNKS2/Wnt/β-catenin signaling axis in ovarian cancer. Front Oncol. 2022;12:940926. doi:10.3389/fonc.2022.940926
12. Li C, Yang S, Ma H, Ruan M, Fang L, Cheng J. Influence of icariin on inflammation, apoptosis, invasion, and tumor immunity in cervical cancer by reducing the TLR4/MyD88/NF-κB and Wnt/β-catenin pathways. Cancer Cell Int. 2021;21(1):206. doi:10.1186/s12935-021-01910-2
13. Zhang F, Yin Y, Xu W, et al. Icariin inhibits gastric cancer cell growth by regulating the hsa_circ_0003159/miR-223-3p/NLRP3 signaling axis. Hum Exp Toxicol. 2022;41:9603271221097363. doi:10.1177/09603271221097363
14. Zhu F, Ren Z. Icariin inhibits the malignant progression of lung cancer by affecting the PI3K/Akt pathway through the miR‑205‑5p/PTEN axis. Oncol Rep. 2022;47(6). doi:10.3892/or.2022.8326
15. Song L, Chen X, Mi L, et al. Icariin-induced inhibition of SIRT6/NF-κB triggers redox mediated apoptosis and enhances anti-tumor immunity in triple-negative breast cancer. Cancer Sci. 2020;111(11):4242–4256. doi:10.1111/cas.14648
16. Wang D, Xu W, Chen X, et al. Icariin induces cell differentiation and cell cycle arrest in mouse melanoma B16 cells via Erk1/2-p38-JNK-dependent pathway. Oncotarget. 2017;8(59):99504–99513. doi:10.18632/oncotarget.20118
17. Li S, Dong P, Wang J, et al. Icariin, a natural flavonol glycoside, induces apoptosis in human hepatoma SMMC-7721 cells via a ROS/JNK-dependent mitochondrial pathway. Cancer Lett. 2010;298(2):222–230. doi:10.1016/j.canlet.2010.07.009
18. Yuan H, Ma Q, Cui H, et al. How can synergism of traditional medicines benefit from network pharmacology? Molecules. 2017;22:7.
19. Pinzi L, Rastelli G. Molecular docking: shifting paradigms in drug discovery. Int J Mol Sci. 2019;20(18):4331. doi:10.3390/ijms20184331
20. Wu Q, Wu W, Jacevic V, Franca TCC, Wang X, Kuca K. Selective inhibitors for JNK signalling: a potential targeted therapy in cancer. J Enzyme Inhib Med Chem. 2020;35(1):574–583. doi:10.1080/14756366.2020.1720013
21. Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15(6):411–421. doi:10.1038/nrm3801
22. Wang H, Wen C, Chen S, et al. ROS/JNK/C-Jun pathway is involved in chaetocin induced colorectal cancer cells apoptosis and macrophage phagocytosis enhancement. Front Pharmacol. 2021;12:729367. doi:10.3389/fphar.2021.729367
23. Han J, Jeon M, Shin I, Kim S. Elevated STC‑1 augments the invasiveness of triple‑negative breast cancer cells through activation of the JNK/c‑Jun signaling pathway. Oncol Rep. 2016;36(3):1764–1771. doi:10.3892/or.2016.4977
24. Castro-Caldas M, Carvalho AN, Rodrigues E, Henderson C, Wolf CR, Gama MJ. Glutathione S-transferase pi mediates MPTP-induced c-Jun N-terminal kinase activation in the nigrostriatal pathway. Mol Neurobiol. 2012;45(3):466–477. doi:10.1007/s12035-012-8266-9
25. Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87(7):1157–1180. doi:10.1007/s00204-013-1034-4
26. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi:10.1038/75556
27. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi:10.1093/nar/28.1.27
28. Clough E, Barrett T. The gene expression omnibus database. Methods Mol Biol. 2016;1418:93–110.
29. Wang X, Chao L, Li X, et al. Elevated expression of phosphorylated c-Jun NH2-terminal kinase in basal-like and “triple-negative” breast cancers. Hum Pathol. 2010;41(3):401–406. doi:10.1016/j.humpath.2009.08.018
30. Xie X, Kaoud TS, Edupuganti R, et al. c-Jun N-terminal kinase promotes stem cell phenotype in triple-negative breast cancer through upregulation of Notch1 via activation of c-Jun. Oncogene. 2017;36(18):2599–2608. doi:10.1038/onc.2016.417
31. Manjunath M, Choudhary B. Triple-negative breast cancer: a run-through of features, classification and current therapies. Oncol Lett. 2021;22(1):512. doi:10.3892/ol.2021.12773
32. Tan HL, Chan KG, Pusparajah P, et al. Anti-cancer properties of the naturally occurring aphrodisiacs: icariin and its derivatives. Front Pharmacol. 2016;7:191. doi:10.3389/fphar.2016.00191
33. Huang S, Xie T, Liu W. Icariin inhibits the growth of human cervical cancer cells by inducing apoptosis and autophagy by targeting mTOR/PI3K/AKT signalling pathway. J buon. 2019;24(3):990–996.
34. Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19(3):1997–2007. doi:10.3892/etm.2020.8454
35. Wen S, Hou Y, Fu L, et al. Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3-p38 MAPK signalling. Cancer Lett. 2019;442:320–332. doi:10.1016/j.canlet.2018.10.015
36. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–950. doi:10.1152/physrev.00026.2013
37. Zhang J, Wang X, Vikash V, et al. ROS and ROS-Mediated Cellular Signaling. Oxid Med Cell Longev. 2016;2016:4350965. doi:10.1155/2016/4350965
38. Yang Y, Karakhanova S, Hartwig W, et al. Mitochondria and Mitochondrial ROS in cancer: novel targets for anticancer therapy. J Cell Physiol. 2016;231(12):2570–2581. doi:10.1002/jcp.25349
39. Galadari S, Rahman A, Pallichankandy S, Thayyullathil F. Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic Biol Med. 2017;104:144–164. doi:10.1016/j.freeradbiomed.2017.01.004
40. Gu ZF, Zhang ZT, Wang JY, Xu BB. Icariin exerts inhibitory effects on the growth and metastasis of KYSE70 human esophageal carcinoma cells via PI3K/AKT and STAT3 pathways. Environ Toxicol Pharmacol. 2017;54:7–13. doi:10.1016/j.etap.2017.06.004
41. Cai J, Du S, Wang H, et al. Tenascin-C induces migration and invasion through JNK/c-Jun signalling in pancreatic cancer. Oncotarget. 2017;8(43):74406–74422. doi:10.18632/oncotarget.20160
42. Zhang D, Jiang Q, Ge X, et al. RHOV promotes lung adenocarcinoma cell growth and metastasis through JNK/c-Jun pathway. Int J Biol Sci. 2021;17(10):2622–2632. doi:10.7150/ijbs.59939
43. Zhou X, Hua L, Zhang W, et al. FRK controls migration and invasion of human glioma cells by regulating JNK/c-Jun signaling. J Neurooncol. 2012;110(1):9–19. doi:10.1007/s11060-012-0933-1
44. Wang K, Gong Q, Zhan Y, et al. Blockage of autophagic flux and induction of mitochondria fragmentation by paroxetine hydrochloride in lung cancer cells promotes apoptosis via the ROS-MAPK pathway. Front Cell Dev Biol. 2019;7:397. doi:10.3389/fcell.2019.00397
45. Almasi S, Kennedy BE, El-Aghil M, et al. TRPM2 channel-mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J Biol Chem. 2018;293(10):3637–3650. doi:10.1074/jbc.M117.817635
46. Ventura JJ, Hübner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell. 2006;21(5):701–710. doi:10.1016/j.molcel.2006.01.018
47. Dhanasekaran DN, Reddy EP. JNK-signaling: a multiplexing hub in programmed cell death. Genes Cancer. 2017;8(9–10):682–694. doi:10.18632/genesandcancer.155
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.