Back to Archived Journals » Hypoxia » Volume 3
Hypoxic regulation of osteoclast differentiation and bone resorption activity
Authors Knowles H ![]()
Received 8 September 2015
Accepted for publication 7 October 2015
Published 11 November 2015 Volume 2015:3 Pages 73—82
DOI https://doi.org/10.2147/HP.S95960
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Helen J Knowles
Botnar Research Centre, NDORMS, University of Oxford, Oxford, Oxfordshire, UK
Abstract: Bone integrity is maintained throughout life via the homeostatic actions of bone cells, namely, osteoclasts, which resorb bone, and osteoblasts, which produce bone. Disruption of this balance in favor of osteoclast activation results in pathological bone loss, which occurs in conditions including osteoporosis, rheumatoid arthritis, primary bone cancer, and cancer metastasis to bone. Hypoxia also plays a major role in these conditions, where it is associated with disease progression and poor prognosis. In recent years, considerable interest has arisen in the mechanisms whereby hypoxia and the hypoxia-inducible transcription factors, HIF-1α and HIF-2α, affect bone remodeling and bone pathologies. This review summarizes the current evidence for hypoxia-mediated regulation of osteoclast differentiation and bone resorption activity. Role(s) of HIF and HIF target genes in the formation of multinucleated osteoclasts from cells of the monocyte–macrophage lineage and in the activation of bone resorption by mature osteoclasts will be discussed. Specific attention will be paid to hypoxic metabolism and generation of ATP by osteoclasts. Hypoxia-driven increases in both glycolytic flux and mitochondrial metabolic activity, along with consequent generation of mitochondrial reactive oxygen species, have been found to be essential for osteoclast formation and resorption activity. Finally, evidence for the use of HIF inhibitors as potential therapeutic agents targeting bone resorption in osteolytic disease will be discussed.
Keywords: ATP, hypoxia-inducible factor, HIF, osteolysis, glycolysis, reactive oxygen species, mitochondrial metabolism
Introduction
Hypoxia is a characteristic microenvironmental component of numerous pathological conditions, where it correlates with disease progression and/or severity. Many of these conditions are also associated with bone loss, including cancer,1,2 rheumatoid arthritis,3,4 osteoporosis,5 and bone fracture,6 as well as extra-skeletal conditions such as obstructive pulmonary disease.7
Bone remodeling during development and bone integrity throughout life are normally regulated by a balance between bone formation, performed by osteoblasts, and bone resorption, performed by osteoclasts. Pathological bone loss occurs when this homeostatic relationship is disturbed. Overactivation of osteoclasts is directly responsible for the resorptive bone loss evident in rheumatoid arthritis,8,9 osteoporosis,10 and cancer metastasis to bone.11
Given that hypoxia and hypoxia-inducible factor (HIF) are also present in these conditions, it is important to understand how hypoxia pathways affect the differentiation and activity of osteoclasts. This review summarizes the research into effects of hypoxia and HIF on bone-resorbing osteoclasts, highlighting potential new targets for the development of antiresorptive therapies.
Hypoxia stimulates osteoclastogenesis in vitro
Osteoclasts form by the fusion of CD14+ monocyte or macrophage precursors, in the presence of macrophage colony stimulating factor (M-CSF) and receptor activator of nuclear factor kappa B ligand (RANKL), to produce mature multinucleated cells.12,13 The mature osteoclasts then attach to mineralized bone and acidify the extracellular environment at the cell-bone interface, releasing bone minerals and exposing the organic matrix to resorption by the secreted acid protease cathepsin K.14,15 Cells of the monocyte–macrophage lineage from which osteoclasts derive have long been known to be activated by hypoxia,16,17 suggesting that monocyte–osteoclast differentiation and osteoclast activity might also be increased at low O2 tensions.
The first study into effects of hypoxia on osteoclast formation used bone marrow cells flushed from mouse long bones as a source of monocytic precursors. When differentiated in vitro in the presence of M-CSF and RANKL, maximal hypoxic stimulation was achieved at 2% O2, which produced a fourfold increase in osteoclast number and a threefold increase in resorption activity per osteoclast at the end of the differentiation period.18 Similar results were found using circulating monocytes from feline19 and human20 peripheral blood mononuclear cells (PBMCs) as osteoclast precursors. Attention to the hypoxia protocol in these studies reveals that the cells were actually exposed to a hypoxia/re-oxygenation schedule, rather than hypoxia per se, during the period of differentiation. The need for re-oxygenation during hypoxic differentiation was confirmed in a separate study; steady-state exposure to 2% O2 in a gloved hypoxic workstation dramatically inhibited osteoclast formation and resorption due to extensive cell death.21
Acute exposure to hypoxia also increases the ability of mature osteoclasts to resorb bone. Resorption is normally measured in vitro by culturing osteoclasts on dentine (elephant ivory) discs and either quantifying the area of resorption tracks or assaying release of cross-linked C-telopeptide of type I collagen (CTXI) into the supernatant. Osteoclasts derived from feline19 or human21–23 PBMC, as well as mature human osteoclasts curetted from the primary bone tumor giant cell tumor of bone,21 exhibited a twofold to fourfold increase in resorption after 24 hours exposure to 2% O2. No effect was observed in primary osteoclasts from disaggregated rat bones.18
The fact that hypoxia can stimulate the formation and activity of human osteoclasts derived from PBMCs, where there is effectively no stromal cell support,20,21 as well as from pure populations of CD14+ monocytes,22,23 suggests that the osteoclastogenic response to hypoxia is an intrinsic property of this cell lineage.
Hypoxia stimulates osteoblast-mediated osteoclastogenesis
Obviously, in the in vivo situation, monocytes and osteoclasts do not exist in isolation but are surrounded by osteoblasts, fibroblasts, and other cellular components of the bone microenvironment that will also be exposed to local hypoxia. Co-culture of monocytes with stromal cells including osteoblasts, fibroblasts, and cancer cells has revealed that hypoxia stimulates local production of pro-osteoclastogenic cytokines including RANKL,24,25 vascular endothelial growth factor (VEGF),24–27 M-CSF,27 insulin-like growth factor 2,28 and growth differentiation factor 15,29 as well as inhibiting production of osteoprotegerin (OPG), a soluble decoy receptor for RANKL that inhibits osteoclast formation and activity.30
Discussion of the regulation and mechanisms of action of the full range of osteoclastogenic cytokines produced by support cells within the hypoxic bone microenvironment is beyond the scope of this review. However, it is clear that hypoxia, both directly and indirectly, has a large impact on osteoclast formation and bone resorption activity (Figure 1). This is supported by a study in mice lacking the Fos-related protein Fra-2, which form giant osteoclasts due to the presence of hypoxia in their long bones.31 Conversely, exposure to hyperbaric oxygen reduces human osteoclast formation and bone resorption in vitro.32,33
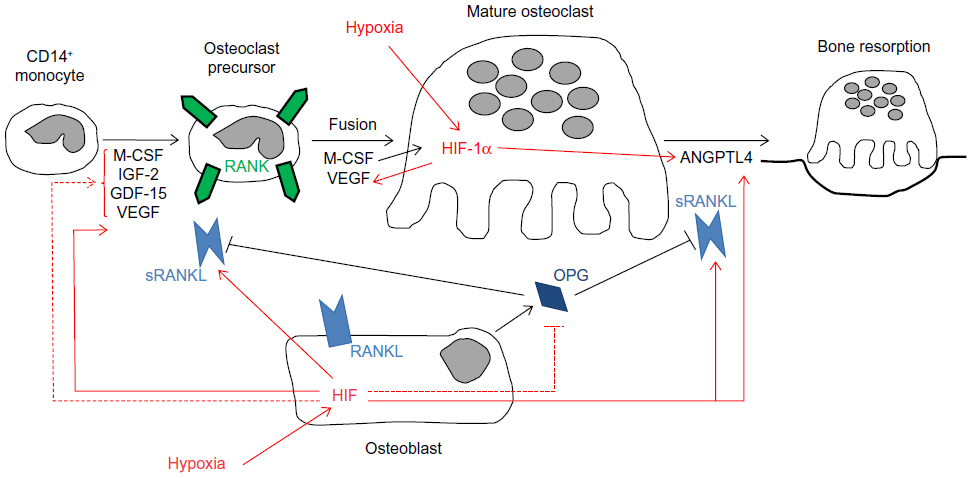 | Figure 1 Cytokine-mediated effects of hypoxia on osteoclast differentiation and bone resorption activity. |
Role of HIF in osteoclast-mediated bone resorption
Expression of HIF-1α and HIF-2α protein by human osteoclasts was first described as recently as 2008.26 Mature human monocyte-derived osteoclasts in vitro have since been shown to stabilize HIF-1α and HIF-2α in response to either hypoxia or hypoxia mimetics such as MG132, CoCl2, desferrioxamine, or dimethyloxalylglycine.20–23,26 HIF expression also increases on exposure to osteoclastogenic cytokines26 and, therefore, during monocyte–osteoclast differentiation.5,20
However, it seems likely that expression of HIF alone is insufficient to stimulate cell autonomous osteoclastogenesis. Formation of murine osteoclasts was shown to be inhibited in the presence of dimethyloxalylglycine or desferrioxamine, or when monocytes were transfected with a constitutively active form of HIF-1α.34 Similarly, differentiation of human PBMC was inhibited in the presence of CoCl2.35 This is in agreement with our unpublished observations that CoCl2 and desferrioxamine inhibit human monocyte–osteoclast differentiation. However, other groups have reported that cobalt stimulates differentiation of murine osteoclasts.36 A specific role for HIF in the process of monocyte–osteoclast differentiation therefore remains to be defined.
On the other hand, HIF does appear to be responsible for the hypoxia-induced increase in bone resorption by mature osteoclasts. HIF-1α siRNA completely ablated the hypoxic increase in resorption by human monocyte-derived osteoclasts, although HIF-2α siRNA had no effect.21,22 Hypoxic osteoclasts show increased secretion of the HIF-regulated osteoclastogenic cytokine VEGF20,21 and resorption-promoting angiopoietin-like 4 (ANGPTL4; Figure 1).22 These were not apparently directly responsible for the HIF-1α-dependent increase in osteoclast activity, however,21,22 suggesting that HIF plays other role(s) to increase osteoclast activity.
HIF and osteoclast metabolism
The bone resorption process involves osteoclasts binding to bone via αvβ3 integrin and then forming an F-actin-rich seal to isolate a resorptive compartment. Active transport of protons across the bone-apposing membrane by vacuolar H+ ATPase along with Na,K-ATPase, Ca-ATPase, and gastric H,K-ATPase37 acidifies this compartment, releasing bone minerals and exposing the organic matrix. The matrix is subsequently resorbed by the secreted acid protease cathepsin K.14 Osteoclasts are also highly motile, all of which accumulates to make osteoclast-mediated bone resorption, an energy-intensive process with a high demand for ATP.15,38 Osteoclasts contain numerous mitochondria,39 associated with high expression of tricarboxylic acid cycle and oxidative phosphorylation enzymes,40 and exhibit high rates of oxygen consumption.41 This implies that high mitochondrial metabolic activity drives ATP production in these cells.
It is therefore interesting to consider how osteoclasts generate sufficient ATP to support the observed increase in bone resorption under hypoxia. Cellular adaptation to hypoxia generally entails switching to anaerobic metabolism – a HIF-mediated survival mechanism involving inhibition of mitochondrial ATP production in order to prevent accumulation of toxic levels of reactive oxygen species (ROS).42,43 This is a multistep process. Initially, HIF increases the efficiency of complex IV of the mitochondrial electron transport chain (ETC) with respect to the amounts of ATP and ROS produced, triggering a switch in expression of cytochrome c oxidase subunits from COX4-1 to COX4-2.44 When this is unable to maintain energy/redox homeostasis, a switch occurs from mitochondrial to purely glycolytic metabolism. HIF stimulates increased expression of glucose transporters and glycolytic enzymes to increase flux through the glycolytic pathway.45 It also increases expression of pyruvate dehydrogenase (PDH) kinase (PDK), which phosphorylates and inactivates PDH, the mitochondrial enzyme responsible for converting pyruvate into acetyl co-enzyme A.46,47 This reduces flux through the mitochondrial tricarboxylic acid cycle and ETC and again reduces accumulation of ROS. As a final response, HIF induces expression of BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3), which initiates mitochondrial autophagy and further reduces accumulation of ROS.48
Glycolysis
The monocyte/macrophage population from which osteoclasts derive, which must also be able to function in hypoxic environments, relies heavily on HIF-1α-mediated transcription of glycolytic genes to produce ATP.49 Despite this already high baseline glycolytic activity, the glycolytic rate, measured either as glucose consumption or lactate production, increases further during monocyte–osteoclast differentiation.41,50
Glucose is the principal energy source necessary for bone degradation.50,51 Within the physiological range, an increased glucose concentration rapidly increases the intracellular ATP:ADP ratio.52 Longer exposure activates transcription of the A-subunit of vacuolar H+ ATPase,53 which interacts directly with the glycolytic enzyme phosphofructokinase-1.54 This interaction is thought to micro-compartmentalize glycolytic ATP generation at the required intracellular location, directly linking glycolysis and osteoclast activation. Indeed, inhibition of glycolysis could be a therapeutic antiresorptive option. Glycolytic inhibitors reduce bone resorption in animal models of disease55,56 and have been shown to induce clinical remission in rheumatoid arthritis.57,58
Hypoxia then elevates the already high basal glycolytic rate of osteoclasts still further. Hypoxic osteoclasts demonstrate increased expression of HIF-regulated glucose transporters (SLC2A1 mRNA23,59 and Glut-1 protein26) and glycolytic enzymes (PGK1, PFKFB4, ALDOC, and LDHA23), resulting in a HIF-1α-dependent increase in glucose consumption.23 Lactate production also increases, although the ratio of glucose consumption to lactate production remained unchanged, suggestive of a hypoxic increase in flux through the glycolytic pathway but not of a switch to anaerobic glycolysis23 (Figure 2).
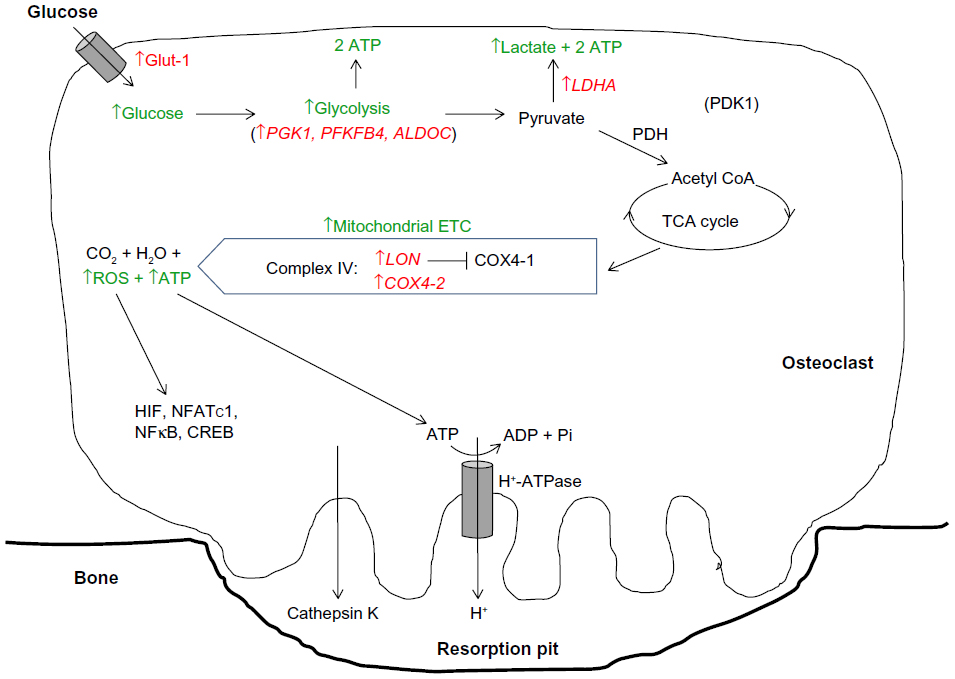 | Figure 2 Modification of the HIF-mediated switch to anaerobic respiration in osteoclasts. |
Glucose uptake remains essential for osteoclast activity in hypoxia as depletion of glucose severely reduced the generation of intracellular ATP by hypoxic osteoclasts.23 Increased glycolysis by actively resorbing hypoxic osteoclasts may also occur in vivo. Positron emission tomography with 2-(fluorine-18)fluoro-2-deoxy-d-glucose (18FDG) in benign primary bone tumors can distinguish those containing many osteoclasts from those where osteoclasts are sparse,60 additionally correlating with markers of hypoxia.61
Mitochondrial metabolism
In line with the generally accepted switch to anaerobic metabolism in hypoxia, most hypoxic cells exhibit reduced concentrations of intracellular ATP and reduced mitochondrial metabolic flux.46,47 However, elevated concentrations of ATP were observed in hypoxic osteoclasts, as well as increased mitochondrial reductase activity within the ETC. There was also no reduction in O2 consumption via the ETC under hypoxia; this remained close to maximal and was even more sensitive to ETC inhibition with rotenone than in the corresponding normoxic cells.23 This hypoxic increase in ETC activity was at least partially dependent on HIF-1α, mediated by apparently selective utilization of components of the classical HIF-mediated metabolic switch to anaerobic respiration that increase or maintain pathway activity (the COX subunit switch, increased glycolytic rate), while neither inhibiting PDH activity nor stimulating BNIP3 production23 (Figure 2).
HIF-1α-mediated induction of PDK1 normally results in phosphorylation of PDH and inhibition of PDH activity. However, in mature human osteoclasts, hypoxia had no effect on either PDK1 expression or PDH activity, nor were these affected by HIF-1α siRNA.23 PDH can also be inhibited by hypoxic phosphorylation and activation of AMP-activated protein kinase (AMPK), via induction of expression of PDK4.62,63 However, in osteoclasts, hypoxia dramatically inhibited AMPK phosphorylation and so inactivated AMPK.23
As an alternative to hypoxia, AMPK can be activated by reduced intracellular ratios of ATP:ADP or ATP:AMP, hypoxic activation occurring via a mechanism independent of changes in intracellular energy status.63,64 It may be that high levels of ATP production in hypoxic osteoclasts increase the intracellular ratio of ATP:AMP and override hypoxic mechanisms of AMPK activation in favor of de-phosphorylation and inactivation. AMPK inhibition would be necessary for hypoxic resorption to occur, as AMPK inhibits osteoclast differentiation and activity.65
The classical hypoxic attenuation of PDH activity is therefore prevented in osteoclasts by blockade of at least two pathways that usually contribute to its inhibition, allowing continued mitochondrial metabolic flux under hypoxia. As would be expected, hypoxic continuation of oxidative phosphorylation leads to accumulation of significant amounts of mitochondrial ROS in hypoxic osteoclasts.23,66,67
Reactive oxygen species
ROS, usually in the form of H2O2 or superoxide, are essential for osteoclast function. During monocyte–osteoclast differentiation, RANKL interacts with RANK on the surface of precursor cells to activate a range of signaling pathways. RANKL signaling involves production of ROS by nicotinamide adenine dinucleotide phosphate oxidase following binding of TNF receptor-associated factor 6 to the cytoplasmic domain of RANK. These ROS then serve as second messengers in the MAPK, NFκB, and Ca2+ mobilization pathways,68,69 which combine to promote expression of transcription factors including nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 1 (NFATc1). NFATc1 is essential for osteoclastogenesis, promoting transcription of genes such as tartrate-resistant acid phosphatase (TRAP), calcitonin receptor, cathepsin K, and pro-fusion genes.70
ROS also regulate effects on osteoclast-mediated bone resorption and survival. For example, following degradation of bone matrix in the resorption compartment by cathepsin K, osteoclasts endocytose the initial degradation products, along with the cathepsin K, then transcytose these vesicles through the osteoclast. TRAP-containing vesicles fuse with the transcytotic vesicles and cathepsin K digests TRAP to activate its ROS-generating activity. These ROS complete the degradation of the matrix components during their transcytosis.71 ROS-induced survival mechanisms are initiated by association of c-Src with Src homology 2 domain-containing phosphatase 1. This association activates c-Src, resulting in degradation of the pro-apoptotic protein Bim.72
However, hypoxia has been shown to specifically result in accumulation of mitochondrial ROS in osteoclasts.23,66,67 Mitochondrial ROS are essential for hypoxic enhancement of osteoclast differentiation and resorption, which is reversed by the mitochondria-specific antioxidant MitoQ. MitoQ also prevents hypoxic induction of NFκB, the calcineurin-NFAT pathway, adenosine 3’,5’-cyclic adenosine monophosphate response element-binding protein and, interestingly, HIF – all key signaling molecules in osteoclast differentiation and activity.67,73 This mitochondrial ROS pathway is proposed to be mediated by mitochondrial respiratory stress signaling, induced by hypoxic release of Ca2+ from the endoplasmic reticulum, which stimulates production of mitochondrial ROS.74 Calcium uptake by mitochondria is another mechanism that could contribute to the observed hypoxic increase in oxidative phosphorylation, having been shown to activate isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, and PDH activity.75
The importance of mitochondrial ROS in osteoclastogenesis has recently been demonstrated in mice overexpressing mitochondria-targeted catalase specifically in osteoclasts. These mice had increased bone mass due to a reduction in osteoclast formation and survival and were also protected from ovariectomy-induced bone loss.76 This demonstrates the importance of mitochondrial ROS to osteoclast activation and provides support for the hypothesis that accumulation of ROS under hypoxia directly promotes bone resorption.
HIF and osteoclast survival
Despite being resistant to short-term exposure to high levels of ROS, osteoclasts are relatively sensitive to hypoxia when compared with other cell types. At the 2% O2 optimal for stimulating osteoclast activity, osteoclast numbers fell by 16% and 35% after 24 hours and 72 hours of exposure, respectively.21,23 After 24-hour exposure to 2% O2, one in five remaining osteoclasts exhibited compromised membrane integrity, as assessed by trypan blue uptake through the aqueous pores associated with membrane damage.21
Strikingly, re-oxygenation reversed the hypoxic increase in membrane permeability and rescued osteoclasts from the early stages of cell death.21 Reversible plasma membrane permeability does occur in other cell types,77,78 and other features of early cell death, including plasma membrane phosphatidylserine exposure79–81 and mitochondrial swelling,78,82 are also reversible. This recovery explains how hypoxia/re-oxygenation is able to stimulate osteoclastogenesis,18–21 whereas continuous hypoxic exposure is inhibitory.21 Hypoxia/re-oxygenation occurs during injury, ischemia, and reperfusion and is the more likely microenvironmental characteristic to be experienced by monocytes and osteoclasts during in vivo differentiation than static hypoxia.
Sensitivity to hypoxia-induced cell death is likely to be the consequence of maintaining high rates of oxidative phosphorylation in a hypoxic environment; eventually, ROS accumulation would be expected to exceed the osteoclasts’ anti-apoptotic capacity. HIF-1α appears to regulate both aspects of this phenomenon. HIF-1α siRNA has been shown to prevent hypoxic induction of bone resorption, block the hypoxic increase in glucose consumption, and reduce hypoxic mitochondrial ETC activity, but it also rescues osteoclasts from cell death induced by chronic hypoxic exposure.21,23
This led to the hypothesis that, in hypoxic osteoclasts, functional HIF-1α-dependent pathways initially increase ATP production and bone resorption, but lack of activation of HIF-1α-dependent survival pathways eventually results in cell death. As osteoclasts are anyway short-lived cells that cannot be allowed to resorb indefinitely, permitting progressive accumulation of ROS under hypoxia may be an adaptive mechanism enabling rapid bone resorption in the short term, while ensuring that the process is halted in the absence of re-oxygenation.
In vivo effects of HIF inhibition
HIF expression by osteoclasts in vivo has only been recently described in ovariectomized (OVX) mice which develop osteoporosis,5 in the multinucleated giant cells associated with giant cell tumor of bone,26 and in resorbing osteoclasts within the rheumatoid synovium.59 However, given that hypoxia and HIF are generally associated with disease progression, HIF inhibition83 had already been approached as a potential method of improving disease severity in a number of bone resorption conditions.
In murine models of osteolytic breast cancer, treatment with 2-methoxyestradiol (2ME) and its analogs induced apoptosis of osteoclasts and their precursors, protected against tumor-induced osteolysis in vivo and inhibited bone resorption in vitro.84,85 Similarly, in murine models of rheumatoid arthritis treatment with endostatin, bortezomib or 2ME improved incidence and severity of arthritis, as well as scores for subchondral bone erosion.86–89 In OVX mice, 2ME or apigenin preserved bone mineral density, improved concentrations of serum markers of bone turnover, and inhibited osteoclast formation ex vivo.5,87,90
Some of this inhibitory effect is likely via direct actions of HIF inhibition on osteoclast activity. Ovariectomy results in estrogen depletion, which has been shown to stabilize HIF-1α protein in osteoporosis-associated osteoclasts in a manner independent of the oxygenation status.5 Using osteoclast-specific conditional HIF-1α knockout mice, it was shown that HIF-1α was essential for the bone loss seen under conditions of estrogen depletion, due to reduced rates of osteoclast formation and reduced bone resorption.5
Another contributory factor is likely to be inhibition of secretion of HIF-regulated osteoclastogenic factors. VEGF and ANGPTL4 stimulate osteoclast differentiation91 and resorption activity,22 respectively. Serum concentrations of both factors are elevated in patients with rheumatoid arthritis, with high serum concentrations being associated with elevated markers of bone resorption.59,92
What about the osteoblasts?
This review has focused on HIF-mediated regulation of osteoclast activity, the data for which indicates HIF inhibition as a good strategy for targeted therapies to prevent or inhibit pathological bone loss in a number of different conditions.
However, as was mentioned in the “Introduction” section, bone remodeling is regulated by a balance between osteoblast-mediated bone formation and osteoclast-mediated bone resorption. The processes of bone formation (osteogenesis) and angiogenesis are tightly linked during bone development and bone repair, and HIF has been shown to regulate this osteogenic–angiogenic coupling. Using mice with osteoblast-specific deletions in either the von Hippel-Lindau gene (VHL) or HIF-1α, it was shown that hypoxia promotes bone formation by osteoblasts in vivo via HIF-1α-mediated induction of pro-angiogenic cytokines such as VEGF.93 Subsequent work confirmed that HIF plays a central role in regulating bone formation during skeletal development.94,95
This, and other, osteoblast-directed research led to the hypothesis that HIF pathway activation might be a therapeutic strategy to ameliorate pathological bone loss. Treatment with either HIF prolyl hydroxylase domain enzyme inhibitors inhibitors or CoCl2 has been described to improve bone health in murine models of fracture96–98 and osteoporosis99,100 by stimulating angiogenesis and concomitant bone formation.
To delve into the reasons behind the apparent discrepancy in these approaches is beyond the remit of this review. Taken at face value, it would seem that osteoclast activity is more sensitive to HIF inhibition and that bone formation is more sensitive to HIF activation, potentially due to HIF-dependent induction of OPG, an inhibitor of osteoclast activity.95,101 Intriguingly, intervention in either direction apparently tips the homeostatic balance back in favor of maintaining or improving bone integrity.
Conclusion
HIF, specifically HIF-1α, is closely involved in both monocyte–osteoclast differentiation and bone resorption by mature osteoclasts, especially within the hypoxic microenvironment so closely associated with many pathological bone resorption conditions. HIF stimulates the expression of cytokines that regulate the differentiation and resorption process. It also increases both the glycolytic and mitochondrial metabolic rate in order to generate sufficient ATP to support the hypoxic increase in bone resorption. Generation of mitochondrial ROS, a by-product of maintaining mitochondrial respiratory activity under hypoxic conditions, is itself necessary to enable this high rate of resorption. These data suggest HIF as an attractive therapeutic target in osteolytic disease, although further research is urgently needed in order to align this with work on osteoblastic aspects of bone disease.
Acknowledgments
This work was supported by grants from Arthritis Research UK, the Rosetrees Trust and the Oxford National Institute of Health Research (NIHR) Musculoskeletal Biomedical Research Unit (BRU). Thanks goes to Professor Nick Athanasou for critically reading the manuscript.
Disclosure
The author reports no conflicts of interest in this work.
References
Papachristou DJ, Basdra EK, Papavassiliou AG. Bone metastases: molecular mechanisms and novel therapeutic interventions. Med Res Rev. 2012;32(3):611–636. | |
Zeng W, Wan R, Zheng Y, Singh SR, Wei Y. Hypoxia, stem cells and bone tumor. Cancer Lett. 2011;313(2):129–136. | |
Kaarela K. Prognostic factors and diagnostic criteria in early rheumatoid arthritis. Scand J Rheumatol. 1985;57:1–54. | |
Treuhaft PS, McCarty DJ. Synovial fluid pH, lactate, oxygen and carbon dioxide partial pressure in various joint diseases. Arthritis Rheum. 1971; 14(4):475–484. | |
Miyauchi Y, Sato Y, Kobayashi T, et al. HIF1alpha is required for osteoclast activation by estrogen deficiency in postmenopausal osteoporosis. Proc Natl Acad Sci U S A. 2013;110(41):16568–16573. | |
Pountos I, Panteli M, Panagiotopoulos E, Jones E, Giannoudis PV. Can we enhance fracture vascularity: what is the evidence? Injury. 2014; 45(Suppl 2):S49–S57. | |
Evans RA, Morgan MD. The systemic nature of chronic lung disease. Clin Chest Med. 2014;35(2):283–293. | |
Bromley M, Woolley DE. Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis Rheum. 1984;27(9):968–975. | |
Gough A, Sambrook P, Devlin J, et al. Osteoclastic activation is the principal mechanism leading to secondary osteoporosis in rheumatoid arthritis. J Rheumatol. 1998;25(7):1282–1289. | |
Tella SH, Gallagher JC. Biological agents in management of osteoporosis. Eur J Clin Pharmacol. 2014;70(11):1291–1301. | |
Krzeszinski JY, Wan Y. New therapeutic targets for cancer bone metastasis. Trends Pharmacol Sci. 2015;36(6):360–373. | |
Fujikawa Y, Quinn JM, Sabokbar A, McGee JO, Athanasou NA. The human osteoclast precursor circulates in the monocyte fraction. Endocrinology. 1996;137(9):4058–4060. | |
Quinn JM, Elliott J, Gillespie MT, Martin TJ. A combination of osteoclast differentiation factor and macrophage-colony stimulating factor is sufficient for both human and mouse osteoclast formation in vitro. Endocrinology. 1998;139(10):4424–4427. | |
Fuller K, Kirstein B, Chambers TJ. Regulation and enzymatic basis of bone resorption by human osteoclasts. Clin Sci (Lond). 2007; 112(11):567–575. | |
Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4(8):638–649. | |
Broxmeyer HE, Cooper S, Lu L, Miller ME, Langefeld CD, Ralph P. Enhanced stimulation of human bone marrow macrophage colony formation in vitro by recombinant human macrophage colony-stimulating factor in agarose medium and at low oxygen tension. Blood. 1990;76(2):323–329. | |
Strehl C, Fangradt M, Fearon U, Gaber T, Buttgereit F, Veale DJ. Hypoxia: how does the monocyte-macrophage system respond to changes in oxygen availability? J Leukoc Biol. 2014;95(2):233–241. | |
Arnett TR, Gibbons DC, Utting JC, et al. Hypoxia is a major stimulator of osteoclast formation and bone resorption. J Cell Physiol. 2003; 196(1):2–8. | |
Muzylak M, Price JS, Horton MA. Hypoxia induces giant osteoclast formation and extensive bone resorption in the cat. Calcif Tissue Int. 2006;79(5):301–309. | |
Utting JC, Flanagan AM, Brandao-Burch A, Orriss IR, Arnett TR. Hypoxia stimulates osteoclast formation from human peripheral blood. Cell Biochem Funct. 2010;28(5):374–380. | |
Knowles HJ, Athanasou NA. Acute hypoxia and osteoclast activity: a balance between enhanced resorption and increased apoptosis. J Pathol. 2009;218(2):256–264. | |
Knowles HJ, Cleton-Jansen AM, Korsching E, Athanasou NA. Hypoxia-inducible factor regulates osteoclast-mediated bone resorption: role of angiopoietin-like 4. FASEB J. 2010;24(12):4648–4659. | |
Morten KJ, Badder L, Knowles HJ. Differential regulation of HIF-mediated pathways increases mitochondrial metabolism and ATP production in hypoxic osteoclasts. J Pathol. 2013;229(5):755–764. | |
Dandajena TC, Ihnat MA, Disch B, Thorpe J, Currier GF. Hypoxia triggers a HIF-mediated differentiation of peripheral blood mononuclear cells into osteoclasts. Orthod Craniofac Res. 2012;15(1):1–9. | |
Bhaskara VK, Mohanam I, Gujrati M, Mohanam S. Intermittent hypoxia effect on osteoclastogenesis stimulated by neuroblastoma cells. PLoS One. 2014;9(8):e105555. | |
Knowles HJ, Athanasou NA. Hypoxia-inducible factor is expressed in giant cell tumour of bone and mediates paracrine effects of hypoxia on monocyte-osteoclast differentiation via induction of VEGF. J Pathol. 2008;215(1):56–66. | |
Kurowska-Stolarska M, Distler JH, Jüngel A, et al. Inhibitor of DNA binding/differentiation 2 induced by hypoxia promotes synovial fibroblast-dependent osteoclastogenesis. Arthritis Rheum. 2009; 60(12):3663–3675. | |
Fukuoka H, Aoyama M, Miyazawa K, Asai K, Goto S. Hypoxic stress enhances osteoclast differentiation via increasing IGF2 production by non-osteoclastic cells. Biochem Biophys Res Commun. 2005; 328(4):885–894. | |
Hinoi E, Ochi H, Takarada T, et al. Positive regulation of osteoclastic differentiation by growth differentiation factor 15 upregulated in osteocytic cells under hypoxia. J Bone Miner Res. 2012;27(4):938–949. | |
Shirakura M, Tanimoto K, Eguchi H, et al. Activation of the hypoxia-inducible factor-1 in overloaded temporomandibular joint, and induction of osteoclastogenesis. Biochem Biophys Res Commun. 2010;393(4):800–805. | |
Bozec A, Bakiri L, Hoebertz A, et al. Osteoclast size is controlled by Fra-2 through LIF/LIF-receptor signalling and hypoxia. Nature. 2008; 454(7201):221–225. | |
Al Hadi H, Smerdon GR, Fox SW. Hyperbaric oxygen therapy suppresses osteoclast formation and bone resorption. J Orthop Res. 2013; 31(11):1839–1844. | |
Hadi HA, Smerdon G, Fox SW. Osteoclastic resorptive capacity is suppressed in patients receiving hyperbaric oxygen therapy. Acta Orthop. 2015;86(2):264–269. | |
Leger AJ, Altobelli A, Mosquea LM, et al. Inhibition of osteoclastogenesis by prolyl hydroxylase inhibitor dimethyloxallyl glycine. J Bone Miner Metab. 2010;28(5):510–519. | |
Andrews RE, Shah KM, Wilkinson JM, Gartland A. Effects of cobalt and chromium ions at clinically equivalent concentrations after metal-on-metal hip replacement on human osteoblasts and osteoclasts: implications for skeletal health. Bone. 2011;49(4):717–723. | |
Patntirapong S, Habibovic P, Hauschka PV. Effects of soluble cobalt and cobalt incorporated into calcium phosphate layers on osteoclast differentiation and activation. Biomaterials. 2009;30(4):548–555. | |
Francis MJ, Lees RL, Trujillo E, Martin-Vasallo P, Heersche JN, Mobasheri A. ATPase pumps in osteoclasts and osteoblasts. Int J Biochem Cell Biol. 2002;34(5):459–476. | |
Väänänen HK, Karhukorpi EK, Sundquist K, et al. Evidence for the presence of a proton pump of the vacuolar H(+)-ATPase type in the ruffled borders of osteoclasts. J Cell Biol. 1990;111(3):1305–1311. | |
Dudley HR, Spiro D. The fine structure of bone cells. J Biophys Biochem Cytol. 1961;11(3):627–649. | |
Czupalla C, Mansukoski H, Pursche T, Krause E, Hoflack B. Comparative study of protein and mRNA expression during osteoclastogenesis. Proteomics. 2005;5(15):3868–3875. | |
Kim JM, Jeong D, Kang HK, Jung SY, Kang SS, Min BM. Osteoclast precursors display dynamic metabolic shifts toward accelerated glucose metabolism at an early stage of RANKL-stimulated osteoclast differentiation. Cell Physiol Biochem. 2007;20(6):935–946. | |
Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. 2007;405(1):1–9. | |
Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40(2):294–309. | |
Fukuda R, Zhang HF, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129(1):111–122. | |
Seagroves TN, Ryan HE, Lu H, et al. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol. 2001;21(10):3436–3444. | |
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3(3):187–197. | |
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–185. | |
Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–10903. | |
Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. | |
Indo Y, Takeshita S, Ishii KA, et al. Metabolic regulation of osteoclast differentiation and function. J Bone Miner Res. 2013;28(11):2392–2399. | |
Williams JP, Blair HC, McDonald JM, et al. Regulation of osteoclastic bone resorption by glucose. Biochem Biophys Res Commun. 1997; 235(3):646–651. | |
Larsen KI, Falany M, Wang W, Williams JP. Glucose is a key metabolic regulator of osteoclasts; glucose stimulated increases in ATP/ADP ratio and calmodulin kinase II activity. Biochem Cell Biol. 2005; 83(5):667–673. | |
Larsen KI, Falany ML, Ponomareva LV, Wang W, Williams JP. Glucose-dependent regulation of osteoclast H(+)-ATPase expression: potential role of p38 MAP-kinase. J Cell Biochem. 2002;87(1):75–84. | |
Su Y, Zhou A, Al-Lamki RS, Karet FE. The a-subunit of the V-type H+-ATPase interacts with phosphofructokinase-1 in humans. J Biol Chem. 2003;278(22):20013–20018. | |
Li B, Yu S. Genistein prevents bone resorption diseases by inhibiting bone resorption and stimulating bone formation. Biol Pharm Bull. 2003; 26(6):780–786. | |
El Hajj Dib I, Gallet M, Mentaverri R, Sevenet N, Brazier M, Kamel S. Imatinib mesylate (Gleevec) enhances mature osteoclast apoptosis and suppresses osteoclast bone resorbing activity. Eur J Pharmacol. 2006;551(1–3):27–33. | |
Pereira I, Fialho S, Castro G, Zimmermann A. Imatinib mesylate induces clinical remission in rheumatoid arthritis. Joint Bone Spine. 2010;77(4):372–373. | |
Ames PR, Aye WW, Beatty C, O’Reilly D. Imatinib treatment of seropositive arthritis in a young woman with chronic myeloid leukemia. J Rheumatol. 2008;35(8):1682. | |
Swales C, Athanasou NA, Knowles HJ. Angiopoietin-like 4 is over-expressed in rheumatoid arthritis patients: association with pathological bone resorption. PLoS One. 2014;9(10):e109524. | |
Aoki J, Watanabe H, Shinozaki T, et al. FDG PET of primary benign and malignant bone tumors: standardized uptake value in 52 lesions. Radiology. 2001;219(3):774–777. | |
Strauss LG, Dimitrakopoulou-Strauss A, Koczan D, et al. 18F-FDG kinetics and gene expression in giant cell tumors. J Nucl Med. 2004; 45(9):1528–1535. | |
Houten SM, Chegary M, Te Brinke H, et al. Pyruvate dehydrogenase kinase 4 expression is synergistically induced by AMP-activated protein kinase and fatty acids. Cell Mol Life Sci. 2009;66(7):1283–1294. | |
Laderoute KR, Amin K, Calaoagan JM, et al. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26(14):5336–5347. | |
Mungai PT, Waypa GB, Jairaman A, et al. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol. 2011;31(17):3531–3545. | |
Lee YS, Kim YS, Lee SY, et al. AMP kinase acts as a negative regulator of RANKL in the differentiation of osteoclasts. Bone. 2010; 47(5):926–937. | |
Srinivasan S, Avadhani NG. Hypoxia-mediated mitochondrial stress in RAW264.7 cells induces osteoclast-like TRAP-positive cells. Ann NY Acad Sci. 2007;1117:51–61. | |
Srinivasan S, Koenigstein A, Joseph J, et al. Role of mitochondrial reactive oxygen species in osteoclast differentiation. Ann NY Acad Sci. 2010;1192(1):245–252. | |
Callaway DA, Jiang JX. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J Bone Miner Metab. 2015;33(4):359–370. | |
Kim MS, Yang YM, Son A, et al. RANKL-mediated reactive oxygen species pathway that induces long lasting Ca2+ oscillations essential for osteoclastogenesis. J Biol Chem. 2010;285(10):6913–6921. | |
Nakashima T, Takayanagi H. New regulation mechanisms of osteoclast differentiation. Ann NY Acad Sci. 2011;1240:E13–E18. | |
Vääräniemi J, Halleen JM, Kaarlonen K, et al. Intracellular machinery for matrix degradation in bone-resorbing osteoclasts. J Bone Miner Res. 2004;19(9):1432–1440. | |
Ke K, Sul OJ, Choi EK, Safdar AM, Kim ES, Choi HS. Reactive oxygen species induce the association of SHP-1 with c-Src and the oxidation of both to enhance osteoclast survival. Am J Physiol Endocrinol Metab. 2014;307(1):E61–E70. | |
Guha M, Srinivasan S, Koenigstein A, Zaidi M, Avadhani NG. Enhanced osteoclastogenesis by mitochondrial retrograde signaling through transcriptional activation of the cathepsin K gene. Ann NY Acad Sci. Epub 2015 Mar 18. | |
Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262(5134):744–747. | |
McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70(2):391–425. | |
Bartell SM, Kim HN, Ambrogini E, et al. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation. Nat Commun. 2014;5:3773. | |
Thelestam M, Mollby R. Survival of cultured cells after functional and structural disorganization of plasma membrane by bacterial haemolysins and phospholipases. Toxicon. 1983;21(6):805–815. | |
Pena C, Zhou Y, Lust D, Pilar G. Restoration of mitochondrial function reverses developmental neuronal death in vitro. J Comp Neurol. 2001; 440(2):156–176. | |
Hammill AK, Uhr JW, Scheuermann RH. Annexin V staining due to loss of membrane asymmetry can be reversible and precede commitment to apoptotic death. Exp Cell Res. 1999;251(1):16–21. | |
Yang MY, Chuang H, Chen RF, Yang KD. Reversible phosphatidylserine expression on blood granulocytes related to membrane perturbation but not DNA strand breaks. J Leukoc Biol. 2002;71(2):231–237. | |
Martin S, Pombo I, Poncet P, David B, Arock M, Blank U. Immunologic stimulation of mast cells leads to the reversible exposure of phosphatidylserine in the absence of apoptosis. Int Arch Allergy Immunol. 2000;123(3):249–258. | |
Minamikawa T, Williams DA, Bowser DN, Nagley P. Mitochondrial permeability transition and swelling can occur reversibly without inducing cell death in intact human cells. Exp Cell Res. 1999; 246(1):26–37. | |
Xia Y, Choi HK, Lee K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur J Med Chem. 2012;49:24–40. | |
Snoeks TJ, Mol IM, Que I, Kaijzel EL, Lowik CW. 2-methoxyestradiol analogue ENMD-1198 reduces breast cancer-induced osteolysis and tumor burden both in vitro and in vivo. Mol Cancer Ther. 2011; 10(5):874–882. | |
Cicek M, Iwaniec UT, Goblirsch MJ, et al. 2-methoxyestradiol suppresses osteolytic breast cancer tumor progression in vivo. Cancer Res. 2007;67(21):10106–10111. | |
Kurosaka D, Yoshida K, Yasuda J, et al. Inhibition of arthritis by systemic administration of endostatin in passive murine collagen induced arthritis. Ann Rheum Dis. 2003;62(7):677–679. | |
Stubelius A, Andréasson E, Karlsson A, et al. Role of 2-methoxyestradiol as inhibitor of arthritis and osteoporosis in a model of postmenopausal rheumatoid arthritis. Clin Immunol. 2011;140(1):37–46. | |
Plum SM, Park EJ, Strawn SJ, Moore EG, Sidor CF, Fogler WE. Disease modifying and antiangiogenic activity of 2-methoxyestradiol in a murine model of rheumatoid arthritis. BMC Musculoskelet Disord. 2009;10:46. | |
Yannaki E, Papadopoulou A, Athanasiou E, et al. The proteasome inhibitor bortezomib drastically affects inflammation and bone disease in adjuvant-induced arthritis in rats. Arthritis Rheum. 2010; 62(11):3277–3288. | |
Goto T, Hagiwara K, Shirai N, Yoshida K, Hagiwara H. Apigenin inhibits osteoblastogenesis and osteoclastogenesis and prevents bone loss in ovariectomized mice. Cytotechnology. 2015;67(2):357–365. | |
Niida S, Kaku M, Amano H, et al. Vascular endothelial growth factor can substitute for macrophage colony-stimulating factor in the support of osteoclastic bone resorption. J Exp Med. 1999;190(2):293–298. | |
Ballara S, Taylor PC, Reusch P, et al. Raised serum vascular endothelial growth factor levels are associated with destructive change in inflammatory arthritis. Arthritis Rheum. 2001;44(9):2055–2064. | |
Wang Y, Wan C, Deng L, et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest. 2007;117(6):1616–1626. | |
Wan C, Shao J, Gilbert SR, et al. Role of HIF-1alpha in skeletal development. Ann NY Acad Sci. 2010;1192:322–326. | |
Wu C, Rankin EB, Castellini L, et al. Oxygen-sensing PHDs regulate bone homeostasis through the modulation of osteoprotegerin. Genes Dev. 2015;29(8):817–831. | |
Shen X, Wan C, Ramaswamy G, et al. Prolyl hydroxylase inhibitors increase neoangiogenesis and callus formation following femur fracture in mice. J Orthop Res. 2009;27(10):1298–1305. | |
Huang J, Liu L, Feng M, et al. Effect of CoCl2 on fracture repair in a rat model of bone fracture. Mol Med Rep. 2015;12(4):5951–5956. | |
Wan C, Gilbert SR, Wang Y, et al. Activation of the hypoxia-inducible factor-1alpha pathway accelerates bone regeneration. Proc Natl Acad Sci U S A. 2008;105(2):686–691. | |
Liu X, Tu Y, Zhang L, Qi J, Ma T, Deng L. Prolyl hydroxylase inhibitors protect from the bone loss in ovariectomy rats by increasing bone vascularity. Cell Biochem Biophys. 2014;69(1):141–149. | |
Peng J, Lai ZG, Fang ZL, et al. Dimethyloxalylglycine prevents bone loss in ovariectomized C57BL/6J mice through enhanced angiogenesis and osteogenesis. PLoS One. 2014;9(11):e112744. | |
Shao J, Zhang Y, Yang T, Qi J, Zhang L, Deng L. HIF-1alpha disturbs osteoblasts and osteoclasts coupling in bone remodeling by up-regulating OPG expression. In Vitro Cell Dev Biol. 2015;51(8):808–814. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.