Back to Journals » Journal of Inflammation Research » Volume 14
Hypothermia Protects Mice Against Ischemic Stroke by Modulating Macrophage Polarization Through Upregulation of Interferon Regulatory Factor-4
Authors Yu X, Feng Y, Liu R, Chen Q
Received 25 January 2021
Accepted for publication 24 March 2021
Published 7 April 2021 Volume 2021:14 Pages 1271—1281
DOI https://doi.org/10.2147/JIR.S303053
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Xinyuan Yu, Yanping Feng, Renzhong Liu, Qianxue Chen
Department of Neurosurgery, Renmin Hospital of Wuhan University, Wuhan, Hubei, 430060, People’s Republic of China
Correspondence: Qianxue Chen
Department of Neurosurgery, Renmin Hospital of Wuhan University, Wuhan, Hubei, 430060, People’s Republic of China
Tel +86 13607141618
Email [email protected]
Background: Therapeutic hypothermia (TH) has been proven to be protective in ischemic stroke (IS) due to its anti-inflammatory capacity. Recently, the interferon regulatory factor 4 (IRF4) has been characterized as a central regulator of neuroinflammation in IS. Here we aim to determine whether IFR4 contributes to the neuroprotective effects of TH in IS.
Methods: In the present study, IRF4 knockout (IRF4−/-) and wild-type (IRF4+/+) mice were treated with or without TH after IS. Cerebral IRF4 expression, the production of pro-inflammatory and anti-inflammatory cytokines and macrophage polarization were determined at 8 hours after reperfusion. In addition, cerebral infarct volume and neurological function were evaluated at 7 days after IS.
Results: TH attenuates IS together with enhanced IRF4 expression as well as reduced production of pro-inflammatory cytokines. In addition, TH increased M2 macrophage polarization while inhibited M1 macrophage polarization. However, IRF4 knockout worsens neurological outcomes of stoke mice. The expression of pro-inflammatory cytokines were markedly increased in IRF4−/- mice as compared with IRF4+/+ mice at 8 h after stroke. Moreover, IRF4 knockout driven the macrophage polarization toward M1phenotype at 8 h after stroke. Most importantly, IRF4 knockout abolished the neuroprotective and anti-inflammatory effects of TH in IS.
Conclusion: Together, we report for the first time that TH attenuates neuroinflammation following IS by modulating M1/M2 macrophage polarization through the upregulation of IRF4 expression.
Keywords: therapeutic hypothermia, interferon regulatory factor 4, ischemic stroke, inflammation, macrophage polarization
Introduction
Stroke is the leading cause of long-term disability and one of the most common causes of death around the world.1 Ischemic stroke (IS) represents the majority of strokes and is currently the main focus of stroke research.2 Cerebral injuries following IS result from a complex series of pathophysiological events including oxidative stress, excitotoxicity, inflammation and apoptosis.3 Despite marked process in understanding the molecular mechanisms of IS, current standard treatments including thrombolytic therapy and endovascular reperfusion are only used for very few stroke patients due to narrow window.4 Therefore, researchers are competing to improve new strategies that guard the brain from ischemic injury. Normobaric oxygenation therapy, hypothermia, and approaches to support collateral flow are promising treatments for stroke.3
Therapeutic hypothermia (TH) is one of the most commonly used neuroprotectants at both laboratory and clinical levels.5 Neuroprotective properties of hypothermia have been established in the past 30 years,6 early implementation of TH will decrease metabolic demand and activities of neurons and thereby prevents shift toward anaerobic metabolism.7 In fact, the protective effects of TH extend beyond the acute phase. During subacute phase of ischemic stroke, TH can prevent cell apoptosis via both caspase-dependent and caspase-independent pathways.8 Most importantly, TH limits inflammatory response following cerebral ischemia that leads to better outcomes.9,10 Current evidences indicate that TH exerts its anti-inflammatory properties mainly through the inhibition of NF-κB and MAPK signaling pathways.11,12 In addition, release of various pro-inflammatory cytokines and chemokines is also reduced by TH.13,14 However, the underlying mechanism by which hypothermia reduces neuroinflammation has not yet been completely understood.
Interferon regulatory factor 4 (IRF4) has been demonstrated as a key transcription factor, playing important roles in modulating inflammatory response.15 In addition, IRFs participate in the regulation of M1/M2 macrophage polarization,16 which plays a critical role in the regulation of neuroinflammation following cerebral ischemia.17 It has been reported that IRF4 regulates M2 macrophage polarization in inflammatory process.18 In addition, a very recent study revealed that IRF4 regulates microglial M2 activation and impact on stroke outcomes.19 Most importantly, IRF4 has been identified as a mediator of neuronal survival in IS.20 Since TH is also able to enhance M2 macrophage polarization,21 it is reasonable to speculate that IRF4 probably contributes to the anti-inflammatory properties of TH in IS. In the present study, we aim to determine the effects of TH on cerebral IRF4 expression and investigate whether TH attenuates neuroinflammation following IS through the regulation of IRF4 expression.
Methods
Animals
Wild-type (IRF4+/+) and IRF4 KO (IRF4−/-) mice are in a C57BL/6 background. IRF4+/+ mice were purchased from the animal laboratory of Hubei Provincial Center for Disease Control and Prevention (Wuhan, China), and the IRF4−/- mice were established by Cyagen (Suzhou, China) as previously described.20 Animals were kept in cages and free to water and food. Animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Research Council (US) Committee in 201121 and were approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University (AUP20190120).
Study Design
To establish a murine IS model, IRF4+/+ and IRF4−/- mice received transient middle cerebral artery occlusion (tMCAO). Once tMCAO was successfully performed, animals were randomly assigned into five following groups: (1) a control group (Control, n=12): healthy wild-type animals received sham operation without treatment; (2) a ischemic stroke group (IS, n=12): wild-type (IRF4+/+) mice received tMCAO for 90 min without treatment; (3) an IRF4−/- stroke group (IS + IRF4 KO, n=12): IRF4 knockout (IRF4−/-) mice received tMCAO for 90 min without treatment; (4) a therapeutic hypothermia group (IS + TH, n=12): IRF4+/+ mice received tMCAO for 90 min with TH; and (5) a IRF4−/- stroke + TH group (IS + TH + IRF4 KO, n=12): IRF4−/- mice received tMCAO for 90 min with TH. TH was initiated at the onset of ischemic stroke and ended at 8 hours after reperfusion. At the end of TH, 6 mice in each group were sacrificed for biochemical analysis. Other animals in each group were used for histological and neurological evaluation at 7 days after stroke. Study diagram is summarized in Figure 1A.
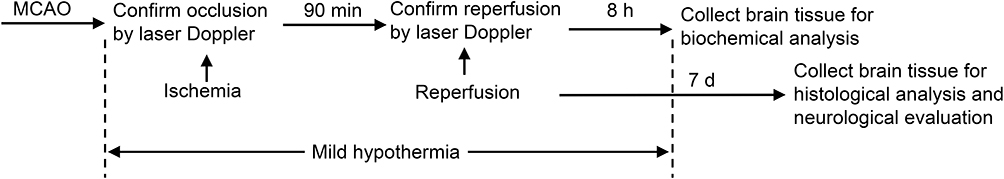 |
Figure 1 Study protocol. Murine model of IS was established by tMCAO. Ischemia and reperfusion were confirmed by laser Doppler. Abbreviations: IS, ischemic stroke; TH, therapeutic hypothermia; tMCAO, transient middle cerebral artery occlusion. |
Cerebral Ischemia/Reperfusion Model
Animals were fasted overnight and anesthetized by an intraperitoneal injection of pentobarbital sodium (50mg/kg, #Y0002194, Sigma–Aldrich, St. Louis, USA). After 15 min of stabilization, the murine IS model was established by tMCAO as previously reported.22 The left cerebral middle artery was blocked by a filament and maintained for 90 min to induce ischemic injury. Sham operation was defined as the exposure of left cerebral middle artery but without blockage. The occlusion and reperfusion were confirmed by laser Doppler (PeriFlux5000, PERIMED, Switzerland).
Induction of Hypothermia
Hypothermia was achieved by using intraperitoneal injection of HPI-201 as previously described.23 Briefly, the first bolus injection of HPI-201 (2 mg/kg body weight) was given at the onset of IS (occlusion). To ensure a constant mild hypothermia at 33°C, additional injections of HPI-201 (1 mg/kg per injection) were given with an interval of at least 1.5 hours. Rectal temperature was monitored with a rectal probe.
2,3,5-Triphenyl-Tetrazolium-Chloride (TTC) Staining
At 7 days after stroke, animals were sacrificed by blood dropping under anesthesia without perfusion. Then, brains were removed quickly and frozen at 0°C for 10 min, then cut into sections (2-mm thickness). Slices were immersed for 10 min into 2% 2,3,5-triphenyl-tetrazolium-chloride (TTC, #T8877, Sigma–Aldrich) at room temperature and were post-fixed in 4% cold paraformaldehyde overnight. Finally, TTC-stained slices were scanned by a photo scanner.
Enzyme-Linked Immunosorbent Assay (ELISA)
The production of pro-inflammatory cytokines including IL-1β (#SMLB00C), IL-6 (#SM6000B) and TNF-α (#SMTA00B) and anti-inflammatory cytokines including IL-4 (#SM4000B), IL-10 (#SM1000B) and TGF-β (#SMB100B) were determined using commercial detection kits (R&D Systems, Minneapolis, MN). Blocking, hybridization, washing conditions, and detection steps were performed according to the manufacturer’s guidelines.
Quantitative Real-Time PCR
Mice were transcardially perfused with cold PBS and the brains were then immediately removed. Total RNA was extracted using TRIZOL reagent (#15596026, Invitrogen, MA, USA) and was reverse-transcribed into cDNA using a Transcriptor First Strand cDNA Synthesis Kit (#11117831001, Roche, Indianapolis, IN, USA). Quantitative real-time PCR analysis was performed with the LightCycler 480 QPCR System (Roche) with following conditions: 95°C for 10 min; 40 cycles of 95°C for 10 s, 60°C for 10 s, 72°C for 20 s and 72°C for 10 min.
Western Blotting
Brain penumbra tissues were collected after transcardial perfusion with cold PBS (pH: 7.4). Then, tissue samples were lysed in lysis buffer containing protease inhibitor (#P8340, Sigma-Aldrich). The supernatant was collected after centrifugation (15,000 g, 10 min, 4°C) and protein concentration was determined using a bicinchoninic acid assay kit (#23228, Pierce Biotechnology, IL, USA). Equivalent amounts of protein (50 μg per well) were separated on SDS-PAGE gels and transferred onto PVDF membranes. The membranes were incubated in blocking buffer for 1 h and then immersed in primary antibodies at 4°C overnight. The membranes were washed with TBST and then incubated with secondary antibodies (#C41110-01, LI-COR Biosciences, NE, USA) for 1 h at room temperature. We used an Odyssey infrared imaging system (LI-COR Biosciences) to detect the blot signals and quantified signals using an Odyssey software (LI-COR). GAPDH served as a loading control.
Immunofluorescence Staining
Brain tissues were fixed in 4% paraformaldehyde and cut into sections of 30 μm in thickness. Sections were incubated overnight with primary antibodies including F4/80 (ab6640, Abcam), iNOS (ab210823, Abcam), or CD206 (Santa Cruz Biotechnology, CA, USA) at 4°C overnight. Then, sections were incubated with secondary antibody for 1 hour at room temperature. Nuclear staining was performed with DAPI (62,248, Thermo Scientific, MA, USA). The fluorescence images were captured by an LSM700 imaging system (Carl Zeiss, Aalen, Germany). The number of total F4/80/iNOS or F4/80/CD206 positive cells were counted using Image J software (National Institutes of Health, MD, USA).
Neurological Function Evaluation
The neurological function of ischemic stroke mice at 7 days after reperfusion was evaluated by two independent investigators who were unaware of the study design. Once a disagreement occurred between two investigators, a third investigator would make the final decision. Neurological deficits were measured using a 9-point scale as previously described.23 The scale was based on the following observations: 0 point: absence of neurological deficits; 1 point: left forelimb flexion upon suspension by the tail or failure to fully extend the right forepaw; 2 points: left shoulder adduction upon suspension by the tail; 3 points: reduced resistance to a lateral push towards the left; 4 points: spontaneous movement in all directions with circling to the left only if pulled by the tail; 5 points: circling or walking spontaneously only to the left; 6 points: walking only when stimulated; 7 points: no response to stimulation; 8 points: death after stoke.
Morris Water Maze Test
The learning and memory abilities of mice were evaluated by Morris water maze test as previously described.24 Water maze test was performed at 7 days after reperfusion. Animals were trained for 4 days and 4 times per day. The time of searching for platform was recorded as escape latency (EL) and the mean value was calculated.
Statistical Analysis
Data were expressed as number, percentage or mean ± standard deviation (SD). Comparisons were made by unpaired Student’s t-test or one-way ANOVA test wherever appropriate. Neurological deficit scores were compared using the Steel-Dwass test followed by the Kruskal–Wallis test. A two-tailed p value less than 0.05 was considered to indicate significant difference. Statistical analysis was performed using GraphPad Prism (GraphPad Software, Inc., CA, US).
Results
TH Enhanced IRF4 Expressions After Ischemic Stroke
Murine IS model was generated by tMCAO with IRF4 wild-type (IRF4+/+) and knockout (IRF4−/-) mice. The left artery occlusion and reperfusion were confirmed by laser Doppler (Figure 1). Brain penumbra tissue samples were collected at 8 hours after reperfusion for the detection of IRF4 expression. The efficiency of IRF4 knockout in mice was confirmed by RT-PCR and Western-blot assays (data not shown). As seen in Figure 2A and B, our results showed that the expression of IRF4 in brain penumbra tissue in the IS group was significantly higher than that of the control group (p<0.001). In addition, IRF4 expression in the IS + TH group was significantly increased as compared with the IS group (p<0.01). These findings indicated that TH was able to enhance IRF4 expression in IS mice. However, cerebral IRF4 expressions in the IS + IRF4 KO and the IS + IRF4 KO + TH groups were markedly decreased as compared with the IS and the IS + TH groups (p<0.01, respectively).
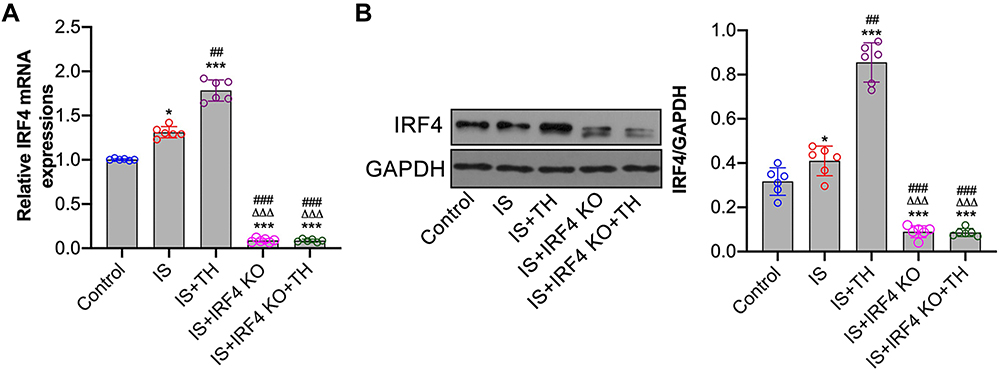 |
Figure 2 Cerebral IRF4 expressions after IS. RT-PCR (A) and Western-blots (B) were used to measure IRF4 mRNA and protein expressions, respectively. n = 6 in each group. Data were expressed as mean±SD. Comparisons of continuous variables between groups were made by unpaired Student’s t-test. *p < 0.05, ***p < 0.001 vs control group; ##p<0.01, ###p<0.001 vs IS group; ΔΔΔp<0.001 vs IS + TH group. Abbreviations: IS, ischemic stroke; TH, therapeutic hypothermia; IRF4 KO, interferon regulatory factor 4 knockout. |
TH Inhibited Neuroinflammation Through the Upregulation of IRF4
Compared to sham-operated mice, the production of pro-inflammatory cytokines including IL-1β, IL-6 and TNF-α was upregulated in IS mice (p<0.01, respectively). TH treatments markedly reduced the production of pro-inflammatory cytokines in brain penumbra tissues (p<0.01, respectively). However, IRF4 knockout resulted in significant increases in the production of pro-inflammatory cytokines including IL-1β, IL-6 and TNF-α (p<0.01, respectively). Most importantly, IRF4 knockout abolished the anti-inflammatory effects of TH in IS mice. In detail, the expression levels of IL-1β, IL-6 and TNF-α were significantly higher in the IS + IRF4 KO + TH group than that of the IS + TH group (p<0.01, respectively). We also measured the expression of anti-inflammatory cytokines including IL-4, IL-10 and TGF-β in ischemic brain tissues. Our results showed that TH enhanced the production of anti-inflammatory cytokines while IRF4 KO markedly reduced the production of IL-4, IL-10 and TGF-β. In addition, the expressions of IL-4, IL-10 and TGF-β were significantly increased in the IS + IRF4 KO + TH group as compared with the IS + TH group (p<0.01, respectively). Data are summarized in Figure 3.
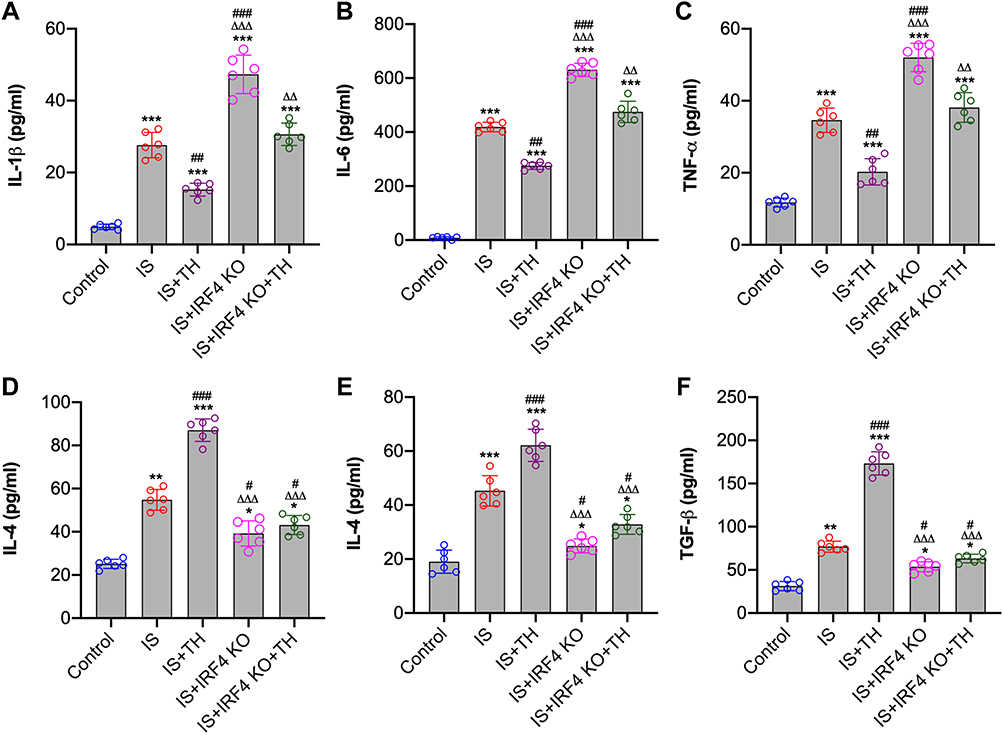 |
Figure 3 Measurement of cytokines. Brain penumbra tissues were collected at 8 hours after IS for the measurements of cytokines. Pro-inflammatory cytokines including IL-1β (A), IL-6 (B) and TNF-α (C) and anti-inflammatory cytokines including IL-4 (D), IL-10 (E) and TGF-β (F) were measured by enzyme-linked immunosorbent assay (ELISA) assays. n = 6 in each group. Data were expressed as mean±SD. Comparisons of continuous variables between groups were made by unpaired Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001 vs control group; #p<0.05, ##p<0.01, ###p<0.001 vs IS group; ΔΔΔp<0.001 vs IS + TH group. Abbreviations: IS, ischemic stroke; TH, therapeutic hypothermia; IRF4 KO, interferon regulatory factor 4 knockout. |
TH Promoted M2 Macrophage Polarization Through the Upregulation of IRF4
Our results showed that IRF4 KO abolished the anti-inflammatory effects of TH in experimental stroke. Next, we aim to determine the underlying mechanisms. As seen in Figure 4A and B, tMCAO induced increases in iNOS and Arg-1 expressions in IS mice as compared with control ones (p<0.01, respectively). However, TH promoted Arg-1 expression and inhibited iNOS expression in IS mice. In addition, immunofluorescence staining showed that the number of F4/80+/CD206+ positive cells increased while the number of F4/80+/iNOS+ positive cells decreased in IS mice after TH treatments (Figure 4C and D). IRF4 KO increased iNOS expression and the number of F4/80+/iNOS+ positive cells. However, cerebral expression of Arg-1 and the number of F4/80+/CD206+ positive cells were downregulated by IRF4 KO. Moreover, IRF4 KO reversed TH-induced macrophage polarization in IS mice as reflected by the changes in macrophage markers and the counts of cell number.
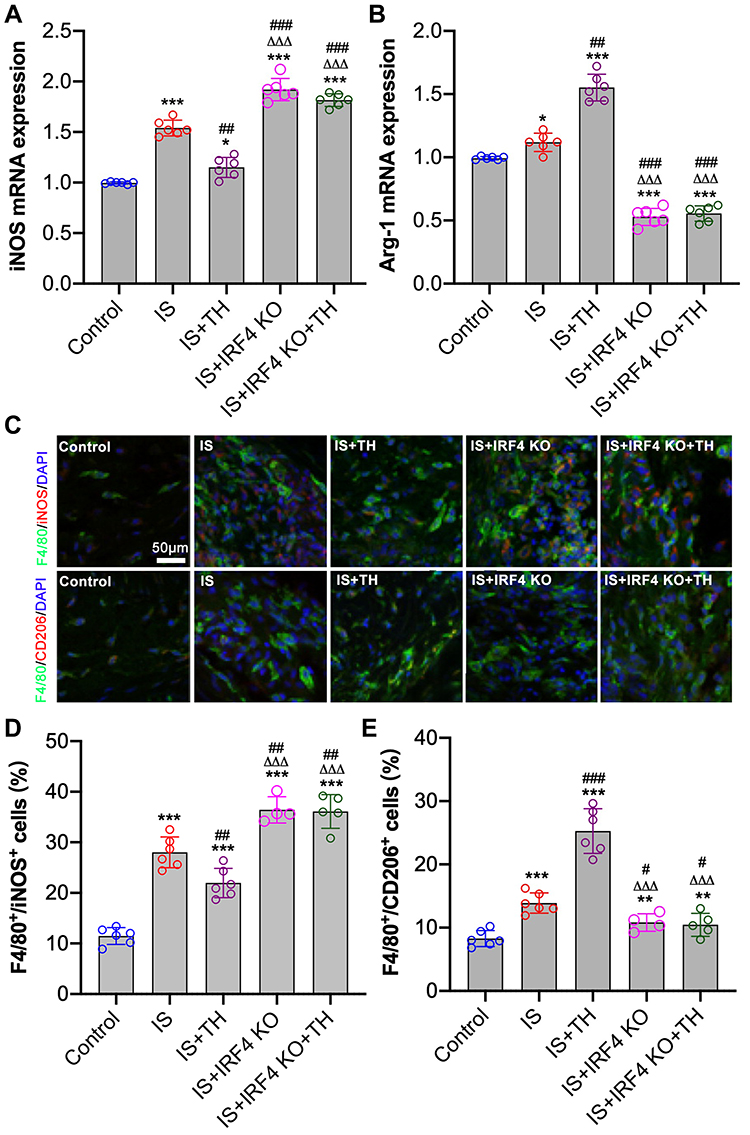 |
Figure 4 Determination of macrophage polarization in ischemic brain. RT-PCR were used to measure the mRNA and protein expressions of iNOS (A) and Arg-1 (B), respectively. Immunofluorescence staining (C) was used to determine the number of F4/80+/iNOS+ positive (D) and F4/80+/CD206+ positive (E) cells in penumbra tissues. n = 6 in each group for mRNA measurements; n = 4 in the IS + IRF4 KO group, n = 5 in the IS + IRF4 KO + TH group, and n = 6 in other groups for immunofluorescence staining. Data were expressed as mean±SD. Comparisons of continuous variables between groups were made by unpaired Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001 vs control group; #p<0.05, ##p<0.01, ###p<0.001 vs IS group; ΔΔΔp<0.001 vs IS + TH group. Abbreviations: IS, ischemic stroke; TH, therapeutic hypothermia; IRF4 KO, interferon regulatory factor 4 knockout; iNOS, inducible nitric oxide synthase; Arg-1, arginase-1. |
TH Reduced Infarct Volume and Improved Neurological Outcomes by Regulating IRF4 Expression
The neuroprotective effects of TH have been well documented. As seen in Figure 5A and B, TTC staining showed that TH protects mice against IS with reduced infarct volume. In contrast, the infarct volume in the IS + IRF4 KO group was significantly higher than that of the IS group (p<0.01, respectively), indicating IRF4 deficiency increased cerebral infarct. Moreover, the infarct volume in the IS + IRF4 KO + TH group was markedly increased as compared with the IS + TH group (p<0.01, respectively). There were no significant differences in cerebral infarct volume between the IS + IRF4 KO + TH and the IS or the IS + IRF4 KO group (p>0.05). Neurological scores (Figure 5C) and water maze test (Figure 5D) also suggested that IRF4 KO abolished the neuroprotective effects of TH in IS.
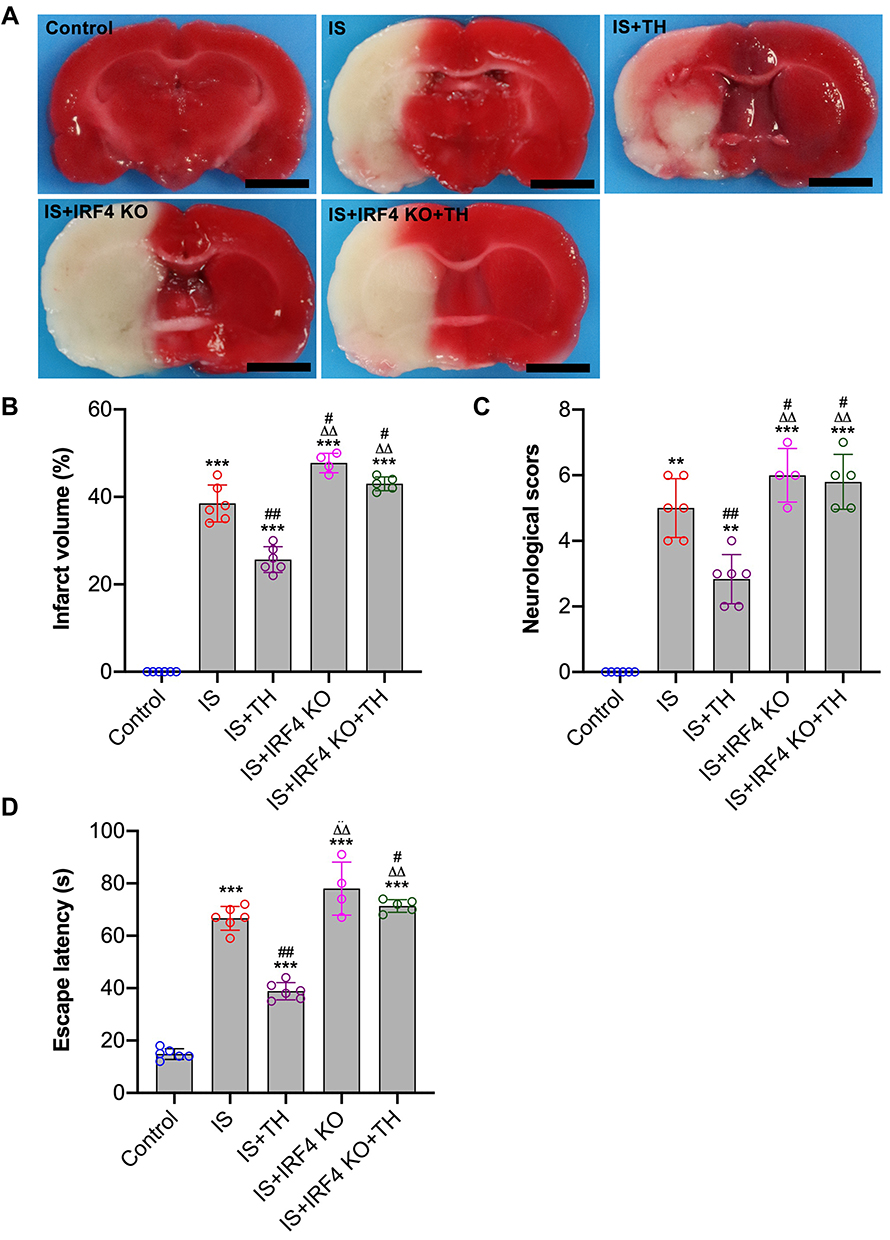 |
Figure 5 Neurological function assessments. Histological and neurological assessments were performed at 7 days after IS. Brain tissues were used for TTC staining (A) for the evaluation of cerebral infarct volume (B); neurological deficits scores (C) and escape latency (D) of water maze test were both determined. n = 4 in the IS + IRF4 KO group, n = 5 in the IS + IRF4 KO + TH group, and n = 6 in other groups. Data were expressed as mean±SD. Comparisons of neurological scores were made by Mann–Whitney test. Comparisons of infarct volume and EL between groups were made by unpaired Student’s t-test. **p<0.051, ***p<0.001 vs IS group; #p<0.05, ##p<0.01 vs IS group. ΔΔp<0.01 vs IS + TH group. Abbreviations: TTC, 2,3,5-triphenyl-tetrazolium-chloride; IS, ischemic stroke; TH, therapeutic hypothermia; IRF4 KO, interferon regulatory factor 4 knockout; iNOS, inducible nitric oxide synthase; Arg-1, arginase-1. |
Discussion
The major findings of this study can be summarized as follows: (1) Cerebral IRF4 expression was upregulated after IS and its deficiency worsens stroke-induced neurological injury with increased production of pro-inflammatory cytokines and macrophage polarization toward M1 phenotype; (2) TH inhibits neuroinflammation, reduces brain infarct volume and improves neurological recovery; (3) TH profoundly increases cerebral IRF4 expression, downregulates the production of pro-inflammatory and upregulates anti-inflammatory cytokine production through the modulation of macrophage polarization; (4) IRF4 deficiency abolished the neuroprotective and anti-inflammatory effects of TH against IS by reversing macrophage polarization. Together, these results indicated the neuroprotective effects TH and its anti-inflammatory properties are probably associated with the modulation of IRF4 expression and subsequent macrophage polarization in brain after IS.
Brain injury following IS results of a complex of pathophysiological events, and inflammation play a critical role in the pathogenesis of IS in both acute and chronic phases.3 Neuroinflammation following IS can be initiated by the activation of various signaling pathways including nuclear factor kappa beta (NF-κB) and mitogen-activated protein kinase (MAPK), which both play critical roles in the activation of inflammatory signals.25 NF-κB can translocate to the nucleus and bind to promoter domains of genes that initiate the transcription of pro-inflammatory cytokines including IL-6, iNOS and TNF. In addition, MAPKs have been proven to mediate the transcriptional process of many pro-inflammatory cytokines.26 A cascade of neuroinflammatory events finally results in activation of apoptosis and cell death. However, numerous studies have indicated that the inhibition of either NF-κB or MAPK and other anti-inflammatory drugs that block the production of pro-inflammatory cytokines markedly improved neurological outcomes after IS with reduced neuroinflammation.27,28 Importantly, the neuroprotective effects of TH in stroke were tightly associated with its inhibitory effects on NF-κB translocation and MAPK activation.29,30 In the present study, we observed that TH protects mice against IS with downregulated production of pro-inflammatory cytokines including IL-6, IL-1β and TNF-α. However, the activation of NF-κB and MAPK signaling has not been determined.
Various types of cells participate in the development of neuroinflammation following IS. Animal experiments and clinical studies have suggested that after IS, apart from the activation of microglial cells, the infiltration of circulating neutrophils, monocytes/macrophages and T cells also contribute to cell death in ischemic brain tissues.31–34 It has been reported that the resident macrophages are activated within few minutes after IS leading to abundant production of pro-inflammatory cytokines.35 In addition, various studies have shown that circulating macrophages infiltrated into ischemic brain tissues at 3–4 hours after stroke.36 The activated macrophages have been defined as either classic (pro-inflammatory; M1) or alternative (anti-inflammatory; M2) under pathophysiological conditions.37 M1 macrophage secretes pro-inflammatory cytokines and exacerbate inflammation and tissue injury. In contrast, M2 macrophage secretes anti-inflammatory cytokines, suppresses inflammation and promote tissue recovery. In the present study, our results suggested that TH promotes macrophage polarization toward M2 phenotype, which finally resulted in reduced neuroinflammation and improved neurological outcomes.
In fact, it has been widely reported that IRFs play a key role in regulating macrophage maturation, phenotypic polarization, phenotypic switch and function.16 Of 9 IRFs, IRF-1, IRF-5, and IRF-8 are involved in the regulation of M1 polarization whereas IRF-3 and IRF-4 control M2 polarization, and the role of IRF-2 is context-dependent. As mentioned above, it has been reported that IRF4 regulates microglial M2 activation and impacts on stroke outcomes.19 In the present study, IRF4 deficiency worsens neurological outcomes of IS mice with reduced M2 polarization and upregulated neuroinflammation. In addition, we observed that IRF4 knockout also enhanced M1 polarization together with increased production of pro-inflammatory cytokines. Moreover, the protective effects of TH against IS have been abolished by IRF4 knockout by reversing macrophage polarization. These results indicated that IRF4 mediates the neuroprotective effects of TH in IS probably through the regulation of macrophage polarization.
This study has some limitations. First, the overexpression of IRF4 has not been performed. In the present study, we demonstrated that IRF4 deficiency worsens neurological outcomes of IS mice. However, it is unclear whether IRF4 overexpression is able to protect mice against ischemic stroke. Second, our results indicated that TH enhances IRF4 expression at both mRNA and protein levels in ischemic stroke mice. But the underlying molecular mechanism by which TH regulates IRF4 transcription has not been investigated. Further studies are required to elucidate the exact molecular mechanism. Third, we observed that both TH treatment and IRF4 knockout induce marked changes in neuroinflammation. However, the infiltration and activation of other types of inflammatory cells have not been determined. Finally, it has been suggested that NF-κB and MAPK signaling pathways contribute to the neuroprotective effects of TH in experimental stroke. We claimed that TH inhibits neuroinflammation and exerts anti-inflammatory effects in IS mice through the modulation of IRF4 expression. However, the relationship between IRF4 expression and the activation of NF-κB and MAPK signaling pathways was unclear. Further studies are required to elucidate the potential association between IRF4 expression and other inflammatory signaling pathways.
In conclusion, our results indicate that IRF4 deficiency drives macrophage polarization toward M1 phenotype and increases the production of pro-inflammatory cytokines, which finally results in worsen neurological outcomes. Importantly, the neuroprotective effects and anti-inflammatory properties of TH in IS are tightly associated with the upregulation of IRF4 in ischemic brain. Together, these findings offer new insights into the mechanism by which TH minimizes neuroinflammation in IS.
Acknowledgments
The authors would like to thank the Animal Experimental Center of Renmin Hospital of Wuhan University for the technical assistance.
Author Contributions
XYY and QXC designed this study; XYY, YPF and RZL performed experiments and collected data; XYY and YPF performed data analysis; XYY wrote the manuscript; QXC revised the manuscript. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Kyu HH, Bachman VF, Alexander LT, et al. Physical activity and risk of breast cancer, colon cancer, diabetes, ischemic heart disease, and ischemic stroke events: systematic review and dose-response meta-analysis for the Global Burden of Disease Study 2013. BMJ. 2016;354:i3857. doi:10.1136/bmj.i3857
2. Wu S, Wu B, Liu M, et al.; China Stroke Study Collaboration. Stroke in China: advances and challenges in epidemiology, prevention, and management. Lancet Neurol. 2019;18(4):394–405. doi:10.1016/S1474-4422(18)30500-3
3. Khoshnam SE, Winlow W, Farzaneh M, Farbood Y, Moghaddam HF. Pathogenic mechanisms following ischemic stroke. Neurol Sci. 2017;38:1167–1186.
4. Sandercock P, Wardlaw JM, Lindley RI, et al.; IST-3 collaborative group. The benefits and harms of intravenous thrombolysis with recombinant tissue plasminogen activator within 6 h of acute ischaemic stroke (the third international stroke trial [IST-3]): a randomised controlled trial. Lancet. 2012;379:2352–2363.
5. Kurisu K, Yenari MA. Therapeutic hypothermia for ischemic stroke; pathophysiology and future promise. Neuropharmacology. 2018;134:302–309. doi:10.1016/j.neuropharm.2017.08.025
6. Busto R, Dietrich WD, Globus MY, et al. Small differences in intraischemic brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab. 1987;7(6):729–738. doi:10.1038/jcbfm.1987.127
7. Kaibara T, Sutherland GR, Colbourne F, Tyson RL. Hypothermia: depression of tricarboxylic acid cycle flux and evidence for pentose phosphate shunt upregulation. J Neurosurg. 1999;90(2):339–347. doi:10.3171/jns.1999.90.2.0339
8. Yenari MA, Han HS. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat Rev Neurosci. 2012;13(4):267–278. doi:10.1038/nrn3174
9. Kawamura N, Schmeichel AM, Wang Y, Schmelzer JD, Low PA. Multiple effects of hypothermia on inflammatory response following ischemia-reperfusion injury in experimental ischemic neuropathy. Exp Neurol. 2006;202(2):487–496. doi:10.1016/j.expneurol.2006.07.012
10. Deng H, Han HS, Cheng D, Sun GH, Yenari MA. Mild hypothermia inhibits inflammation after experimental stroke and brain inflammation. Stroke. 2003;34(10):2495–2501. doi:10.1161/01.STR.0000091269.67384.E7
11. Yenari MA, Han HS. Influence of hypothermia on post-ischemic inflammation: role of nuclear factor kappa B (NFkappaB). Neurochem Int. 2006;49(2):164–169. doi:10.1016/j.neuint.2006.03.016
12. Choi JS, Park J, Suk K, et al. Mild hypothermia attenuates intercellular adhesion molecule-1 induction via activation of extracellular signal-regulated kinase-1/2 in a focal cerebral ischemia model. Stroke Res Treat. 2011;2011:846716.
13. Meybohm P, Gruenewald M, Zacharowski KD, et al. Mild hypothermia alone or in combination with anesthetic post-conditioning reduces expression of inflammatory cytokines in the cerebral cortex of pigs after cardiopulmonary resuscitation. Crit Care. 2010;14(1):R21. doi:10.1186/cc8879
14. Lee JH, Wei ZZ, Cao W, et al. Regulation of therapeutic hypothermia on inflammatory cytokines, microglia polarization, migration and functional recovery after ischemic stroke in mice. Neurobiol Dis. 2016;96:248–260. doi:10.1016/j.nbd.2016.09.013
15. Eames HL, Corbin AL, Udalova IA. Interferon regulatory factor 5 in human autoimmunity and murine models of autoimmune disease. Transl Res. 2016;167(1):167–182. doi:10.1016/j.trsl.2015.06.018
16. Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN, Bobryshev YV. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology. 2018;223(1):101–111. doi:10.1016/j.imbio.2017.10.005
17. Xiong XY, Liu L, Yang QW. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog Neurobiol. 2016;142:23–44.
18. Iwanowycz S, Wang J, Altomare D, Hui Y, Fan D. Emodin bidirectionally modulates macrophage polarization and epigenetically regulates macrophage memory. J Biol Chem. 2016;291(22):11491–11503. doi:10.1074/jbc.M115.702092
19. Al Mamun A, Chauhan A, Yu H, Xu Y, Sharmeen R, Liu F. Interferon regulatory factor 4/5 signaling impacts on microglial activation after ischemic stroke in mice. Eur J Neurosci. 2018;47(2):140–149. doi:10.1111/ejn.13778
20. Guo S, Li ZZ, Jiang DS, et al. IRF4 is a novel mediator for neuronal survival in ischaemic stroke. Cell Death Differ. 2014;21(6):888–903. doi:10.1038/cdd.2014.9
21. National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals.
22. Chiang T, Messing RO, Chou WH, Mouse model of middle cerebral artery occlusion. J Vis Exp. 2011;(48):2761. doi:10.3791/2761
23. Wang L, Lu Y, Guan H, et al. Tumor necrosis factor receptor-associated factor 5 is an essential mediator of ischemic brain infarction. J Neurochem. 2013;126(3):400–414. doi:10.1111/jnc.12207
24. Zhang HM, Liu P, Jiang C, et al. Notch signaling inhibitor DAPT provides protection against acute craniocerebral injury. PLoS One. 2018;13(2):e0193037. doi:10.1371/journal.pone.0193037
25. Baeuerle PA, Henkel T. Function and activation of NF-kappaB in the immune system. Annu Rev Immunol. 1994;12(1):14–79. doi:10.1146/annurev.iy.12.040194.001041
26. Yu X, Quan J, Long W, et al. LL-37 inhibits LPS-induced inflammation and stimulates the osteogenic differentiation of BMSCs via P2X7 receptor and MAPK signaling pathway. Exp Cell Res. 2018;372(2):178–187. doi:10.1016/j.yexcr.2018.09.024
27. Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19(8):819–834. doi:10.1097/00004647-199908000-00001
28. Herrmann O, Baumann B, de Lorenzi R, et al. IKK mediates ischemia-induced neuronal death. Nat Med. 2005;11(12):1322–1329. doi:10.1038/nm1323
29. Han HS, Karabiyikoglu M, Kelly S, Sobel RA, Yenari MA. Mild hypothermia inhibits nuclear factor-κB translocation in experimental stroke. J Cereb Blood Flow Metab. 2003;23(5):589–598. doi:10.1097/01.WCB.0000059566.39780.8D
30. Li J, Benashski S, McCullough LD. Post-stroke hypothermia provides neuroprotection through inhibition of AMP-activated protein kinase. J Neurotrauma. 2011;28(7):1281–1288. doi:10.1089/neu.2011.1751
31. Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2003;183(1):25–33. doi:10.1016/S0014-4886(03)00082-7
32. Tanaka R, Komine-Kobayashi M, Mochizuki H, et al. Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia/macrophage into the mouse brain following permanent focal ischemia. Neuroscience. 2003;117(3):531–539. doi:10.1016/S0306-4522(02)00954-5
33. Price C, Menon D, Peters A, et al. Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: an imaging-based study. Stroke. 2004;35(7):1659–1664. doi:10.1161/01.STR.0000130592.71028.92
34. Buck BH, Liebeskind DS, Saver JL, et al. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke. 2008;39(2):355–360. doi:10.1161/STROKEAHA.107.490128
35. Taylor RA, Sansing LH. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol. 2013;2013:746068. doi:10.1155/2013/746068
36. Kokovay E, Li L, Cunningham LA. Angiogenic recruitment of pericytes from bone marrow after stroke. J Cereb Blood Flow Metab. 2006;26(4):545–555. doi:10.1038/sj.jcbfm.9600214
37. Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int J Mol Sci. 2017;18(10):2135. doi:10.3390/ijms18102135
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.