")
Back to Journals » International Journal of General Medicine » Volume 15
Hypocomplementemia in Primary Sjogren’s Syndrome: A Retrospective Study of 120 Treatment-Naive Chinese Patients
Received 25 October 2021
Accepted for publication 24 December 2021
Published 8 January 2022 Volume 2022:15 Pages 359—366
DOI https://doi.org/10.2147/IJGM.S346188
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Meiju Zhou, Fang Yuan
Department of Rheumatology and Immunology, Affiliated Zhejiang Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang Province, People’s Republic of China
Correspondence: Fang Yuan
Department of Rheumatology and Immunology, Affiliated Zhejiang Hospital, Zhejiang University School of Medicine, 1229 Gudun Road, Hangzhou, Zhejiang Province, 310030, People’s Republic of China
Email [email protected]
Objective: To determine the prevalence of hypocomplementemia in primary Sjogren’s syndrome (pSS) patients and compare the clinical characteristics of patients with and without hypocomplementemia.
Methods: A retrospective study was conducted in 120 treatment-naive Chinese patients that met the 2012 American College of Rheumatology Classification Criteria for pSS and were followed up for 3 to 24 months. Based on the complement results, patients were divided into four groups: only low C3, only low C4, both low C3 and C4 (double low), normal group. The data on patient demographics, clinical manifestations, laboratory results, disease activity and pharmacologic therapy were collected and compared among the four groups.
Results: The prevalence of only low C3, only low C4, both low C3 and C4 in pSS patients was 21.7%, 16.7%, and 10%, respectively. The mean age of the four groups was significantly different. Unlike rampant caries and parotitis, the prevalence of dry eyes and dry mouth differed among the four groups. The proportion of patients with anemia, leukocytopenia, lymphadenopathy, hematological involvement and fatigue was significantly higher in the double low group and lower in the normal complement group. The proportion of patients with increased serum IgG was higher in the only low C4 group than in the other groups. Logistic regression revealed that hypocomplementemia was an independent risk factor for lymphadenopathy and leukopenia. The double low group had a significant history of exposure to glucocorticoids and cyclophosphamide, compared with other groups.
Conclusion: Our study found that the clinical characteristics of pSS patients with hypocomplementemia differed from those without hypocomplementemia. Hypocomplementemia in pSS was associated with hematological involvement, hyper-IgG, lymphadenopathy, and fatigue, contributing to more significant exposure to glucocorticoid and cyclophosphamide.
Keywords: primary Sjogren’s syndrome, hypocomplementemia, complement C3, complement C4
Introduction
Sjogren’s syndrome (SS) is a multisystemic autoimmune disease characterized by hypofunction of salivary and lacrimal glands and possible systemic multi-organ manifestations, such as interstitial lung disease, kidney diseases, and neurological, hematological and musculoskeletal involvements.1 It is characterized by lymphocytic infiltration of exocrine glands and other organs, leading to dry eyes, dry mouth and various extra-glandular symptoms.2 SS is subcategorized into primary SS (pSS), which is not associated with other well-defined connective tissue diseases (CTDs), and secondary SS, which is associated with other well-defined CTDs.2,3
The complement system is a part of natural immunity, composed of more than 30 soluble and membrane-bound proteins activated in a sequential enzymatic cascade. Importantly, it plays a critical role in immune regulation and formation of the Membrane Attack Complex to kill targeted cells.4 The complement system can be activated via the classical, alternative and mannose-binding protein pathways.5,6 Nowadays, serum complement screening is important in diagnosing and treating systemic lupus erythematosus. However, in pSS patients, complement levels are not routinely measured, and the clinical significance of hypocomplementemia has been understudied, especially in the Chinese population.7 Clinically, C3 and C4 hypocomplementemia are found at pSS diagnosis in only 1–15% of patients and 20% of cases during the disease course.8,9 Interestingly, it has been shown that pSS patients with C3 and C4 hypocomplementemia could represent a subset of patients with a poorer disease course and outcome.10 During clinical practice, C3 and C4 hypocomplementemia are not frequently used as serologic markers in pSS patients. This study aimed to investigate the differences in the clinical features of hypocomplementemia in pSS patients, focusing on the relationship between C3 and C4 hypocomplementemia and clinical manifestations, immunological features, and treatment.
Materials and Methods
Patients
One hundred and twenty pSS patients who met the 2012 American College of Rheumatology (ACR) criteria for pSS1 and were followed up for 3 to 24 months (median follow-up: 7 months) were enrolled in this study. All patients visited Zhejiang hospital between August 2018 and October 2020 and never received corticosteroid or immunosuppressive treatment.
Patients with a history of other rheumatic diseases in addition to pSS were excluded from this study. Demographic, clinical, and laboratory data were collected from the electronic hospital information system. All procedures were approved by the Ethics Committee of Zhejiang hospital (2020101K) and followed the tenets of the Declaration of Helsinki. Written informed consent was obtained from all subjects prior to participation in the study.
Data and Definitions
Clinical data, serologic and imaging results were obtained from the electronic hospital information system, including age, gender, disease duration, symptoms, laboratory data, and disease activity scores.
At the inception of the study, the presence of the following symptoms or signs were determined: persistent dry mouth and eyes for at least 3 months, arthritis, purpura rash, parotitis, rampant caries, fatigue, renal tubular acidosis, interstitial lung disease (ILD), lymphadenopathy, psychosomatic disease, anemia (hemoglobin < 110 g/L), leukopenia (white blood cell < 4×109/L), thrombocytopenia (platelet < 100×109/L), hypergammaglobulinemia (> 16 g/L), hematological involvement (at least one of anemia, thrombocytopenia, or leukopenia). Psychosomatic disorders were diagnosed by psychiatrists.
C3/C4 hypocomplementemia was defined as C3/C4 levels below the lower limit of the reference range. The normal range for serum C3 and C4 were 0.9–1.8 g/L and 0.1–0.4 g/L, respectively. Complement C3 and C4 were measured by an immunochemical assay. pSS patients were divided into four groups according to the serum C3 and C4 values: only low C3, only low C4, both low C3 and C4 (double low), and neither low C3 nor C4 (normal C3 and C4).
Lymphadenopathy was defined as lymph node length > 1 cm, or reduced ratio of the long to short diameter, using ultrasound or high-resolution computed tomography (HRCT).11 ILD was identified using chest radiographs or HRCT.12 Fatigue was measured with the fatigue visual analog scale (fVAS), a widely used and accepted generic and unidimensional fatigue instrument: an fVAS score >1 was defined as having fatigue symptoms.13
Disease activity was assessed with the EULAR Sjogren’s Syndrome Disease Activity Index (ESSDAI).14 Patient-reported symptoms were assessed with the EULAR Sjogren’s Syndrome Patient-Reported Index (ESSPRI).15 The Health Assessment Questionnaire (HAQ) was used.16 Schirmer tests were conducted by an ophthalmologist. For one pSS patient, we took the minimum value of both eyes in this study. All serologic results, imagological examinations, and disease activity assessments were measured upon admission. IgG, IgA, and IgM were measured by enzyme-linked immunosorbent assay (ELISA). Auto-antibodies were measured by indirect immunofluorescence. Rheumatoid factor was measured by latex agglutination turbidimetry.
Statistical Analysis
Data were analyzed using SPSS V23.0 for Windows. Descriptive statistics were described as mean ± standard deviation (SD). The Kolmogorov–Smirnov test determined whether the data followed a normal distribution. Kruskal–Wallis and Dunn’s tests were used to assess the statistical significance of differences between groups. Discrete variables were evaluated using the chi-square test and Bonferroni test. Parameters that were statistically significant during univariate analysis and one particular pSS subtype were selected for multivariate analysis. Odds ratios (ORs) were obtained using logistic regression. A P-value < 0.05 was statistically significant.
Results
Clinical Characteristics of pSS Patients
The clinical characteristics of the included pSS patients on admission are shown in Table 1. A total of 120 patients with pSS were included, consisting predominantly of females (n = 109, 90.8%). The mean age was 53 ± 13.8 years (range: 17–88 years). Patients were divided into four groups according to the serum C3 and C4 levels. The prevalence of only low C3, only low C4, and double low in the included pSS patients was 21.7% (n = 26), 16.7% (n = 20), and 10% (n = 12), respectively. 71.7% (n= 86) patients had normal C3 and C4 levels.
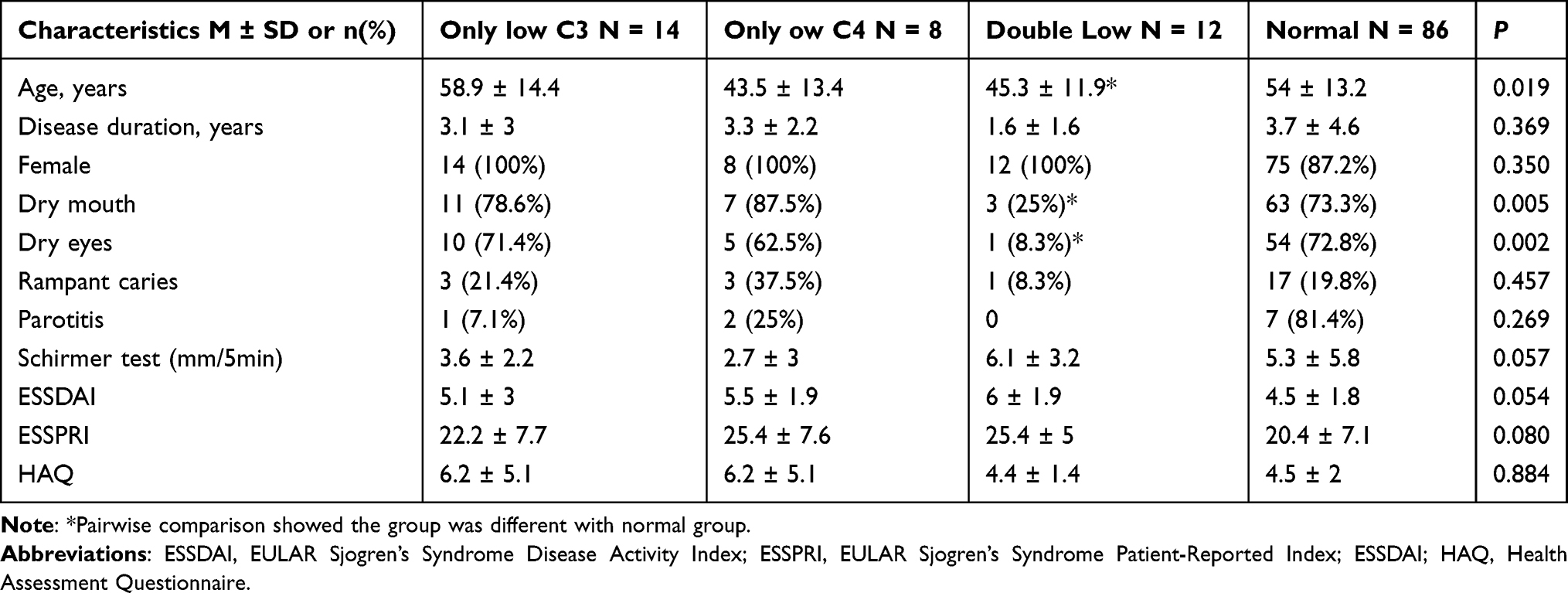 |
Table 1 Baseline Clinical Characteristics of 120 pSS Patients |
There was no difference in disease activity scores, Schirmer test values, disease duration, sex, and the proportion of rampant caries and parotitis, while the proportion of dry eyes and dry mouth was significantly different among the four groups (P = 0.005 and P = 0.002, respectively). The mean age of the four groups was significantly different (P = 0.019). Pairwise comparisons suggested that parameters such as age, dry eyes and dry mouth, were significantly different between the double low group and the normal complement group.
Serological Parameters
Table 2 shows the serological parameters of pSS patients on admission. The proportions of pSS patients with anemia, leukocytopenia, and increased IgG were 50%, 64.3%, and 57.1% in the low C3 group, 37.5%, 37.5% and 87.5% in the low C group, 66.7%, 83.3% and 83.3% in the double low group, and 24.4%, 24.4% and 48.9% in the normal complement group (P = 0.01, P < 0.0001 and P = 0.034), respectively. There was no difference in the proportion of patients with thrombocytopenia, increased IgA, increased IgM, anti- SSA antibody, anti- SSB antibody, anti- RO52 antibody, and rheumatoid factor among the four groups. Intriguingly, the platelet count in the double low group was the lowest among the four groups (P = 0.008). Pairwise comparisons showed that the hemoglobin, platelet count, and white blood cell count were significantly different in the double low group and low C3 group, compared with the normal complement group. IgG levels in the double low group and low C4 group were significantly different from the normal complement group.
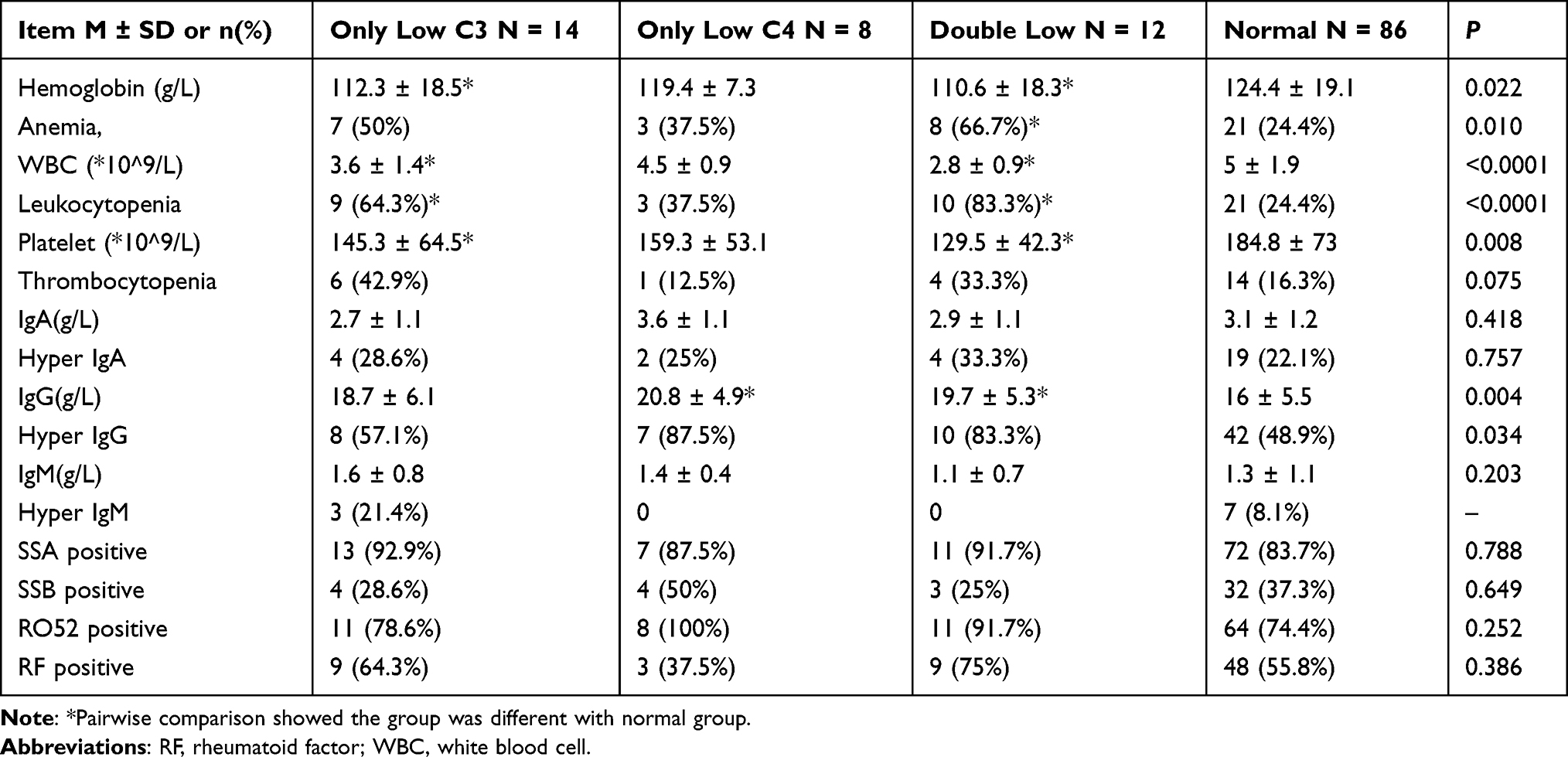 |
Table 2 Baseline Serological Parameters of 120 pSS Patients |
Organ Involvement
As seen in Table 3, extra-glandular involvement and complications were included: interstitial lung disease (n = 14, 11.7%), articular involvement (n = 64, 53.3%) and fatigue (n = 64, 53.3%), lymphadenopathy (n = 36, 30%), hematological involvement (n = 66, 55%), skin involvement (n = 12, 10%), renal involvement (n = 6, 5%), autoimmune hepatitis (n = 7, 5.8%), and psychiatric disease (n = 23, 19.1%). The proportion of pSS patients with lymphadenopathy, hematological involvement and fatigue was higher in the double low group (P < 0.0001, P < 0.0001 and P = 0.001, respectively). Pairwise comparisons suggested that the incidences of lymphadenopathy and hematological involvement in the double low, only low C3 and only low C4 group were significantly different from the normal complement group. The incidence of fatigue in the double low group was significantly different from the normal group. No differences in the articular involvement, skin involvement, renal involvement, autoimmune hepatitis, and psychiatric disease were found among the four groups.
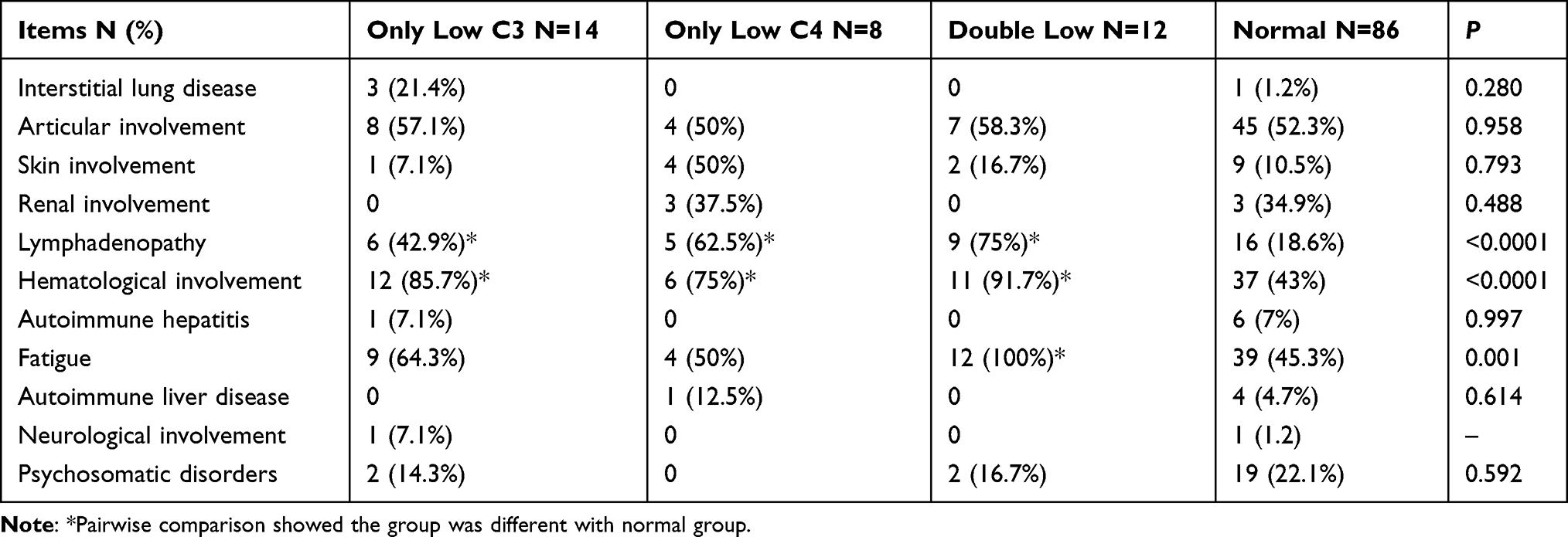 |
Table 3 Extra-Glandular Major Visceral Involvement and Complications in 120 pSS Patients |
Features Associated with Hypocomplementemia
Binary logistic regression was performed to identify the factors associated with hypocomplementemia. We found only low C3, only low C4, both low C3 and C4 were identified as independent risk factors for lymphadenopathy (OR = 5.517, OR = 7.317, OR = 2.451, P = 0.010, P = 0.008, P = 0.031, respectively) and leukopenia (OR = 4.585, OR = 5.333, OR = 6.160, P = 0.004, P = 0.046, P = 0.049, respectively). The other parameters, including autoantibodies, anemia, thrombocytopenia, ILD, fatigue, hypergammaglobulinemia, were not significant.
Pharmacologic Treatment
Pharmacologic treatments are shown in Table 4. pSS Patients with only low C3, only low C4, and double low relatively more exposed to glucocorticoid (53.8%, 50%, 75%, vs 23.3%, P = 0.006, P = 0.034, P = 0.0008, respectively) and cyclophosphamide (15.4%, 15%, 16.7%, vs 1.2%, P = 0.011, P = 0.023, P = 0.043, respectively) treatment than those with normal C3 and C4. Pairwise comparisons showed more pSS patients in double low group were treated with glucocorticoid and cyclophosphamide, compared with the normal group. No significant differences were found for other drugs, including cyclosporine, methotrexate, hydroxychloroquine, and tacrolimus among the four subgroups.
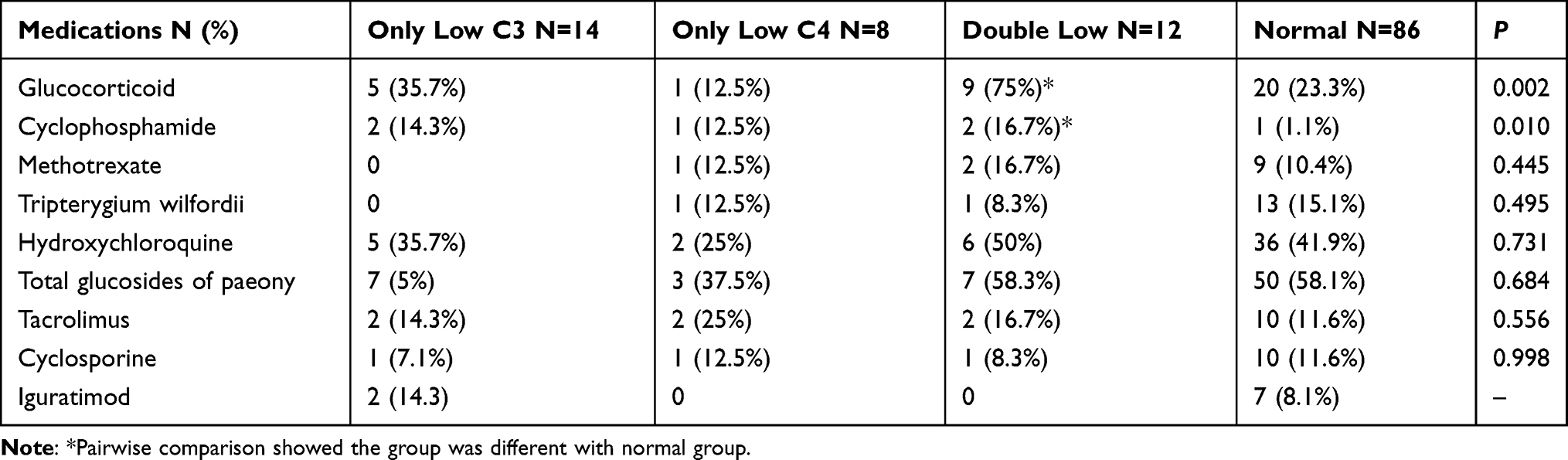 |
Table 4 Pharmacologic Treatment in pSS Patients with and without Hypocomplementemia |
Discussion
pSS is a complex autoimmune disease characterized by dysfunction of secretory epithelia that can be managed only by palliative therapy. The prevalence of hypocomplementemia in pSS is heterogeneous, and little is currently known about its role. This study aimed to determine the prevalence of hypocomplementemia in pSS and the difference in clinical characteristics of treatment-naive pSS patients with and without hypocomplementemia. The prevalence of C3, C4 and C3 and C4 hypocomplementemia were 21.7%, 16.7%, and 10%, respectively, similar to the literature.8,9,17,18 However, the incidence of hypocomplementemia varied in different studies and areas. Skopouli et al8 documented C3 and C4 hypocomplementemia rates of 2% and 17% in Greek pSS patients. In a multicenter study including 601 Greek pSS patients by Ioannidis JP et al.9 C3 and C4 hypocomplementemia rates were 3% and 20%, respectively. Moreover, Theander et al18 reported C3 and C4 hypocomplementemia rates of 25% and 27% in Swedish pSS patients (n = 484), higher than any other European study. These inter-study differences can be attributed to the heterogeneity in study areas, subject race, classification criterion and cut-off levels for hypocomplementemia used.
In the present study, the immunological and clinical characteristics of pSS patients with hypocomplementemia significantly differed from those with normal complement levels. Hypocomplementemia was associated with a high concentration of IgG and a low platelet count. The proportion of patients with anemia and leukocytopenia in the double low group was significantly higher than other groups. As for extra-glandular involvement, the proportion of fatigue, lymphadenopathy and hematological system involvement was significantly higher in the double low group. Importantly, hypocomplementemia was an independent risk factor for lymphadenopathy and leukopenia.
Our findings were similar to the results of Ramos-Casals et al10 to a certain extent. Lymphadenopathy is widely acknowledged as a clinical feature of pSS, and non-Hodgkin lymphoma is the most severe complication of pSS, occurring in around 5% of patients.19 Lymphoproliferation is an early characteristic of lymphoma, the predominant cause of death for pSS patients. Theander et al20 confirmed the close association between hypocomplementemia and lymphoma and found that hypocomplementemia could predict an increased risk of lymphoma for pSS patients. Interestingly, Ramos-Casals et al10 found that pSS patients with C4 hypocomplementemia had a higher lymphadenopathy prevalence than the normal C4 subset (18% vs 5%, P = 0.01). Solans-Laque et al21 reported that hypocomplementemia at pSS diagnosis was a strong predictor for lymphocytopenia, and survival was reduced in patients with hypocomplementemia. A multicenter study was conducted in 5 centers (n = 661) and found low C4 (relative-risk ratio = 8.3) were the variables distinguishing B-cell non-Hodgkin’s lymphoma Group from pSS patients without NHL or prelymphomatous conditions.22 Quartuccio et al23 studied 548 pSS patients, and found pSS negative for anti-SSA and anti- SSB antibodies appears to be characterized by a lower risk of lymphoma and by a lower level of B-cell expansion. Additionally, hypocomplementemia and focus score resulted in variables associated with lymphoma at univariate analysis, and focus score appeared to be associated with lymphoma independently on complement fraction concentrations.24 This requires attention to the histological features of pSS patients. In this study, patients only with seven months median follow-up, we did not observe any individuals developed lymphoma.
As previously reported by Sjogren Big Data Project,25 C3 and C4 hypocomplementemia was tightly correlated with the hematologic system. In this regard, Zhao et al7 found that pSS patients with hypocomplementemia had a lower mean age at diagnosis and a higher frequency of anemia and multivariate analysis showed that anemia was an independent predictor, similar to our study results. Jordán-González et al17 found no significant relationship between low complement and the hematological involvement in Puerto Rican patients with pSS (n = 94). In contrast, Brito-Zerón et al25 studied 10,500 pSS patients from 22 countries (78% White), and found that pSS patients with C4 hypocomplementemia had a higher incidence and more severe xerophthalmia and xerostomia. In the present study, no significant association between low complement and autoantibodies was found, inconsistent with previous studies.10,17 It is highly conceivable that the clinical manifestations of pSS are significantly influenced by the epidemiological characteristics, systemic involvement, or the immunological profile at diagnosis. Furthermore, the results varied according to the study areas and subjects’ races.
The Italian cohort study included 1115 pSS patients, and found extra-glandular manifestations were detectable in 15% of the patients and were represented by active synovitis (11%), axonal sensory-motor neuropathy (2%), severe leukocytopenia (14%), cutaneous vasculitis (6%) and non-Hodgkin’s lymphoma (4.5%).26 There is slightly difference from our study. Maybe, the different reference value of variables, different ethnicities and different inclusion criterion contribute to the results. The pattern of extra-glandular involvement is different among different ethnicities, however no studies have compared the disease phenotype among ethnicities. An enhanced risk of pulmonary and renal involvement has been reported in an Asian cohort study.27 Brito-Zerón et al28 were the first to report significant geoepidemiological variations in the prevalence of xerostomia, xerophthalmia, abnormal diagnostic tests and positivity of immunological marker.
Hematological involvement, fatigue syndrome, lymphadenopathy, and serum IgG contribute to high ESSDAI score. However, no relationship was found between hypocomplementemia and the ESSDAI score. All ten criteria (constitutional and lymphadenopathy, glandular, articular, cutaneous, pulmonary, renal, muscular, peripheral nervous system, central nervous system, hematological) proposed by the ACR/EULAR committee14 have been included in the ESSDAI score. In addition, the extent of organ involvement or presence of subjective symptoms such as fatigue and pain can influence the physician’s evaluation of disease activity. In this study, all pSS patients enrolled were hospitalized and exhibited heterogeneous degrees of disease severity. In additional, ESSDAI is a useful tool to assess disease activity, and ClinESSDAI seems to be another valid tool, which is independently of B-cell biomarkers and also to allow clinical assessment when some laboratory features are not available. Maybe, we can combine ESSDAI and ClinESSDAI assessment to accurately evaluate disease activity.29 Jordán-González et al17 reported higher ESSDAI scores in pSS patients with C4 hypocomplementemia; however, there was no difference found in the C3 hypocomplementemia group, which could be attributed to the small sample size.
Moreover, we found that pSS patients with low complement levels were more likely to receive glucocorticoid and cyclophosphamide treatment. Moreover, pSS patients with hypocomplementemia at diagnosis may represent a subset of patients with a worse clinical outcome when compared with patients with normal complement levels.10 In a Spanish cohort of 1580 patients with pSS, 65% of pSS patients with hypocomplementemia received glucocorticoid treatment.30 Even though glucocorticoid is commonly used in pSS patients with glandular, joint, cutaneous, lung, hematological, renal, neurological involvement, no reliable evidence is available to support the use of glucocorticoids in pSS, which is still based on case reports, case studies, retrospective or prospective studies and a small number of randomized controlled trials.31
In addition, compared with previous studies,7,10,17,24 there are some difference in this study. In our study, pSS patients were divided into four groups: only low C3, only low C4, both low C3 and C4 (double low), and normal C3 and C4, which is different from other studies reported in the literature.7,10,17,24 The difference in the grouping strategy could potentially account for the difference in results. Moreover, it should be borne in mind that other factors such as the patient ethnicity, study areas and different clinical criteria impact the results. Ethnicity is a key influencer of the clinical phenotype and outcomes of pSS.32 Recent studies have analyzed the potential role of ethnicity in pSS phenotypic expression. It has been established that ethnicity strongly influences the age at diagnosis33,34 as the expression of sicca symptoms is enhanced in White patients and decreased in Black/African American and Asian patients.7,33,35 The variability in pSS features was strongly related to individual demographics, such as age, gender, ethnicity and place of residence, which should be emphasized for new patients diagnosed with pSS. Moreover, in the present study, the median follow-up was only seven months, which could have limited our study results. The heterogeneity in results from studies in the literature can be attributed to the small number of patients included and the different classification criteria used.
Several limitations were present in this study. First of all, this was a retrospective study designed to analyze data obtained from medical records, which may have resulted in recall bias. Further studies are necessary to confirm the relevance of our findings in large populations. Moreover, the fact that participants in our study were predominantly southern Chinese could limit the generalization of our results in other populations. In addition, differences in physician assessment can contribute to the heterogeneity in disease scores.
Statement of Ethics
This study was approved by the Ethics Committee of Zhejiang hospital (No. 2020101K). All the patients provided written informed consent.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Shiboski CH, Shiboski SC, Seror R, et al. 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for primary Sjogren's syndrome: a consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheumatol. 2017;69(1):35–45. doi:10.1002/art.39859
2. Tsuboi H, Hagiwara S, Asashima H, et al. Comparison of performance of the 2016 ACR-EULAR classification criteria for primary Sjögren’s syndrome with other sets of criteria in Japanese patients. Ann Rheum Dis. 2017;76(12):1980–1985. doi:10.1136/annrheumdis-2016-210758
3. Nocturne G, Virone A, Ng WF, et al. Rheumatoid factor and disease activity are independent predictors of lymphoma in primary Sjögren’s syndrome. Arthritis Rheumatol. 2016;68(4):977–985. doi:10.1002/art.39518
4. Brodsky RA. Complement in hemolytic anemia. Blood. 2015;126(22):2459–2465. doi:10.1182/blood-2015-06-640995
5. Clarke AE, Weinstein A. Evaluation of the economic benefit of earlier Systemic Lupus Erythematosus (SLE) diagnosis using a Multivariate Assay Panel (MAP). Nov. 2020;2(11):629–639. doi:10.1002/acr2.11177
6. Merrill SA, Brodsky RA. Complement-driven anemia: more than just paroxysmal nocturnal hemoglobinuria. Hematology Am Soc Hematol Educ Program. 2018;2018(1):371–376. doi:10.1182/asheducation-2018.1.371
7. Zhao Y, Li Y, Wang L, et al. Primary Sjögren syndrome in Han Chinese: clinical and immunological characteristics of 483 patients. Medicine. 2015;94(16):e667. doi:10.1097/MD.0000000000000667
8. Skopouli FN, Dafni U, Ioannidis JP, Moutsopoulos HM. Clinical evolution, and morbidity and mortality of primary Sjögren’s syndrome. Semin Arthritis Rheum. 2000;29(5):296–304. doi:10.1016/s0049-0172(00)80016-5
9. Ioannidis JP, Vassiliou VA, Moutsopoulos HM. Long-term risk of mortality and lymphoproliferative disease and predictive classification of primary Sjögren’s syndrome. Arthritis Rheum. 2002;46(3):741–747. doi:10.1002/art.10221
10. Ramos-Casals M, Brito-Zerón P, Yagüe J, et al. Hypocomplementaemia as an immunological marker of morbidity and mortality in patients with primary Sjogren’s syndrome. Rheumatology. 2005;44(1):89–94. doi:10.1093/rheumatology/keh407
11. Wang Y, Xu X, Ran M, Guo X, Zhou L. The Enlargement of Abdominal Lymph Nodes Is a Characteristic of Autoimmune Liver Disease. Mediators Inflamm. 2020;2020:3631625. doi:10.1155/2020/3631625
12. Vij R, Strek ME. Diagnosis and treatment of connective tissue disease-associated interstitial lung disease. Chest. 2013;143(3):814–824. doi:10.1378/chest.12-0741
13. Hewlett S, Dures E, Almeida C. Measures of fatigue: Bristol Rheumatoid Arthritis Fatigue Multi-Dimensional Questionnaire (BRAF MDQ), Bristol Rheumatoid Arthritis Fatigue Numerical Rating Scales (BRAF NRS) for severity, effect, and coping, Chalder Fatigue Questionnaire (CFQ), Checklist Individual Strength (CIS20R and CIS8R), Fatigue Severity Scale (FSS), Functional Assessment Chronic Illness Therapy (Fatigue) (FACIT-F), Multi-Dimensional Assessment of Fatigue (MAF), Multi-Dimensional Fatigue Inventory (MFI), Pediatric Quality Of Life (PedsQL) Multi-Dimensional Fatigue Scale, Profile of Fatigue (ProF), Short Form 36 Vitality Subscale (SF-36 VT), and Visual Analog Scales (VAS). Arthritis Care Res. 2011;63 Suppl 11:S263–286. doi:10.1002/acr.20579
14. Seror R, Ravaud P, Bowman SJ, et al. EULAR Sjogren’s syndrome disease activity index: development of a consensus systemic disease activity index for primary Sjogren’s syndrome. Ann Rheum Dis. 2010;69(6):1103–1109. doi:10.1136/ard.2009.110619
15. Vitali C, Palombi G, Baldini C, et al. Sjögren’s Syndrome Disease Damage Index and disease activity index: scoring systems for the assessment of disease damage and disease activity in Sjögren’s syndrome, derived from an analysis of a cohort of Italian patients. Arthritis Rheum. 2007;56(7):2223–2231. doi:10.1002/art.22658
16. Bruce B, Fries JF. The Health Assessment Questionnaire (HAQ). Clin Exp Rheumatol. 2005;23(5 Suppl 39):S14–18.
17. Jordán-González P, Gago-Piñero R. Characterization of a subset of patients with primary Sjögren’s syndrome initially presenting with C3 or C4 hypocomplementemia. Eur J Rheumatol. 2020;7(3):112–117. doi:10.5152/eurjrheum.2020.19132
18. Theander E, Manthorpe R, Jacobsson LT. Mortality and causes of death in primary Sjögren’s syndrome: a prospective cohort study. Arthritis Rheum. 2004;50(4):1262–1269. doi:10.1002/art.20176
19. Alunno A, Leone MC, Giacomelli R, Gerli R, Carubbi F. Lymphoma and lymphomagenesis in primary Sjögren’s syndrome. Front Med. 2018;5:102. doi:10.3389/fmed.2018.00102
20. Theander E, Henriksson G, Ljungberg O, Mandl T, Manthorpe R, Jacobsson LT. Lymphoma and other malignancies in primary Sjögren’s syndrome: a cohort study on cancer incidence and lymphoma predictors. Ann Rheum Dis. 2006;65(6):796–803. doi:10.1136/ard.2005.041186
21. Solans-Laqué R, López-Hernandez A, Bosch-Gil JA, Palacios A, Campillo M, Vilardell-Tarres M. Risk, predictors, and clinical characteristics of lymphoma development in primary Sjögren’s syndrome. Semin Arthritis Rheum. 2011;41(3):415–423. doi:10.1016/j.semarthrit.2011.04.006
22. Quartuccio L, Isola M, Baldini C, et al. Biomarkers of lymphoma in Sjögren’s syndrome and evaluation of the lymphoma risk in prelymphomatous conditions: results of a multicenter study. J Autoimmun. 2014;51:75–80. doi:10.1016/j.jaut.2013.10.002
23. Quartuccio L, Baldini C, Bartoloni E, et al. Anti-SSA/SSB-negative Sjögren’s syndrome shows a lower prevalence of lymphoproliferative manifestations, and a lower risk of lymphoma evolution. Autoimmun Rev. 2015;14(11):1019–1022. doi:10.1016/j.autrev.2015.07.002
24. Carubbi F, Alunno A, Cipriani P, et al. A retrospective, multicenter study evaluating the prognostic value of minor salivary gland histology in a large cohort of patients with primary Sjögren’s syndrome. Lupus. 2015;24(3):315–320. doi:10.1177/0961203314554251
25. Brito-Zerón P, Acar-Denizli N, Ng WF, et al. How immunological profile drives clinical phenotype of primary Sjögren’s syndrome at diagnosis: analysis of 10,500 patients (Sjögren Big Data Project). Clin Exp Rheumatol. 2018;36 Suppl 112(3):102–112.
26. Baldini C, Pepe P, Quartuccio L, et al. Primary Sjogren’s syndrome as a multi-organ disease: impact of the serological profile on the clinical presentation of the disease in a large cohort of Italian patients. Rheumatology. 2014;53(5):839–844. doi:10.1093/rheumatology/ket427
27. Sandhya P, Danda D. Primary Sjögren’s syndrome in Asia: Yin and Yang? Int J Rheum Dis. 2017;20(10):1309–1312. doi:10.1111/1756-185X.13201
28. Brito-Zerón P, Acar-Denizli N, Ng WF, et al. Epidemiological profile and north-south gradient driving baseline systemic involvement of primary Sjögren’s syndrome. Rheumatology. 2020;59(9):2350–2359. doi:10.1093/rheumatology/kez578
29. Quartuccio L, Baldini C, Bartoloni E, et al. Correlation between ESSDAI and ClinESSDAI in a real-life cohort of patients with Sjögren’s syndrome. Clin Exp Rheumatol. 2017;35(3):546–547.
30. Flores-Chávez A, Kostov B, Solans R, et al. Severe, life-threatening phenotype of primary Sjögren’s syndrome: clinical characterisation and outcomes in 1580 patients (GEAS-SS Registry). Clin Exp Rheumatol. 2018;36 Suppl 112(3):121–129.
31. Priori R, Mastromanno L, Izzo R. What about glucocorticoids in primary Sjögren’s syndrome? Clin Exp Rheumatol. 2020;38 Suppl 126(4):237–244.
32. Ramos-Casals M, Solans R, Rosas J, et al. Primary Sjögren syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine. 2008;87(4):210–219. doi:10.1097/MD.0b013e318181e6af
33. Brito-Zerón P, Acar-Denizli N, Zeher M, et al. Influence of geolocation and ethnicity on the phenotypic expression of primary Sjögren’s syndrome at diagnosis in 8310 patients: a cross-sectional study from the big data Sjögren project consortium. Annals of the Rheumatic Diseases. 2017;76(6):1042–1050. doi:10.1136/annrheumdis-2016-209952
34. Maldini C, Seror R, Fain O, et al. Epidemiology of primary Sjögren’s syndrome in a French multiracial/multiethnic area. Arthritis Care Res. 2014;66(3):454–463. doi:10.1002/acr.22115
35. Billings M, Dye BA, Iafolla T, Baer AN, Grisius M, Alevizos I. Significance and implications of patient-reported xerostomia in Sjogren's syndrome: findings from the national institutes of health cohort. EBioMedicine. 2016;12:270–279. doi:10.1016/j.ebiom.2016.09.005
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.