Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Hyperbaric oxygen therapy attenuates neuronal apoptosis induced by traumatic brain injury via Akt/GSK3β/β-catenin pathway
Received 12 August 2018
Accepted for publication 27 October 2018
Published 25 January 2019 Volume 2019:15 Pages 369—374
DOI https://doi.org/10.2147/NDT.S183632
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yu-Ping Ning
Hui He,1 Xiufang Li,2 Yuling He1
1Department of Emergency, Zhuji People’s Hospital of Zhejiang Province, Zhuji, Zhejiang, China; 2Department of Pathology, Zhuji People’s Hospital of Zhejiang Province, Zhuji, Zhejiang, China
Background: Given that the therapeutic effect of hyperbaric oxygen (HBO) therapy on traumatic brain injury (TBI) has been debated for a long time, it is necessary to clarify the mechanism underlying the effect of HBO on acute TBI.
Methods: This study investigated the effect of HBO therapy on neuronal apoptosis induced by acute TBI using the mouse model of TBI. The number of apoptotic cells and expression of apoptosis-associated factors (including caspase 3, pAkt/Akt, pGSK3β/GSK3β, and β-catenin) in pericontusional cortices of mice exposed to sham, TBI, and TBI + HBO treatment were measured and analyzed using TUNEL assay, quantitative reverse-transcription PCR, and Western blot.
Results: Results showed that acute TBI increased the number of apoptotic neurons and mRNA expression and activated caspase 3 protein. With regard to proteins, acute TBI also resulted in decreased levels of pAkt/Akt, pGSK3β/GSK3β, and β-catenin, which facilitates neuronal apoptosis. This study shows that HBO therapy reversed these changes of pAkt/Akt, pGSK3β/GSK3β, and β-catenin induced by acute TBI and attenuated the apoptotic process in the pericontusional cortex.
Conclusion: This study demonstrates the beneficial effect of HBO therapy on neuronal apoptosis caused by acute TBI. Furthermore, the mechanism underlying the therapeutic effect of HBO on acute TBI partly involves the Akt/GSK3β/β-catenin pathway.
Keywords: hyperbaric oxygen, TBI, apoptosis, Akt, GSK3β, β-catenin
Introduction
Traumatic brain injury (TBI) is a growing public health problem in the world and the leading cause of death in Chinese adults aged <40 years.1 Oxygen supply to the brain is often insufficient after TBI, and then results in decreased energy production, which leads to neuronal apoptosis.2 Therefore, maintaining brain-oxygenation status is the main goal of treatment for TBI.3 As a nondrug and noninvasive treatment, hyperbaric oxygen (HBO) therapy has been used as a treatment for TBI since 1960s.4 However, the efficiency of HBO therapy remains extremely controversial.5–7 Several studies have reported that HBO therapy improved the neurological deficits and cognitive impairments of patients in the acute phase of severe TBI.8,9 In contrast, other studies have demonstrated that HBO therapy had no effect on patients with mild and chronic TBI.10 The therapeutic effect of HBO on multiple types of TBI is still widely debated. Therefore, it is necessary to clarify the mechanisms underlying the effect of HBO on TBI. Furthermore, the neuronal mechanism underlying the effect of HBO therapy on TBI is still obscure. A weight-drop model of closed-head injury has been proven to simulate acute TBI and has been widely adopted to investigate the mechanism involved in acute TBI.11 This study was conducted to verify the effect and mechanism of HBO therapy on acute TBI.
Facilitating an apoptotic cascade is one of the primary pathogenic mechanisms of acute TBI. It has been demonstrated that the number of apoptotic neurons and the levels of proapoptotic factors increase significantly after TBI.12 In the present study, the effect of HBO therapy on neuronal apoptosis after acute TBI was investigated by measuring the number of apoptotic neurons and the levels of apoptosis-related factors. Activated caspase 3 protein (C-caspase 3) is the most commonly used biomarker for apoptosis.13,14 The G-protein-independent Akt/GSK3β/β-catenin pathway is an intracellular signaling pathway and plays a critical role in cell apoptosis.15,16 For instance, β-catenin prevents apoptosis, whereas the downregulation of β-catenin is caused by the phosphorylation of GSK3β (pGSK3β), which is induced by activated/phosphorylated Akt (pAkt).17 Therefore, this study investigated the effect of HBO therapy on the neuronal apoptosis induced by acute TBI and the role played by the Akt/GSK3β/β-catenin pathway in the effect of HBO therapy.
Methods
Animals
Adolescent male C57BL/6 mice (6 weeks postnatal, weight 90±5 g) were used in this study. Mice were group-housed (four per cage) in a controlled environment (23°C±2°C, 55%±10% humidity, 12/12-hour light/dark cycle) with food and water ad libitum for 7 days before the experiment. These mice were randomly assigned into three groups: sham group (n=18), TBI (n=18), and HBO (n=18). Mice in the sham group received a sham operation. Mice in the TBI group were treated by simulated TBI operation. Mice in the HBO group were treated by simulated TBI operation and HBO therapy 1 hour after receiving simulated TBI operation. Mice were killed 1 day after receiving the sham or simulated TBI operation. For the six mice in each group, brains were collected for quantitative real-time (qRT) PCR, Western blot, or TUNEL staining. The study was approved by Zhuji People’s Hospital of Zhejiang Province. All procedures were performed in accordance with the National Institutes of Health’s Guide for the Use and Care of Laboratory Animals.
Mouse TBI model
The mouse model of TBI was created as described previously.11 After being anesthetized by 10% sodium pentobarbital (50 mg/kg, intraperitoneal injection), the mice were fixed in the TBI apparatus. The skull was exposed via a sagittal incision. For the simulated TBI operation, the injury was caused by a free-falling steel weight (200 g, with a blunt tip radius of 4 mm) from a height of 4 cm, producing focal trauma centered at 2 mm posterior to the bregma and 2 mm right of the midline of the skull. For the sham operation, no injury operation was conducted. Then, the scalp incision was sutured. After all procedures, the mice were returned to their cages. Their heart rate and blood pressure were monitored, and their body temperature maintained using a heating pad.
Hyperbaric oxygen treatment
Mice in the HBO group were placed in a research hyperbaric chamber (Hongyuan Oxygen Industry, Yantai, China), in which atmospheric air was flushed with 100% O2. The mice were placed inside for 90 minutes’ exposure to HBO (100% O2) at 2.8 atmospheres absolute (ATA) according to protocols previously reported.18 Chamber pressure was increased to 2.8 ATA in 10 minutes and gradually decreased to normal pressure (100% O2) in 15 minutes at the end of the session.
TUNEL assay
A thoracic cavity was opened for each mouse. A gage catheter for venous drainage was inserted in the left ventricle until the tip of the catheter had been placed inside the aorta. Then, the right auricle was rifted and perused with 0.01 M PBS (4°C). As the supernatant liquid had effused from the right auricle, the mice were killed by decapitation. The brain tissue including the pericontusional cortex (coronal section, 0–4 mm posterior to bregma and 0–4 mm right of midline of the brain) was collected, immersed in 4% paraformaldehyde for 24 hours at 4°C, and then sectioned (5 μm) by cryostat. Brain sections were analyzed using a DeadEnd colorimetric TUNEL system (Promega, Madison, WI, USA) according to the manufacturer’s instructions. After that, apoptotic nuclei were identified by brown staining. The five regions (magnification 10×) were selected randomly from the pericontusional cortex of the three continuous sections for each animal, and then the total neurons and TUNEL-positive neurons were counted. Apoptosis levels are expressed by apoptosis index (AI), which is the ratio of the percentage of the number of TUNEL-positive nuclei to the total number of neurons counted.
Quantitative real-time PCR
Pericontusional cortices of the mice were obtained within 3 minutes after death and frozen on dry ice. RNA was extracted with Trizol reagent (TaKaRa Biotechnology, Dalian, China) as per the manufacturer’s recommendations. cDNA was synthesized and RT-PCR carried out in 25 μL reaction volume containing 1 ng total RNA using an SYBR green one-step qRT-PCR kit (Thermo Fisher Scientific, Waltham, MA, USA) in a 7,500 sequence-detection system (Thermo Fisher Scientific) according to the manufacturer’s guidelines. Primers of the autism-associated genes are presented in Table 1. Relative mRNA expression levels of genes were normalized to GAPDH and calculated by the ΔΔCt analytical method. All experiments were performed in triplicate.
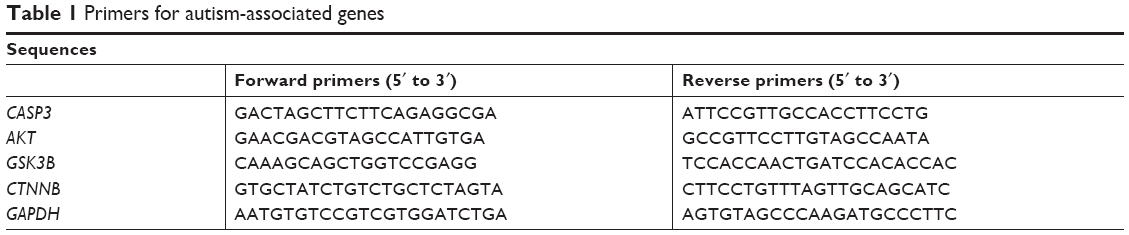 | Table 1 Primers for autism-associated genes |
Western blot
Pericontusional cortices of the mice were obtained and protein was extracted. Protein concentrations were estimated using a BCA protein assay kit (Thermo Fisher Scientific). Proteins (20 μg) were loaded on each lane of SDS–polyacrylamide gels, subjected to electrophoresis for 80 minutes at a constant voltage of 100 V, and then transferred to a polyvinylidene difluoride membrane for 90 minutes at 350 mA. Membranes were blocked with 5% nonfat milk and then incubated overnight at 4°C with primary antibodies against 1:1,000 cleaved/activated caspase 3 (Abcam), 1:1,000 Akt (Cell Signaling Technology), 1:1,000 pAkt (Cell Signaling Technology), 1:1,000 GSK3β (Cell Signaling Technology), 1:1,000 pGSK3β (Cell Signaling Technology), and 1:2,000 GAPDH (Abcam) proteins. Then, the membranes were incubated with secondary antibodies conjugated with horseradish peroxidase at room temperature for 1 hour. Protein bands were detected using an enhanced chemiluminescence system and quantified by ImageJ software.
Statistical analyses
Data were analyzed using SPSS 19.0 (IBM, Armonk, NY, USA). Data normality was assessed by the Kolmogorov–Smirnov test. Data showing normal distribution are summarized by means ± SD, and data showing abnormal distribution are summarized by medians. Means of two comparative groups were compared with Student’s t-test.
Results
Comparative analysis of apoptotic cells
Compared with the TBI group, apoptotic cells (AI) in the pericontusional cortex were significantly higher than in the sham group (sham vs TBI, 1.76±0.46 vs 23.37±5.59; P<0.01) and was significantly lower in the HBO group (HBO vs TBI, 9.81±1.33 vs 23.37±5.59; P<0.05; Figure 1).
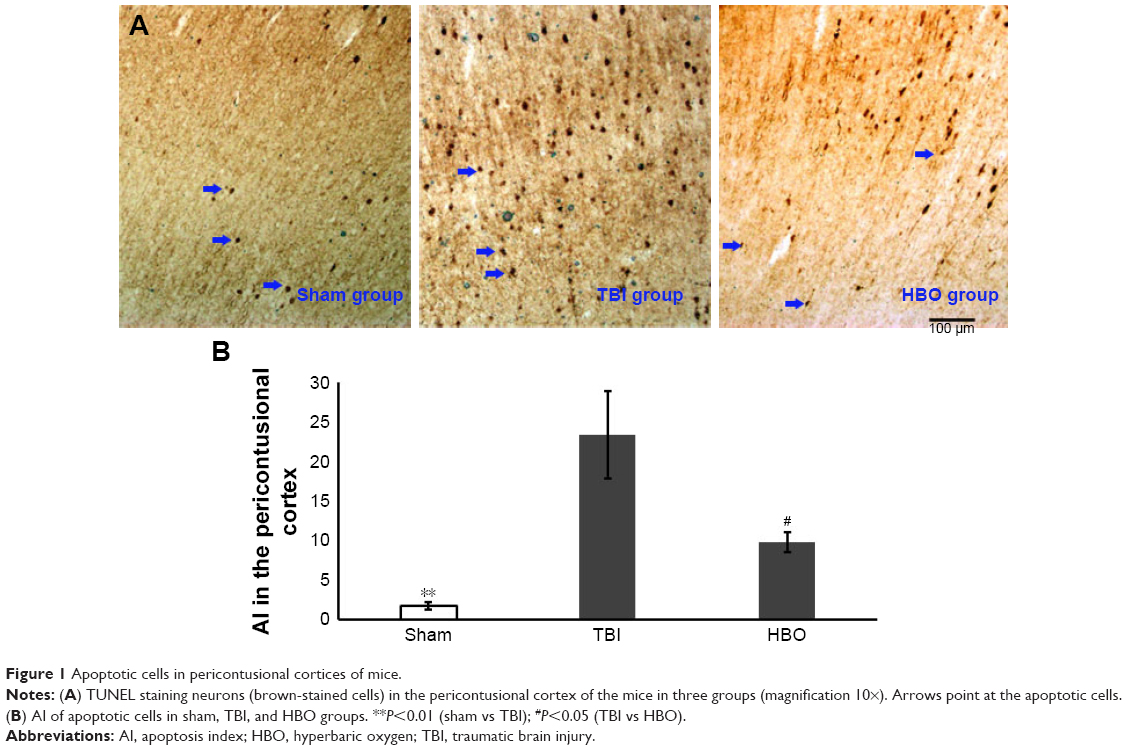 | Figure 1 Apoptotic cells in pericontusional cortices of mice. |
Comparative analysis of relative mRNA levels in affected brain hemisphere
Compared with TBI group, mRNA levels of caspase 3 were significantly lower in the sham group (sham vs TBI, 0.41±0.11 vs 1.24±0.20; P<0.05) and the HBO group (HBO vs TBI, 0.72±0.11 vs 1.24±0.20; P<0.05; Figure 2). mRNA levels of Akt, GSK3β, and β-catenin were not significantly different between the sham and TBI groups or between the TBI and HBO groups (P>0.05).
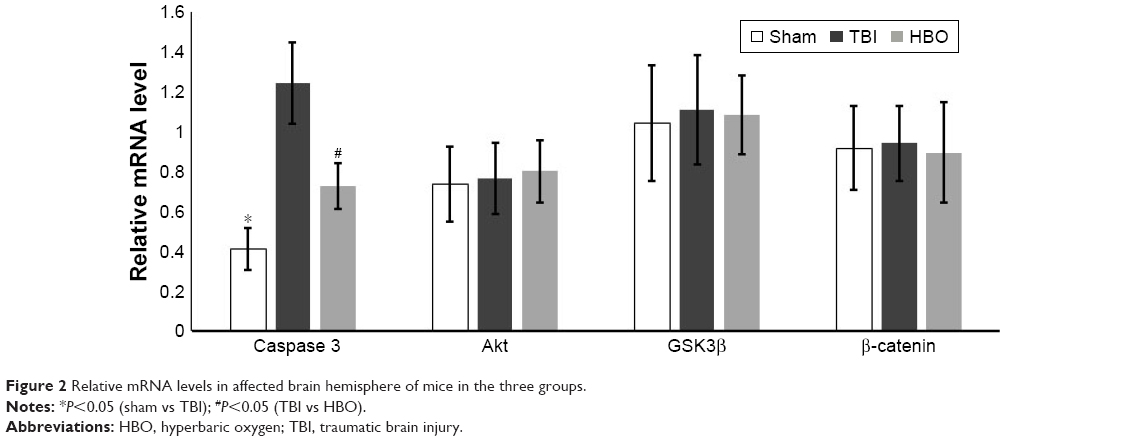 | Figure 2 Relative mRNA levels in affected brain hemisphere of mice in the three groups. |
Comparative analysis of relative protein levels in affected brain hemisphere
Compared with the sham group, C-caspase 3 levels were also significantly higher in the TBI group (sham vs TBI, 1.03±0.2 vs 1.62±0.33; P<0.05); however, pAkt/Akt levels (sham vs TBI, 1.01±0.09 vs 0.39±0.08; P<0.05), pGSK3β/GSK3β (sham vs TBI, 1.02±0.08 vs 0.35±0.07, P<0.05), and β-catenin (sham vs TBI, 1.17±0.38 vs 0.58±0.06; P<0.01) were significantly lower in the TBI group (Figure 3). Compared with the TBI group, C-caspase 3 levels were significantly lower (HBO vs TBI, 1.20±0.26 vs 1.62±0.33; P<0.05) in the HBO group, and pAkt/Akt levels (HBO vs TBI, 0.86±0.09 vs 0.39±0.08; P<0.05), pGSK3β/GSK3β (HBO vs TBI, 0.83±0.09 vs 0.35±0.07; P<0.05) and β-catenin (HBO vs TBI, 0.86±0.20 vs 0.58±0.06; P<0.05) were significantly higher in the HBO group (Figure 3).
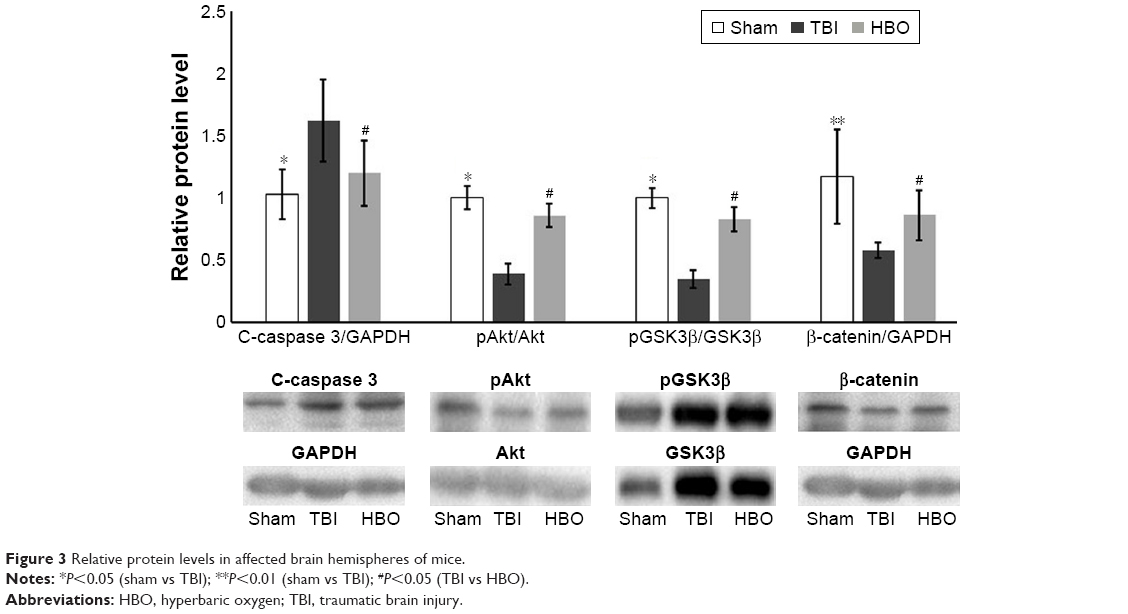 | Figure 3 Relative protein levels in affected brain hemispheres of mice. |
Discussion
Exploring potential treatments for TBI has been a research focus for a long time. TBI damage is primarily directly induced by physical damage and is secondarily due to hypoxia, overincreased oxidative stress, and inflammation, which all lead to neuron apoptosis.19 Because of the influence on oxidative stress, HBO therapy promotes restoration of oxygen supply, expression of antioxidant genes, alleviation of inflammation, inhibition of apoptosis, and promotion of angiogenesis and neurogenesis.20–23
In the present study, mice were treated by HBO therapy after simulated TBI operation. One day after TBI, the number of apoptotic neurons and mRNA and protein expression levels of apoptosis-associated factors in the pericontusional cortex were measured and analyzed. Neuronal apoptosis contributes to the overall pathology of TBI, and the percentage of apoptotic neurons has been used as an index for evaluating the severity of TBI.24,25 Caspase 3 plays a major role in the neuronal apoptosis pathway, and C-caspase 3 has been demonstrated to increase the number of neurons after TBI.26,27 This study demonstrated an increase in the number of apoptotic neurons after TBI. Furthermore, the mRNA level of caspase 3 and protein level of C-caspase 3 increased after acute TBI. However, the number of apoptotic neurons, mRNA level of caspase 3 and protein level of C-caspase 3 were significantly decreased in mice treated by HBO after acute TBI. This result proved that HBO therapy suppressed caspase 3 activity and attenuated the neuronal apoptosis induced by acute TBI. It is consistent with previous studies.28,29
In order to explore the mechanism of HBO therapy further, protein expression levels of components in the Akt–GSK3–β-catenin pathway were analyzed. The results suggested that pGSK3β/GSK3β and pAkt/Akt levels were significantly decreased after acute TBI, and the level of β-catenin was significantly decreased after acute TBI. Studies have proven decreased levels of pAkt, pGSK3β, and β-catenin after TBI.30–32 These data proved that the Akt/GSK3β/β-catenin pathway plays an important role in the pathological mechanisms of acute TBI. In addition, the comparative analysis between the HBO and TBI groups suggested that pGSK3β/GSK3β and pAkt/Akt levels were significantly increased in mice treated by HBO therapy. This result proved that HBO treatment can alleviate the negative changes of the Akt/GSK3β/β-catenin pathway after acute TBI. Considering the role played by the Akt/GSK3β/β-catenin pathway in cell apoptosis, it is reasonable to speculate that the effect of HBO therapy on inhibiting the neuronal apoptosis induced by acute TBI partly involves the Akt/GSK3β/β-catenin pathway.
It is notable that there was no significant difference in mRNA levels of Akt, GSK3β, or β-catenin between the mice in the TBI and HBO groups. This implies that HBO therapy has no direct effect on the RNA levels of Akt/GSK3β/β-catenin. Therefore, further study is needed to investigate in more depth the effect of HBO therapy on protein levels of the Akt/GSK3β/β-catenin pathway.
In conclusion, this study supported the therapeutic effect of HBO therapy on neuronal apoptosis after acute TBI. Moreover, this study primarily verified the role played by the Akt/GSK3β/β-catenin pathway in the mechanism underlying the effect of HBO therapy on neuronal damage induced by acute TBI. These results suggest that HBO therapy is a potential treatment for neural damage caused by TBI and should be investigated further.
Author contributions
Yuling He designed and managed this study. Yuling He and Hui He executed the study and selected the data. Hui He and Xiufang Li analysed the data. Hui He and Xiufang Li wrote the first draft of the manuscript. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Wu X, Hu J, Zhuo L, et al. Epidemiology of traumatic brain injury in eastern China, 2004: a prospective large case study. J Trauma. 2008;64(5):1313–1319. | ||
Senapathi TGA, Wiryana M, Sinardja K, et al. Jugular bulb oxygen saturation correlates with Full Outline of Responsiveness score in severe traumatic brain injury patients. Open Access Emerg Med. 2017 Aug 28;9:69–72. | ||
Verweij BH, Muizelaar JP, Vinas FC, Peterson PL, Xiong Y, Lee CP. Impaired cerebral mitochondrial function after traumatic brain injury in humans. J Neurosurg. 2000;93(5):815–820. | ||
Coe JE, Hayes TM. Treatment of experimental brain injury by hyperbaric oxygenation. Preliminary report. Am Surg. 1966;32(7):493–495. | ||
Wang F, Wang Y, Sun T, Yu HL. Hyperbaric oxygen therapy for the treatment of traumatic brain injury: a meta-analysis. Neurol Sci. 2016;37(5):693–701. | ||
Bennett MH, Trytko B, Jonker B. Hyperbaric oxygen therapy for the adjunctive treatment of traumatic brain injury. Cochrane Database Syst Rev. 2012;12:Cd004609. | ||
Hu Q, Manaenko A, Xu T, Guo Z, Tang J, Zhang JH. Hyperbaric oxygen therapy for traumatic brain injury: bench-to-bedside. Med Gas Res. 2016;6(2):102–110. | ||
Daly S, Thorpe M, Rockswold S, et al. Hyperbaric Oxygen Therapy in the Treatment of Acute Severe Traumatic Brain Injury: A Systematic Review. J Neurotrauma. 2018;35(4):623–629. | ||
Rockswold SB, Rockswold GL, Defillo A. Hyperbaric oxygen in traumatic brain injury. Neurol Res. 2007;29(2):162–172. | ||
Cifu DX, Hoke KW, Wetzel PA, Wares JR, Gitchel G, Carne W. Effects of hyperbaric oxygen on eye tracking abnormalities in males after mild traumatic brain injury. J Rehabil Res Dev. 2014;51(7):1047–1056. | ||
Yu Z, Li H, Yan HY, et al. Expression and Cell Distribution of SENP3 in Brain Tissue After Traumatic Brain Injury in Mice: A Pilot Study. Cell Mol Neurobiol. 2015;35(5):733–740. | ||
Ray SK, Dixon CE, Banik NL. Molecular mechanisms in the pathogenesis of traumatic brain injury. Histol Histopathol. 2002;17(4):1137–1152. | ||
Nathoo N, Narotam PK, Agrawal DK, et al. Influence of apoptosis on neurological outcome following traumatic cerebral contusion. J Neurosurg. 2004;101(2):233–240. | ||
Lorente L. Biomarkers Associated with the Outcome of Traumatic Brain Injury Patients. Brain Sci. 2017;7(11):142. | ||
Polakis P. The oncogenic activation of beta-catenin. Curr Opin Genet Dev. 1999;9(1):15–21. | ||
Dai P, Mao Y, Sun X, et al. Attenuation of Oxidative Stress-Induced Osteoblast Apoptosis by Curcumin is Associated with Preservation of Mitochondrial Functions and Increased Akt-GSK3β Signaling. Cell Physiol Biochem. 2017;41(2):661–677. | ||
Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol. 2006;79(4):173–189. | ||
Liu H, Yang M, Pan L, Liu P, Ma L. Hyperbaric Oxygen Intervention Modulates Early Brain Injury after Experimental Subarachnoid Hemorrhage in Rats: Possible Involvement of TLR4/NF-x03BA; B-Mediated Signaling Pathway. Cell Physiol Biochem. 2016;38(6):2323–2336. | ||
Eve DJ, Steele MR, Sanberg PR, Borlongan CV. Hyperbaric oxygen therapy as a potential treatment for post-traumatic stress disorder associated with traumatic brain injury. Neuropsychiatr Dis Treat. 2016;12:2689–2705. | ||
Thom SR. Oxidative stress is fundamental to hyperbaric oxygen therapy. J Appl Physiol. 2009;106(3):988–995. | ||
Godman CA, Joshi R, Giardina C, Perdrizet G, Hightower LE. Hyperbaric oxygen treatment induces antioxidant gene expression. Ann N Y Acad Sci. 2010;1197:178–183. | ||
Ding Z, Tong WC, Lu XX, Peng HP. Hyperbaric oxygen therapy in acute ischemic stroke: a review. Interv Neurol. 2014;2(4):201–211. | ||
Liu W, Khatibi N, Sridharan A, Zhang JH. Application of medical gases in the field of neurobiology. Med Gas Res. 2011;1(1):13. | ||
Raghupathi R, Graham DI, McIntosh TK. Apoptosis after traumatic brain injury. J Neurotrauma. 2000;17(10):927–938. | ||
Wong J, Hoe NW, Zhiwei F, Ng I. Apoptosis and traumatic brain injury. Neurocrit Care. 2005;3(2):177–182. | ||
Eldadah BA, Faden AI. Caspase pathways, neuronal apoptosis, and CNS injury. J Neurotrauma. 2000;17(10):811–829. | ||
Yakovlev AG, Ota K, Wang G, et al. Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J Neurosci. 2001;21(19):7439–7446. | ||
Xing P, Ma K, Li L, Wang D, Hu G, Long W. The protection effect and mechanism of hyperbaric oxygen therapy in rat brain with traumatic injury. Acta Cir Bras. 2018;33(4):341–353. | ||
Baratz-Goldstein R, Toussia-Cohen S, Elpaz A, Rubovitch V, Pick CG. Immediate and delayed hyperbaric oxygen therapy as a neuroprotective treatment for traumatic brain injury in mice. Mol Cell Neurosci. 2017;83:74–82. | ||
Cho JH, Sung JH, Cho EH, et al. Gingko biloba Extract (EGb 761) prevents ischemic brain injury by activation of the Akt signaling pathway. Am J Chin Med. 2009;37(3):547–555. | ||
Hong Y, Shao A, Wang J, et al. Neuroprotective effect of hydrogen-rich saline against neurologic damage and apoptosis in early brain injury following subarachnoid hemorrhage: possible role of the Akt/GSK3β signaling pathway. PLoS One. 2014;9(4):e96212. | ||
Zhao S, Fu J, Liu X, Wang T, Zhang J, Zhao Y. Activation of Akt/GSK-3beta/beta-catenin signaling pathway is involved in survival of neurons after traumatic brain injury in rats. Neurol Res. 2012;34(4):400–407. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.