Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Hyperbaric oxygen therapy as a potential treatment for post-traumatic stress disorder associated with traumatic brain injury
Authors Eve DJ ![]() , Steele MR, Sanberg PR, Borlongan CV
, Steele MR, Sanberg PR, Borlongan CV
Received 8 April 2016
Accepted for publication 12 July 2016
Published 20 October 2016 Volume 2016:12 Pages 2689—2705
DOI https://doi.org/10.2147/NDT.S110126
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Roger Pinder
David J Eve,1 Martin R Steele,2 Paul R Sanberg,1 Cesar V Borlongan1
1Department of Neurosurgery and Brain Repair, Center of Excellence for Aging and Brain Repair, Morsani College of Medicine, 2Veterans Reintegration Steering Committee, Veterans Research, University of South Florida, Tampa, FL, USA
Abstract: Traumatic brain injury (TBI) describes the presence of physical damage to the brain as a consequence of an insult and frequently possesses psychological and neurological symptoms depending on the severity of the injury. The recent increased military presence of US troops in Iraq and Afghanistan has coincided with greater use of improvised exploding devices, resulting in many returning soldiers suffering from some degree of TBI. A biphasic response is observed which is first directly injury-related, and second due to hypoxia, increased oxidative stress, and inflammation. A proportion of the returning soldiers also suffer from post-traumatic stress disorder (PTSD), and in some cases, this may be a consequence of TBI. Effective treatments are still being identified, and a possible therapeutic candidate is hyperbaric oxygen therapy (HBOT). Some clinical trials have been performed which suggest benefits with regard to survival and disease severity of TBI and/or PTSD, while several other studies do not see any improvement compared to a possibly poorly controlled sham. HBOT has been shown to reduce apoptosis, upregulate growth factors, promote antioxidant levels, and inhibit inflammatory cytokines in animal models, and hence, it is likely that HBOT could be advantageous in treating at least the secondary phase of TBI and PTSD. There is some evidence of a putative prophylactic or preconditioning benefit of HBOT exposure in animal models of brain injury, and the optimal time frame for treatment is yet to be determined. HBOT has potential side effects such as acute cerebral toxicity and more reactive oxygen species with long-term use, and therefore, optimizing exposure duration to maximize the reward and decrease the detrimental effects of HBOT is necessary. This review provides a summary of the current understanding of HBOT as well as suggests future directions including prophylactic use and chronic treatment.
Keywords: mild moderate and severe TBI, clinical trials, reactive oxygen species, acoustic startle response, elevated plus maze, cutoff behavioral criteria, prophylactic use, preconditioning, inflammation, acute and chronic treatment
Traumatic brain injury (TBI)
The recent excursions in Iraq and Afghanistan in which US soldiers have increasingly been exposed to improvised exploding devices (IEDs), along with improvements in personal protective equipment (ie, body armor) and medical technology, have led to an increase in the number of soldiers surviving TBI. TBI is typified by psychological and neurological symptoms arising due to physical damage to the brain as a consequence of an insult, which in the case of returning veterans is frequently blast-related. A transient loss of consciousness is also a hallmark of TBI, although there is a debate as to whether this is a necessary component of its definition, which can lead to it being described as a postconcussion syndrome (PCS) in a quarter of TBI patients.1 TBI can be classified as mild, moderate, or severe depending on the extent of the damage to the brain. This can be difficult to determine directly, except for at post mortem; however, brain imaging by computed tomography, magnetic resonance imaging (MRI), single-photon emission computed tomography (SPECT), and positron emission tomography scans can help reveal damage, particularly in the more severe cases. Neurological severity scales such as the Glasgow Coma Scale (GCS) are therefore frequently used to classify the degree of injury along with a detailed neurological examination. In general, mild TBI (mTBI) is diagnosed with only short-term (<30 min) loss of consciousness, confusion, or disorientation and the presence of frequently subtle cognitive problems that include headaches, memory and attention deficits, problematic cognition, mood swings, and frustration and may develop over time. The GCS score will be ≥13. Brain imaging will frequently not reveal any problems or very minor damage in mTBI patients. A diagnosis of moderate TBI is made in patients with between 30 min and 6 hours of unconsciousness and a GCS score <13, while severe TBI refers to a longer period of unconsciousness and likely >24 hours of memory loss and has a 40% mortality rate and more extensive damage. Additional characteristics span the gamut of impaired higher-level cognitive function through to a comatose state depending on the degree and location of injury. While those suffering from a moderate to severe TBI are relatively easily diagnosed, showing major impairment in, for example, motor responsiveness, verbal performance, and eye reflex (as characterized by the GCS), the diagnosis of mTBI is more difficult, with patients appearing relatively normal on this scale. Detailed neurological examination would be required, and this does not always reveal any injury. This is further compounded by an influx of returning veterans exhibiting symptoms that are suggestive of a mTBI, even though they were not previously diagnosed.2 The pathophysiology of TBI is biphasic with the initial presence of contusions, hematomas, and diffuse cell injury followed by secondary occurrences such as edema, hypoxia, inflammation, and finally cell loss (reviewed by Tanev et al).3 Impairment of the prefrontal, parietal, temporal, and regional neural networks and white matter tract damage frequently results in high-level cognitive dysfunction. Progressive atrophy of gray and white matter in many areas of the brain can be observed in moderate and severe TBI. Hippocampal dysfunction is also common.
In addition to the severity of the injury, the time after the injury (ie, acute vs chronic) will also dictate the observed symptoms to some extent. Favorable recovery has been observed in 80%–90% of mTBI cases within the first 3 months of injury, and the remaining cases are termed as the “miserable minority.”4 The reasons for these patients not recovering are unclear, but any acute study of mTBI should consider this, and ideally chronic studies of mTBI should commence only when the “miserable minority” remain so as to not have “normal improvements” confounding results.
Post-traumatic stress disorder (PTSD)
PTSD is defined as an anxiety disorder triggered by exposure to a stressful event and the memories thereof.5 An exaggerated startle response to both “normal” and trauma-related stimuli is frequently observed along with problems in concentrating, anxiety, perturbed fear conditioning, disrupted sleep, lack of extinction of traumatic memories, and hyperarousal. Epidemiological evidence suggests that over half the general population are exposed to a serious traumatic event during their lifetime, but only a proportion of these people (~7%) are expected to develop PTSD. Many people may suffer from an acute form of PTSD that lasts <3 months, but the number that is expected to progress to the chronic form with symptoms that can wax and wane is relatively small. The reasons for the small proportion that progress from traumatic event exposure to PTSD are unknown, but may be related to trauma severity and predispositions such as a family or prior individual history of psychiatric disorders.6–8 In addition, there is also a delayed onset form of PTSD which takes several months after the initial trauma to appear. Neurobiological characteristics of PTSD show multiple regions of the brain being affected. These include hippocampal atrophy and altered activity of the insular cortex as well as hypoactivity of the hypothalamic–pituitary–adrenal axis (reviewed in Whitaker et al).9 Alterations in the activity of the ventromedial prefrontal cortex, which projects to the central amygdala and the hypothalamic–pituitary–adrenal axis, have also been identified in PTSD.3,9,10 Hippocampal dysfunction has been detected in association with the inability to distinguish between safe and unsafe situations.3,9,10 The medial prefrontal cortex has been shown to be involved in a number of processes including the interpretation of stimuli. Neuroimaging studies suggest that this region may be dysfunctional in PTSD which may implicate this region in several of the processes altered in PTSD such as fear conditioning and cognitive and emotional interactions.11,12
Are TBI and PTSD related?
Although both mTBI and PTSD can be seen in the same soldier (as well as in different soldiers), there is a considerable debate over whether in people with both mTBI and PTSD, the PTSD is a consequence of the mTBI, or just an unconnected disorder (reviewed in Elder et al,1 Hoffman and Harrison,13 and McAllister14). One theory postulates that since TBI is frequently associated with traumatic amnesia, this prevents the patient from remembering the traumatic event(s), which could result in PTSD, thus suggesting that they are not related.15–17 However, many studies have shown that between 10% and 20% of returning soldiers exhibit mTBI symptoms (reviewed in Elder et al1 and Tanielian and Jaycox2) and also exhibit psychological dysfunction including depression or PTSD. Studies suggest that PTSD does occur after TBI even if there is little or no recollection of the period in which the injury was sustained,18 as well as the existence of patients in which TBI appears to be influencing PTSD and vice versa have previously been identified (reviewed by Tanev et al).3 In addition, Elder et al have demonstrated PTSD-like symptoms in an animal model of TBI in which the animals underwent anesthesia during the time of injury, so that they would not “remember” the injury.19 Reger et al also showed that in a rat model following mTBI, fear-conditioning procedures led to an enhanced fear response compared with noninjured animals providing additional evidence of PTSD-like symptoms (ie, exaggerated startle/fear response) after TBI.20 There is still some debate, since some studies of patients with both TBI and PTSD show greater re-experiencing, whereas other studies show fewer intrusive memories (reviewed by Tanev et al).3 In addition, both civilians and military personnel with a TBI seem to show an increased likelihood of PTSD symptoms in several studies, whereas another study in military service members showed a decreased presence of PTSD symptoms (reviewed by Tanev et al).3 There is also some discussion over whether the psychological symptoms associated with TBI should be classified as PTSD rather than a TBI symptom.1 Some of the symptoms of mTBI such as fatigue, irritability, sleep disturbance, and concentration problems are also observed in PTSD. Depression, alcoholism, and emotional alterations also frequently occur in both mTBI and PTSD patients, demonstrating that there is an overlap between the two disorders. Hippocampal damage is observed in both chronic TBI and PTSD, which means that TBI may predispose an individual to PTSD. It is important to determine whether TBI and PTSD are related because a treatment for PTSD with some degree of success is selective serotonin reuptake inhibitors, which could have adverse effects if given to TBI patients.
Evidence of inflammation has been observed in clinical cases of both TBI and PTSD (reviewed by Hinson et al and Passos et al, respectively).21,22 However, it is not clear whether this inflammation is a specific modality for the disorders, since Tursich et al suggest that any trauma exposure can result in inflammation.23 Additionally, Eraly et al imply that inflammation (determined by plasma C-reactive protein levels) may increase the likelihood of a person developing PTSD.24
In a previous study, moderate TBI was induced by controlled cortical impact (CCI) and PTSD induced by predator exposure and social instability in rats, thus treating them as two separate entities in the same animals.25 The results suggested that PTSD did not influence the degree of inflammation and suppressed cell proliferation that was observed in the TBI rats. However, no behavioral analysis was performed; hence, it is possible that the TBI only animals may also have exhibited PTSD symptoms and therefore the contribution of PTSD is still unclear. We therefore intend to induce mild to moderate TBI in rats and then assay the animals for PTSD (and TBI) characteristics over time. These characteristics can be monitored behaviorally in animals using the increased startle response, extinction of classical fear conditioning, reduced social interaction and activity, and impairment of hippocampal-dependent tasks.26–30
One possible therapy that has been proposed for TBI (and PTSD) is hyperbaric oxygen therapy (HBOT) normally defined as exposure to 100% oxygen at ≥1.5 absolute atmosphere (ATA), although the evidence for this is mixed (see “HBOT clinical trials” section). Therefore this review explores this in an animal model of TBI.
HBOT
Under normal conditions, the partial pressure of oxygen is ~95 mmHg with the majority of oxygen being carried by the hemoglobin (97% capacity) and only a small amount being dissolved in the blood itself. Under hypoxic conditions (essentially lack of oxygen, which is frequently associated with brain injuries), the partial pressure of oxygen is greatly reduced resulting in impairment of cell function due to reduced energy production. Potential therapies could seek to restore the presence of oxygen and thus restore the energy balance. However, increasing the concentration of oxygen inhaled from 20% to 100% (normobaric oxygen therapy; NBOT) has only a relatively small effect on the partial pressure of oxygen since the only way to increase oxygen capacity is by promoting the amount of oxygen dissolved in the blood. On the other hand, exposure to higher than normal atmospheric pressure (normal =1 ATA), also known as HBOT, can substantially increase the amount of oxygen dissolved in the blood and thus could be a potential therapy for disorders where hypoxia is involved (eg, 1,053 mmHg at 1.5 ATA and >2,000 mmHg at 2.5 ATA).31,32 Increasing pressure, but not oxygen, for example, 1.3 ATA normal air inspiration, increases the partial pressure of oxygen to 148 mmHg.33 HBOT has been approved by the Food and Drug Administration for enhancing wound and burn healing, carbon monoxide poisoning, gangrene, and radiation injuries and is being tested for other disorders with varied results, which can be open to interpretation.
HBOT clinical trials
Reviews of the literature do not convincingly show that HBOT is effective in the treatment of brain disorders such as TBI,34 stroke,35 multiple sclerosis,36 or cerebral palsy,33,37 although in each case, further more detailed studies are necessary to elucidate whether there is any clinically relevant potential benefit. The results of clinical trials of HBOT and TBI/PTSD are discussed later in the text, and Table 1 highlights the current status of clinical trials using HBOT for brain injury and psychological disorders listed at www.ClinicalTrials.gov (search terms: hyperbaric oxygen therapy filtered by brain injury or psychological disorders ran on June 13, 2016). Note that not all of the referenced studies below have a clear corresponding entry at www.ClinicalTrials.gov.
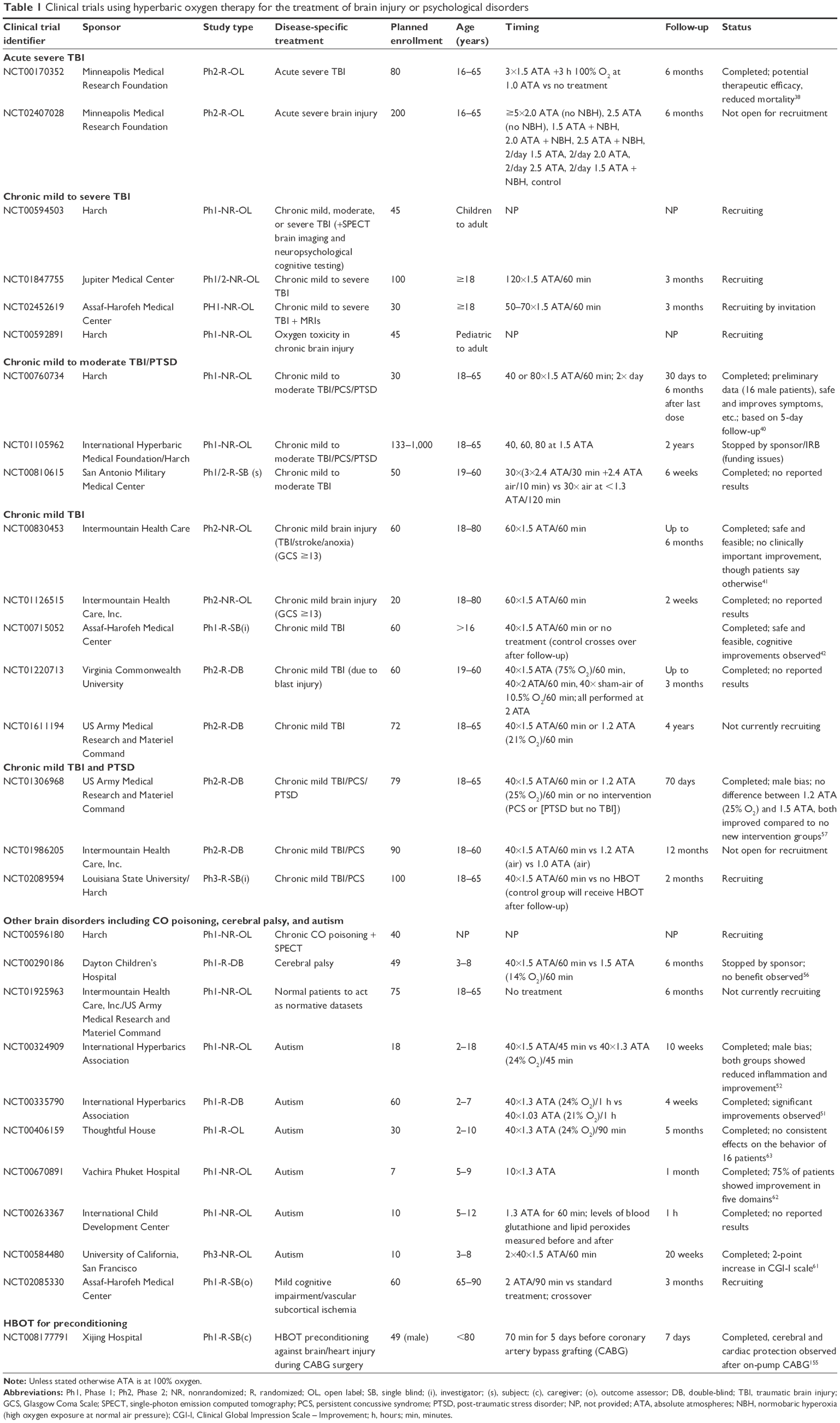 | Table 1 Clinical trials using hyperbaric oxygen therapy for the treatment of brain injury or psychological disorders |
Several clinical trials have been performed using HBOT to treat TBI and PTSD with varied results that are open to interpretation. Acute severe TBI treatment with HBOT (compared with no treatment) in a randomized open label trial (NCT00170352) demonstrated reduced mortality as well as potential therapeutic effects.38 A second randomized open label clinical trial exploring different ATAs and NBOT is underway (NCT02407028). However, the absence of a placebo group undermines the importance of these two studies, which are the only two acute TBI studies listed at www.ClinicalTrials.gov. Several other published studies suggest that mortality from an acute TBI is improved following HBOT treatment, although methodological shortcomings (such as nonrandomized, no placebo, and no blinding) and the limited number of patients dampen enthusiasm to some extent (reviewed by Bennett et al).34 In a case study involving a 27-year-old male who suffered a traumatic car/bicycle accident and was experiencing signs of emotional distress that could develop into PTSD, 7 treatments of HBOT (2.4 ATA for 90 min) commencing 7 days after injury seemed to resolve his emotional distress, suggesting that HBOT may be able to alleviate the development of PTSD.39 However, this is in only one patient, and the placebo effect cannot be discounted, but combined with the other studies; it is an encouraging evidence for a potential benefit. The ability of HBOT to modify acute injury and reduce mortality is therefore worth exploring further in well-designed studies, ideally in a randomized controlled trial that is currently lacking. The benefit of HBOT against acute mild or moderate TBI also requires investigation.
The remaining studies considered chronic TBI (≥3 months since injury). Harch et al exposed 14–16 male military service personnel suffering from mild to moderate TBI/PCS and PTSD for over a year to 40 sessions of 1.5 ATA in 100% oxygen for 60 min, and they determined that the procedure was safe and observed significant improvements in symptoms, cognitive testing, and quality of life measures (NCT00760734).40 However, this was a nonrandom open-label trial without a placebo group; hence, its relevance is unclear when other more rigorous studies with additional groups and randomization are considered. While before and after treatment statistical comparison showed no significant differences in another nonrandom open-label clinical trial for HBOT in chronic mTBI, stroke, and anoxia patients (predominantly male; NCT00830453), the patients themselves reported improvements, and the treatment appeared to be safe and feasible.41 However, since this was solely a treatment group, the positive effect of receiving a treatment cannot be excluded. In a randomized, investigator (but not patient)-blinded crossover trial (ie, control group gets treatment afterwards) for chronic mTBI (NCT00715052), cognitive improvements were observed in both the groups (after crossover), again suggesting that there may be beneficial effects of HBOT, although again the “treatment” effect cannot be excluded.42 By contrast, Wolf et al performed a more rigorous randomized controlled clinical trial on 50 military service personnel (48 male), who suffered from an mTBI 3–71 months ago, by exposing them to either 1.3 ATA (room air) or 2.4 ATA (100% oxygen) for 30 sessions.43 This may correspond to www.ClinicalTrials.gov record NCT00810615, although there are discrepancies (such as the clinical trial describes mild to moderate TBI while the paper only includes mTBI) and the official record at www.ClinicalTrials.gov currently reports no results. The exposure to 1.3 ATA was considered a sham treatment, and no significant differences were observed between the sham and experimental 2.4 ATA treatment. However, it is worth pointing out that both the treatment groups showed improvement in this study, and Harch suggested that this validated the use of HBOT since 1.3 ATA is not a true sham treatment and that “both treatments” showed relatively equal improvement.44 Although this is true to some extent, 1.3 ATA is also not true HBOT since the oxygen concentration was not 100% but that of normal air, and therefore, it is hyperbaric therapy (HBT) rather than HBOT. It is believed that the atmospheric pressure needs to be increased (to 1.3 ATA) for the placebo group so that the patients cannot determine whether they are receiving an active treatment. Breathing normal air at 1.3 ATA is said to be the equivalent of 27%–28% oxygen at 1 ATA and is thus only an exposure to a small increase in oxygen concentration over the normal condition; hence, it is unclear whether it would be expected to also be beneficial.45 Several animal studies do suggest that air under pressure (HBT) also has a biological influence on brain,46 endothelial cells,47 bone,48,49 and fibroblasts.50 Wolf et al proposed a number of possible explanations for the benefits observed in both the “sham” and treatment groups, apart from HBT, including normal recovery that has been observed over time (although all patients were treated at least 3 months after injury), the change in routine and location for the patients, or the Hawthorne effect.43 In addition, in an optimal clinical trial (randomized, double-blind, and controlled) for autism (NCT00335790), the HBT treatment of 1.3 ATA (24% O2) was used as a treatment group compared with 1.03 ATA (21% O2) and was found to induce significant improvement.51 In an earlier nonrandom open-label trial of autism (NCT00324909), the effects of the HBT treatment (1.3 ATA, 24% O2) were comparable with an HBOT treatment (1.5 ATA).52 These studies therefore support the proposal that 1.3 ATA air cannot be considered a true sham group and may also be beneficial. A randomized, double-blind study including 61 male marines suffering from mTBI-induced PCS for 3–36 months, who were exposed to 40×60 min 2 ATA with normal air (“control”), the 1.5 ATA oxygen equivalent (75% oxygen), or 100% oxygen was performed by Cifu et al, and they observed no significant improvements with HBOT compared to the “control.”53 It is worth pointing out that again even the control group is undergoing HBT, and all the patients showed improvements in working, delayed verbal and visual memory, and executive function. In a similar study of a different cohort of 60 male marines suffering from mTBI/PCS for 3–36 months, again no significant changes between the three groups were seen.54 However, a significant improvement was observed in two of the measures of, as well as the, total Post-traumatic Disorder Checklist-Military Version (PCL-M) score in the 2.0 ATA group, suggesting possible improvement in some PTSD symptoms. It is unclear whether the Cifu et al studies mentioned earlier correspond to www.ClinicalTrials.gov record NCT01220713 since there are discrepancies (since the studies were performed in Florida and not Virginia), and the official record does not reveal any reported results.53,54 Another report by Wolf et al of 50 patients exposed to 30 sessions of either 1.3 ATA air or 2.4 ATA oxygen again revealed no significant differences between the groups, but both the groups showed improvements, and some subgroups defined by concussion history and some individual test components such as visual memory and reaction time showed significant differences,55 suggesting that there may be some benefits of HBOT compared with no treatment. A randomized double-blind controlled study of HBOT (40×1.5 ATA [100% O2]) versus HBT (40×1.5 ATA [14% O2]) for cerebral palsy (NCT00290186) was stopped since no benefits were observed.56 However, again there is no true control group. In another male-biased military mTBI/PCS/PTSD study using an HBOT treatment group, an HBT control group, and a no new intervention group (NCT01306968), no significant differences were observed between the HBOT and HBT groups, but both showed significant improvement compared with the no new treatment group.57 Note that in this study, the PTSD group was not a comorbidity with TBI and did not receive HBOT and therefore acted as a second no-treatment group. The nonrandomized open-label study (NCT00760734) saw improvements in PTSD symptoms, while one further trial has been stopped for financial reasons (NCT01105962). Therefore, only a few studies have explored PTSD symptoms in mTBI; hence, the effect of HBOT is unclear.
NBOT may also be beneficial for psychiatric disorders, such as PTSD, since Bloch et al showed positive effects on memory and attention in schizophrenic patients.58 There is also some limited evidence from nonrandom open-label trials for an effect in treating autism (NCT00584480, NCT00670891),59–62 though at least one random open-label study shows no consistent benefit in a small population (NCT00406159).63 The validity of HBOT to treat TBI/PTSD is therefore still unclear, and tweaking of the treatment regime may be required to maximize any effect. Therefore, it has been proposed to perform preclinical studies in rats to determine whether 1) PTSD in rats undergoing TBI can be detected and 2) whether HBOT improves TBI and/or PTSD symptoms in these animals.
Table 1 shows that there are a number of ongoing clinical trials for which the results will be of interest in determining the effectiveness of HBOT against mild to severe TBI and PTSD. Of particular interest will be NCT01986205 which includes HBOT, HBT, and normal air treatment and so has suitable controls – although the patients may know that they are in the normal air group making it difficult to rule out any kind of treatment-specific versus any treatment effect. Several of the other studies are a single treatment group (NCT01847755) and so will not be as useful as other more rigorous studies that have HBT and/or normal air and pressure as a control group (NCT01611194). The crossover design of NCT02089594 may also be revealing with regard to any benefit of HBOT against mTBI. There is also a lack of chronic severe TBI studies, which should also be explored.
Does HBOT improve TBI and PTSD symptoms?
The aforementioned mixed clinical data make it difficult to determine how effective HBOT is, since several of the most scientifically valid randomized controlled trials suggest no benefit of HBOT compared to an HBT “control” for chronic mTBI and PTSD, while there is some indication of improvement compared with no treatment. Acute TBI treatment may also show some benefit, although more rigorous randomized controlled trials are required to determine its effectiveness. Currently, there are no data for HBOT in the treatment of chronic severe TBI as the studies that included chronic severe TBI are still recruiting. Although some studies suggest that mortality from acute severe TBI can be reduced by HBOT, it is unclear whether the improved mortality would actually translate to a better life for the patients or just increase the number that were comatosed.34 Further research is therefore required to elucidate whether any changes in mortality are actually therapeutically relevant.
There are more studies considering HBOT in chronic mild and moderate TBI, although their clinical validity is marred in some instances because of the lack of random group assignment, lack of an appropriate control, and the lack of blindness for the participants and researchers. There are several ongoing randomized controlled trials that may provide more insight to help to resolve this (eg, NCT01611194 [HBT vs HBOT], NCT01986205 [sham vs HBT vs HBOT]). A retrospective study of 20 HBO-treated and 20 no HBOT subacute/chronic TBI patients (1–6 months after injury) demonstrated a greater improvement in the HBO-treated patients, particularly those who were the most impaired.64 Two football players injured during play and diagnosed with TBI (or repetitive TBIs known as chronic traumatic encephalopathy) showed improved symptoms following HBOT.65 However, since there were only two cases that also exhibited differences in age (15 and early 50s) and injury durations (3 months and 30 odd years), this evidence was really anecdotal at best.
Regarding PTSD, there are very few trials that directly consider concomitant PTSD and mTBI. A nonrandomized open-label trial suggests benefit (NCT00760734),40 while the Cifu et al studies demonstrated improved Post-traumatic Disorder Checklist-Military Version measures (and overall) in the 2 ATA HBOT group.54 This therefore suggests that more studies are needed to determine whether PTSD is improved by HBOT.
The increased presence of oxygen in the blood circulation and tissues following HBOT may help to provide oxygen to areas of the brain that have reduced blood supply due to damage and edema following a TBI. In an acute rat model of moderate TBI, edema was shown to be decreased in the hippocampus following 2 weeks of HBOT. This was also associated with improved spatial learning and memory.66 This benefit is most likely to be seen in treating acute TBI, since in a chronic patient, hypoxia and edema are likely to have stabilized and so the effect of HBOT may be negligible in this regard. Therefore, any improvements seen in the treatment of acute TBI are not likely to translate to chronic TBI (and vice versa).
Many animal studies are considering acute TBI/PTSD, while the majority of the clinical trials in Table 1 are chronic studies. In addition, a large number of the animal studies do not distinguish between mild, moderate, and severe TBI, which makes their interpretation with regard to clinical translation difficult. It is impossible to use the GCS in animals; therefore, a different measure such as the size of injury (or size of impact used to generate injury) is required to determine the injury classification. This is further compounded by the various forms of available injury models that are reviewed by Xiong et al.67 Despite these pitfalls, in the next section, possible mechanisms of action for HBOT to work in the treatment of TBI/PTSD are discussed.
Possible mechanisms of action
There are a number of theories as to why HBOT could be beneficial ranging from restoration of oxygen supply, stem cell migration, alleviation of inflammation, inhibition of apoptosis, modulation of cerebral blood flow and brain metabolism, and promotion of angiogenesis and neurogenesis (reviewed by Ding et al and Liu et al).68,69 One of the most likeliest modes of action would be restoration of the oxygen supply to previously deprived areas, although of course if the region has been deprived for too long, then the oxygen would not be able to get to the damaged site, unless angiogenesis was also triggered to ensure that blood flow (and hence the oxygen) reached the damaged area or its close surroundings. In the chronic situation, it is possible that no cells remain in the damaged area and so increased blood flow/oxygen is unlikely to be beneficial, although the suggestion of “idling neurons” within the ischemic penumbra may mean that this is not necessarily the case.70 Oxygen can diffuse ~100–200 μm within tissues; hence, a localized supply is necessary.69 Since oxygen is an important component of the final step of the electron transport chain, restoration of oxygen to tissues will facilitate energy (adenosine triphosphate) production within the mitochondria of cells – if they are still viable (reviewed by Sjoberg and Singer).32 Following TBI, the cells within the injury are metabolically dysfunctional, which is often shown by mitochondrial impairment that can lead to apoptosis.71 Several studies have already shown that HBOT modifies apoptosis, and it may therefore also affect the mitochondria. In an acute rat model of TBI, the substantial loss of mitochondrial transmembrane potential was reversed by HBOT. Increased survival of perilesional neurons was also observed, along with significantly reduced activities of both caspase 3 and 9.72 Mitochondrial changes have also been observed in PTSD with the evidence of gene dysregulation in human patients and mitochondrial-induced apoptosis in the hippocampus and dorsal raphe.73–79 Mitochondrial dysfunction in wobbler mice was also improved by HBOT.80 In contrast to the aforementioned studies, Weber et al demonstrated in vitro using Jurkat T cells that HBOT induced apoptosis in a mitochondrially dependent fashion.81 This disparity may relate to it being an in vitro study and/or because only one cell type (Jurkat T cells) was investigated. These studies suggest that HBOT can influence the mitochondria and restore the energy imbalance that occurs following injury. An early study by Holbach et al demonstrated restoration of the cerebral energy balance with glycolysis occurring at almost normal levels.82 A blast-induced TBI model in rabbits demonstrated increased local neuron metabolism, reduced edema, reduced inflammation, and maintenance of the BBB following HBOT.83 Several studies suggest that HBOT can stimulate the homing and differentiation of circulating stem cells culminating in neovascularization with regard to wound healing (reviewed by Sjoberg and Singer and Ding et al),32,68 and a similar process could occur within the brain. Inflammation can be both beneficial and detrimental depending on the circumstances and time frame, and HBOT has been shown to reduce the infiltration of neutrophils to the damaged site. An inhibitory action on cyclooxygenase 2 signaling which normally promotes inflammation via cytokines has also been observed along with the suppression of matrix metalloproteinase 9 induction, an enzyme critical to both normal and detrimental tissue remodeling (reviewed by Ding et al).68 Inhibitory effects on the activation of microglia and proliferation of astrocytes by TBI have also been observed following HBOT (reviewed by Ding et al).68 Two hours after TBI, HBOT in Sprague Dawley rats was demonstrated to improve neurological function, reduce apoptosis and inflammatory cytokines, as well as inhibit the Toll-like receptor 4/nuclear factor kappa-light-chain-enhancer of activated B cells pathway that is activated by TBI.84 During TBI, the integrity of the blood–brain barrier (BBB) is frequently impaired (reviewed by Shetty et al).85 Although some studies suggest that the BBB is impaired by HBOT,86 a number of other studies suggest that BBB integrity following injury is maintained by HBOT.83,87 An increased density of blood vessels, which correlated with improved spatial learning, was observed in rats modeling a chronic TBI (as a result of a focal cortical weight drop impact) following treatment with HBOT.88 This suggests that HBOT may promote angiogenesis, and this is further supported by a retrospective clinical study that demonstrated significant increases in cerebral blood flow, cerebral blood volume, and the global cognitive scores following HBOT of 10 chronic TBI patients.89 In addition, levels of excitotoxic metabolites such as glutamate, pyruvate, and lactate seem to be reduced following HBOT.32,90,91 Yang et al showed in rats that HBOT after TBI led to an inhibition of hippocampal cell apoptosis and a reduction in BBB dysfunction and hypoxia-inducible factor 1 expression, although no behavioral analysis was performed to show that these changes led to functional improvements.92 Efrati et al demonstrated that HBOT of chronic stroke patients led to the restoration of brain activity, as detected by SPECT, in brain areas that had previously exhibited very low activity, and this coincided with the functional improvements, suggesting the induction of neuroplasticity.93
Although there are some evidences for decreased nitric oxide production (as indicated by global arginine bioavailability) in male PTSD patients,94 the ability of HBOT to counteract this is unclear. Some studies suggest that HBOT may enhance endothelial nitric oxide synthase expression, but reduce inducible nitric oxide synthase activity and likely inflammation, though not all HBOT studies agree on this.95–100 Conversely, an increase in neuronal nitric oxide activity has been seen in TBI,101 and so inhibition of nitric oxide synthase by HBOT could be useful in treating TBI.102 Nitric oxide may also influence the BBB since inducible nitric oxide synthase-generated nitric oxide from perivascular macrophages seems to maintain the integrity of the BBB with regard to inflammatory cells through a negative feedback loop, while a second study suggests that endothelial nitric oxide synthase may help restore integrity during periods of permeability, although it has no effect under normal conditions.103,104 Several other studies suggest that the increased nitric oxide expression coupled with peroxynitrite production (a metabolite of nitric oxide formed after reacting with superoxide molecules, the production of which could be a potential side effect of excessive HBOT) facilitates BBB dysfunction in stroke and TBI.105–107 Excessive peroxynitrite production reduces the amount of freely available nitric oxide that can react with glutathione to generate S-nitrosoglutathione an anti-inflammatory antioxidant.101 Since HBOT may increase the presence of antioxidants such as glutathione (reviewed by Ding et al)68 and inhibit neuronal nitric oxide, it may thus promote the generation of S-nitrosoglutathione in preference to the toxic peroxynitrite molecule. The precise effects of HBOT on nitric oxide production may therefore be of interest so as to determine whether this could be a potentially beneficial mode of action for HBOT in the treatment of PTSD and TBI.
Many of the mechanisms of action mentioned earlier essentially culminate in a more favorable environment for cell survival. This includes the reduction of inflammation and reactive glia as well as promotion of angiogenesis and restoration of energy reserves.108,109 Under these conditions, replacement of dying or dead neural cells is also a possible mechanism of action, and therefore, the effect of HBOT on neurogenesis in treating TBI/PTSD could also be investigated since, as previously mentioned, HBOT potentiates astrocyte proliferation. Although neurogenesis does not necessarily culminate in mature cell integration, the immature cells generated during the process can secrete factors that will further promote survival and contribute to a favorable environment for repair. Lin et al reported observing decreased inflammation and increased angiogenesis and neurogenesis in rats with TBI (by fluid percussion injury) following 3 days of 2×1 h 2.0 ATA HBOT exposure, as determined by decreased brain myeloperoxidase activity, bromodeoxyuridine endothelial cells, and vascular endothelial growth factor-labeled cells, and bromodeoxyuridine and neuronal nuclei (NeuN) co-labeled cells, respectively.109 Alternatively, evidence of neurogenesis could be demonstrated by considering doublecortin expression as a marker of the generation of new immature neurons following HBOT. In a long-term study (3 weeks of HBOT), neurological improvement, increased neurogenesis, and bone marrow stem cell homing to the infarcted area were observed in rats modeling an ischemic stroke.110 Several studies have shown that neuroinflammation is an important component of TBI and PTSD.21,22,25,111–119 HBOT seems to reduce different aspects of neuroinflammation in various disease models including TBI and ischemia.83,108–110,120–126 Vlodavsky et al124 observed a decreased apoptosis, neutrophilic infiltration (myeloperoxidase staining and hence neuroinflammation), and matrix metalloproteinase 9 expression following 6 sessions of HBOT in an acute rat model of TBI (3 h after injury). This was compared to both a no treatment and a normobaric oxygen treatment, suggesting that oxygen at increased pressure was necessary for any benefit.124 Reduced neuroinflammation (myeloperoxidase) was confirmed in another acute rat TBI model by Lin et al, as well as reduced cell loss and gliosis and stimulation of angiogenesis and neurogenesis and overproduction of the anti-inflammatory cytokine interleukin 10 (IL-10).109 The important role of IL-10 was confirmed in IL-10 knockout mice which did not show any neuroprotection by HBOT in an acute TBI model, but neuroprotection was seen in normal mice.127 Another group showed that multiple doses of HBOT were better than a single dose in ischemic rats and also showed increased IL-10, along with decreased inflammation and oxidative damage.120 In an in vitro culture of neutrophils and macrophages, exposure to HBOT increased the phagocytosis of neutrophils, particularly of cells that were apoptotic, suggesting that HBOT may promote the clearance of neutrophils and dying cells and thus modify the inflammatory response.128 Acute HBOT treatment of TBI in rats was also shown to reduce microglial activation and reactive gliosis.108,123 In addition, the expression of the group of proteins that comprise the inflammasome was also shown to be increased following TBI and reduced after subsequent HBOT.121 These studies help provide support for an important influence of HBOT over the inflammatory response in acute TBI.
A related possible mechanism of action is the involvement of endoplasmic reticulum (ER) stress. ER stress has been implicated in TBI with an increase in CCAAT-enhancer-binding protein homologous protein expression (CHOP) detected.129,130 ER stress has also been shown to be the cause of apoptotic cell death in the medial prefrontal cortex and hippocampus and is active in the dorsal raphe, of PTSD modeling rats.131–134 Mitochondrial-dependent apoptosis has also been seen in these brain regions in PTSD and TBI.71–80 Although ER stress does not seem to have been studied in the treatment of TBI or PTSD by HBOT, a rat spinal cord injury model does show overexpression of CHOP, which is reversed by HBOT.135 This may therefore be an additional mode of action for HBOT in treating TBI and PTSD, which is yet to be investigated.
Side effects of HBOT
HBOT has some potential side effects. At high pressures (>3 ATA), prolonged oxygen exposure can cause convulsions, though whether this is detrimental is under debate since the seizures abate on removal of the high pressure oxygen. Animal studies suggest that the seizures may result from increased nitric oxide synthesis.98 Fortunately, these observations are at higher pressures (3 ATA) than those being used in the study; hence, with limited exposure times (60–90 min at a time), this should reduce the likelihood of convulsions as a side effect. In animals exposed to 6 ATA, an increase in apoptotic markers within the hippocampus was observed 1 h and 7 days after seizures, while a decrease was observed in the cortex at 1 h.136 Further study showed that MRI of the hippocampus and cortex showed a small significant decrease in T2 values after HBOT which had recovered by 7 days, although contrast injection was enhanced at 7 days, suggesting that there may be delayed BBB impairment.137 Transient cognitive deficits were also observed, suggesting that potentially reversible damage does occur after HBOT-induced seizures.138
Another potential side effect of HBOT is middle-ear barotrauma (ranging from “fullness” to rupture of the eardrum [tympanic membrane]). A wide incidence range has been reported, and therefore, pointers to determine who will likely suffer from middle-ear barotrauma have been investigated. Lehm and Bennett observed that if the tympanic membrane did not move during the Valsalva manoeuver (determined by otoscopy), this increased the likelihood of a patient exhibiting middle-ear barotrauma.139
Other potential side effects include claustrophobia because of being in a confined space and rare severe side effects include progressive myopia or pulmonary dyspnea, which tend to recede after ceasing treatment.140 Wolf et al reported on the side effects in a randomized, controlled clinical trial and observed negligible incidences of side effects including headache, nausea, or numbness.141 This suggests that under short repetitive periods of HBOT, side effects are normally minor, and therefore the therapy is safe. The clinical trials reported in Table 1 seem to support this conclusion.
Current and future directions
Although females are in general more likely to suffer from PTSD than males,142 a study of post-deployment veterans suggested women were less likely to suffer from PTSD, but more likely to suffer from depression or a different anxiety disorder.143 It is worth noting that the size of the male population was 18 times greater than the female population examined in this study (11,951 vs 654), which may be contributing to the findings. Gender differences in depression and facial allodynia in a mTBI animal model have also been reported recently,144 but we feel that initially studying male mice will help to eliminate uncontrolled variables such as hormonal changes, which could affect their recovery and thus increase variability in the results. The need to perform this in females at a later stage is however clear. A CCI model will be used to induce TBI. The left frontoparietal cortex will be impacted for 150 ms by a 3-mm pneumatically operated metal impactor at 6 m/s to a depth of 1–2 mm (moderate to severe TBI), while sham-treated rats will only receive a craniectomy as previously described.25,145–151 TBI will be confirmed behaviorally using the elevated body swing test, rotorod, and Bederson’s neurological scoring scale, as well as histologically using hematoxylin and eosin staining of the cortex after sacrifice. The expected induction of PTSD in at least 20%–30% of the rats (based on similar occurrences in humans undergoing TBI) will be tested using the elevated plus maze and the acoustic startle response which encompass the proposed cutoff behavioral criteria (CBC) model for PTSD described by Cohen et al27 and use of the social interaction test. The CBC model characterizes the animals’ response as either extreme behavioral response (clearly PTSD), minimal behavioral response (comparatively normal), or partial behavioral response (an intermediate degree of PTSD symptoms) depending on the degree of behavioral disruption.27,152
All the clinical studies mentioned previously involved multiple exposures to HBOT, and this was also used in several animal studies,92,109,153 hence, a single versus multiple exposure (≥3) to HBOT (1.5 ATA for 90 min) will initially be performed immediately after TBI. The optimal exposure determined from the single versus multiple dosing will then be repeated in separate animals 0, 14, or 28 days after TBI, because the timing of the treatment is also likely to be very important.92,153 This is of course also significant with regard to treating acute or chronic TBI. Yang et al compared three different time points (early, delayed, and both early and delayed treatment) and found that the combined treatment was the most effective, followed by the early treatment.92 Therefore, both acute and chronic treatment should also be investigated. Behavioral testing will be performed at baseline (ie, prior to TBI) and 1, 7, and 14 days after TBI/HBOT in the first group with sacrifice after 14 days for histological evaluation. The second group will undergo weekly behavioral testing up to 56 days after TBI before sacrifice.
At the levels of HBOT that are being used, neither pulmonary oxygen toxicity nor central nervous system oxygen toxicity are expected, because any oxidative stress induced by the higher oxygen concentration will be offset by the concomitant enhancement of antioxidants over the short time period of exposure (reviewed by Ding et al).68 Long-term exposure to HBOT is however more likely to result in uncontrolled oxidative stress due to the continued presence of oxygen-free radicals and hence pulmonary and/or central nervous system toxicity.
Although we initially propose to study male animals acutely after TBI, both chronic TBI and females will be considered for later studies since some evidence of benefit against chronic TBI has been observed in some clinical trials41,154 and there is a suggestion of an increased likelihood of PTSD in females.144 These future studies will allow us to determine the optimal time frame for the treatment and whether there are gender differences. Interestingly, Li et al reported that HBOT preconditioning prior to coronary artery bypass graft surgery afforded neuroprotection,155 while Ohguri et al saw decreased white matter injury in patients who were preconditioned with HBOT before stereotactic radiosurgery for brain metastases.156 HBOT preconditioning also seems to be beneficial in reducing the infarct size in animal models of stroke,157 reducing neural apoptosis after spinal cord injury158 and ischemia.159 A recent study demonstrated that HBOT preconditioning may modify neuroinflammation by altering the microglial activity in an animal model of intracerebral hemorrhage.126 One study considered HBOT preconditioning in TBI-treated rats at high altitude, and improvements in neurological function and decreased matrix metalloproteinase 9 expression were observed.160 Reduced anxiety and cognitive impairment were also observed in rats modeling PTSD that had been preconditioned by HBOT.161 These studies therefore suggest that HBOT preconditioning may have therapeutic potential against TBI/PTSD; hence, this should also be explored. Determining how soon prior to injury preconditioning is necessary would be important to find the possible clinical application of this therapy. Treating soldiers before they enter the combat zone may be able to offer them some protection against blast-induced TBI and/or PTSD. Ideally, HBOT would be performed away from the site of action to ameliorate a potentially explosive situation. For the same reason, a chronic TBI therapy would be preferred to an acute TBI therapy, although logistically it could be possible to treat soldiers either prophylactically or immediately following injury close to the combat zone.
Conclusion
There is considerable evidence for some beneficial effects of HBOT in treating TBI or PTSD in animal models, although this does not seem to have translated well to human clinical trials. This may relate to problems with appropriate controls and other factors rather than a lack of efficacy. It is therefore timely to perform HBOT in an animal study of TBI and PTSD to determine an optimal treatment regime and whether a possible mode of action is that it influences neurogenesis and inflammation. Prophylactic use may also be of benefit to help minimize TBI damage in our troops from IEDs.
Acknowledgment
This work was supported by USF #140 Health Veteran PTSD and Traumatic Brain Injury Study.
Disclosure
The authors report no conflicts of interest in this work.
References
Elder GA, Stone JR, Ahlers ST. Effects of low-level blast exposure on the nervous system: is there really a controversy? Front Neurol. 2014;5:269. | ||
Tanielian T, Jaycox LH, editors. Invisible Wounds of War: Psychological and Cognitive Injuries, Their Consequences, and Services to Assist Recovery. Santa Monica, CA: RAND Corporation; 2008. | ||
Tanev KS, Pentel KZ, Kredlow MA, Charney ME. PTSD and TBI co-morbidity: scope, clinical presentation and treatment options. Brain Inj. 2014;28(3):261–270. | ||
Ruff R. Two decades of advances in understanding of mild traumatic brain injury. J Head Trauma Rehabil. 2005;20(1):5–18. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association Publishing; 2013. | ||
Breslau N, Peterson EL, Poisson LM, Schultz LR, Lucia VC. Estimating post-traumatic stress disorder in the community: lifetime perspective and the impact of typical traumatic events. Psychol Med. 2004;34(5):889–898. | ||
Breslau N, Troost JP, Bohnert K, Luo Z. Influence of predispositions on post-traumatic stress disorder: does it vary by trauma severity? Psychol Med. 2013;43(2):381–390. | ||
Javidi H, Yadollahie M. Post-traumatic stress disorder. Int J Occup Environ Med. 2012;3(1):2–9. | ||
Whitaker AM, Gilpin NW, Edwards S. Animal models of post-traumatic stress disorder and recent neurobiological insights. Behav Pharmacol. 2014;25(5–6):398–409. | ||
Andero R, Ressler KJ. Fear extinction and BDNF: translating animal models of PTSD to the clinic. Genes Brain Behav. 2014;11(5):503–512. | ||
Liberzon I, Sripada CS. The functional neuroanatomy of PTSD: a critical review. Prog Brain Res. 2008;167:151–169. | ||
Pitman RK, Rasmusson AM, Koenen KC, et al. Biological studies of post-traumatic stress disorder. Nat Rev Neurosci. 2012;13(11):769–787. | ||
Hoffman SW, Harrison C. The interaction between psychological health and traumatic brain injury: a neuroscience perspective. Clin Neuropsychol. 2009;23(8):1400–1415. | ||
McAllister TW. Psychopharmacological issues in the treatment of TBI and PTSD. Clin Neuropsychol. 2009;23(8):1338–1367. | ||
Joseph S, Masterson J. Posttraumatic stress disorder and traumatic brain injury: are they mutually exclusive? J Trauma Stress. 1999;12(3):437–453. | ||
Gil S, Caspi Y, Ben-Ari IZ, Koren D, Klein E. Does memory of a traumatic event increase the risk for posttraumatic stress disorder in patients with traumatic brain injury? A prospective study. Am J Psychiatry. 2005;162(5):963–969. | ||
Harvey AG, Brewin CR, Jones C, Kopelman MD. Coexistence of posttraumatic stress disorder and traumatic brain injury: towards a resolution of the paradox. J Int Neuropsychol Soc. 2003;9(4):663–676. | ||
McMillan TM, Williams WH, Bryant R. Post-traumatic stress disorder and traumatic brain injury: a review of causal mechanisms, assessment, and treatment. Neuropsychol Rehabil. 2003;13(1–2):149–164. | ||
Elder GA, Dorr NP, De Gasperi R, et al. Blast exposure induces post-traumatic stress disorder-related traits in a rat model of mild traumatic brain injury. J Neurotrauma. 2012;29(16):2564–2575. | ||
Reger ML, Poulos AM, Buen F, Giza CC, Hovda DA, Fanselow MS. Concussive brain injury enhances fear learning and excitatory processes in the amygdala. Biol Psychiatry. 2012;71(4):335–343. | ||
Hinson HE, Rowell S, Schreiber M. Clinical evidence of inflammation driving secondary brain injury: a systematic review. J Trauma Acute Care Surg. 2015;78(1):184–191. | ||
Passos IC, Vasconcelos-Moreno MP, Costa LG, et al. Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry. 2015;2(11):1002–1012. | ||
Tursich M, Neufeld RW, Frewen PA, et al. Association of trauma exposure with proinflammatory activity: a transdiagnostic meta-analysis. Transl Psychiatry. 2014;4:e413. | ||
Eraly SA, Nievergelt CM, Maihofer AX, et al. Assessment of plasma C-reactive protein as a biomarker of posttraumatic stress disorder risk. JAMA Psychiatry. 2014;71(4):423–431. | ||
Acosta SA, Diamond DM, Wolfe S, et al. Influence of post-traumatic stress disorder on neuroinflammation and cell proliferation in a rat model of traumatic brain injury. PLoS One. 2013;8(12):e81585. | ||
Berardi A, Trezza V, Palmery M, Trabace L, Cuomo V, Campolongo P. An updated animal model capturing both the cognitive and emotional features of post-traumatic stress disorder (PTSD). Front Behav Neurosci. 2014;8:142. | ||
Cohen H, Matar MA, Zohar J. Maintaining the clinical relevance of animal models in translational studies of post-traumatic stress disorder. ILAR J. 2014;55(2):233–245. | ||
Daskalakis NP, Yehuda R. Principles for developing animal models of military PTSD. Eur J Psychotraumatol. 2014;5. | ||
Daskalakis NP, Yehuda R, Diamond DM. Animal models in translational studies of PTSD. Psychoneuroendocrinology. 2013;38(9):1895–1911. | ||
Goswami S, Rodriguez-Sierra O, Cascardi M, Pare D. Animal models of post-traumatic stress disorder: face validity. Front Neurosci. 2013;7:89. | ||
Jain KK. Textbook of Hyperbaric Medicine. 5th ed. Cambridge, MA: Hogrefe Publishing; 2009. | ||
Sjoberg F, Singer M. The medical use of oxygen: a time for critical reappraisal. J Intern Med. 2013;274(6):505–528. | ||
Collet JP, Vanasse M, Marois P, et al. Hyperbaric oxygen for children with cerebral palsy: a randomised multicentre trial. HBO-CP Research Group. Lancet. 2001;357(9256):582–586. | ||
Bennett MH, Trytko B, Jonker B. Hyperbaric oxygen therapy for the adjunctive treatment of traumatic brain injury. Cochrane Database Syst Rev. 2012;12:CD004609. | ||
Bennett MH, Weibel S, Wasiak J, Schnabel A, French C, Kranke P. Hyperbaric oxygen therapy for acute ischaemic stroke. Cochrane Database Syst Rev. 2014;11:CD004954. | ||
Bennett M, Heard R. Hyperbaric oxygen therapy for multiple sclerosis. CNS Neurosci Ther. 2010;16(2):115–124. | ||
McDonagh MS, Morgan D, Carson S, Russman BS. Systematic review of hyperbaric oxygen therapy for cerebral palsy: the state of the evidence. Dev Med Child Neurol. 2007;49(12):942–947. | ||
Rockswold SB, Rockswold GL, Zaun DA, Liu J. A prospective, randomized phase II clinical trial to evaluate the effect of combined hyperbaric and normobaric hyperoxia on cerebral metabolism, intracranial pressure, oxygen toxicity, and clinical outcome in severe traumatic brain injury. J Neurosurg. 2013;118(6):1317–1328. | ||
Eovaldi B, Zanetti C. Hyperbaric oxygen ameliorates worsening signs and symptoms of post-traumatic stress disorder. Neuropsychiatr Dis Treat. 2010;6:785–789. | ||
Harch PG, Andrews SR, Fogarty EF, et al. A phase I study of low-pressure hyperbaric oxygen therapy for blast-induced post-concussion syndrome and post-traumatic stress disorder. J Neurotrauma. 2012;29(1):168–185. | ||
Churchill S, Weaver LK, Deru K, et al. A prospective trial of hyperbaric oxygen for chronic sequelae after brain injury (HYBOBI). Undersea Hyperb Med. 2013;40(2):165–193. | ||
Boussi-Gross R, Golan H, Fishlev G, et al. Hyperbaric oxygen therapy can improve post concussion syndrome years after mild traumatic brain injury – randomized prospective trial. PLoS One. 2013;8(11):e79995. | ||
Wolf G, Cifu D, Baugh L, Carne W, Profenna L. The effect of hyperbaric oxygen on symptoms after mild traumatic brain injury. J Neurotrauma. 2012;29(17):2606–2612. | ||
Harch PG. Hyperbaric oxygen therapy for post-concussion syndrome: contradictory conclusions from a study mischaracterized as sham-controlled. J Neurotrauma. 2013;30(23):1995–1999. | ||
Mitchell SJ, Bennett MH. Unestablished indications for hyperbaric oxygen therapy. Diving Hyperb Med. 2014;44(4):228–234. | ||
Malek M, Duszczyk M, Zyszkowski M, Ziembowicz A, Salinska E. Hyperbaric oxygen and hyperbaric air treatment result in comparable neuronal death reduction and improved behavioral outcome after transient forebrain ischemia in the gerbil. Exp Brain Res. 2013;224(1):1–14. | ||
Tokunaga O, Watanabe T. Properties of endothelial cell and smooth muscle cell cultured in ambient pressure. In Vitro Cell Dev Biol. 1987;23(8):528–534. | ||
Al Hadi H, Smerdon GR, Fox SW. Hyperbaric oxygen therapy accelerates osteoblast differentiation and promotes bone formation. J Dent. 2015;43(3):382–388. | ||
Al Hadi H, Smerdon GR, Fox SW. Hyperbaric oxygen therapy suppresses osteoclast formation and bone resorption. J Orthop Res. 2013;31(11):1839–1844. | ||
Oh S, Lee E, Lee J, Lim Y, Kim J, Woo S. Comparison of the effects of 40% oxygen and two atmospheric absolute air pressure conditions on stress-induced premature senescence of normal human diploid fibroblasts. Cell Stress Chaperones. 2008;13(4):447–458. | ||
Rossignol DA, Rossignol LW, Smith S, et al. Hyperbaric treatment for children with autism: a multicenter, randomized, double-blind, controlled trial. BMC Pediatr. 2009;9:21. | ||
Rossignol DA, Rossignol LW, James SJ, Melnyk S, Mumper E. The effects of hyperbaric oxygen therapy on oxidative stress, inflammation, and symptoms in children with autism: an open-label pilot study. BMC Pediatr. 2007;7:36. | ||
Cifu DX, Walker WC, West SL, et al. Hyperbaric oxygen for blast-related postconcussion syndrome: three-month outcomes. Ann Neurol. 2014;75(2):277–286. | ||
Cifu DX, Hart BB, West SL, Walker W, Carne W. The effect of hyperbaric oxygen on persistent postconcussion symptoms. J Head Trauma Rehabil. 2014;29(1):11–20. | ||
Wolf EG, Baugh LM, Kabban CM, Richards MF, Prye J. Cognitive function in a traumatic brain injury hyperbaric oxygen randomized trial. Undersea Hyperb Med. 2015;42(4):313–332. | ||
Lacey DJ, Stolfi A, Pilati LE. Effects of hyperbaric oxygen on motor function in children with cerebral palsy. Ann Neurol. 2012;72(5):695–703. | ||
Miller RS, Weaver LK, Bahraini N, et al. Effects of hyperbaric oxygen on symptoms and quality of life among service members with persistent postconcussion symptoms: a randomized clinical trial. JAMA Intern Med. 2015;175(1):43–52. | ||
Bloch Y, Applebaum J, Osher Y, et al. Normobaric hyperoxia treatment of schizophrenia. J Clin Psychopharmacol. 2012;32(4):525–530. | ||
Halepoto DM, Al-Ayadhi LY, Salam AA. Therapeutic use of hyperbaric oxygen therapy for children with autism spectrum disorder. J Coll Physicians Surg Pak. 2014;24(7):508–514. | ||
Martin R, Srivastava T, Lee J, Raj N, Koth KA, Whelan HT. Using hyperbaric oxygen for autism treatment: a review and discussion of literature. Undersea Hyperb Med. 2015;42(4):353–359. | ||
Bent S, Bertoglio K, Ashwood P, Nemeth E, Hendren RL. Brief report: hyperbaric oxygen therapy (HBOT) in children with autism spectrum disorder: a clinical trial. J Autism Dev Disord. 2012;42(6):1127–1132. | ||
Chungpaibulpatana J, Sumpatanarax T, Thadakul N, Chantharatreerat C, Konkaew M, Aroonlimsawas M. Hyperbaric oxygen therapy in Thai autistic children. J Med Assoc Thai. 2008;91(8):1232–1238. | ||
Jepson B, Granpeesheh D, Tarbox J, et al. Controlled evaluation of the effects of hyperbaric oxygen therapy on the behavior of 16 children with autism spectrum disorders. J Autism Dev Disord. 2011;41(5):575–588. | ||
Sahni T, Jain M, Prasad R, Sogani SK, Singh VP. Use of hyperbaric oxygen in traumatic brain injury: retrospective analysis of data of 20 patients treated at a tertiary care centre. Br J Neurosurg. 2012;26(2):202–207. | ||
Stoller KP. Hyperbaric oxygen therapy (1.5 ATA) in treating sports related TBI/CTE: two case reports. Med Gas Res. 2011;1(1):17. | ||
Liu S, Liu Y, Deng S, Guo A, Wang X, Shen G. Beneficial effects of hyperbaric oxygen on edema in rat hippocampus following traumatic brain injury. Exp Brain Res. 2015;233(12):3359–3365. | ||
Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14(2):128–142. | ||
Ding Z, Tong WC, Lu XX, Peng HP. Hyperbaric oxygen therapy in acute ischemic stroke: a review. Interv Neurol. 2014;2(4):201–211. | ||
Liu W, Khatibi N, Sridharan A, Zhang JH. Application of medical gases in the field of neurobiology. Med Gas Res. 2011;1(1):13. | ||
Neubauer RA. Idling neurons. Lancet. 1990;335(8699):1217. | ||
Hiebert JB, Shen Q, Thimmesch AR, Pierce JD. Traumatic brain injury and mitochondrial dysfunction. Am J Med Sci. 2015;350(2):132–138. | ||
Palzur E, Zaaroor M, Vlodavsky E, Milman F, Soustiel JF. Neuroprotective effect of hyperbaric oxygen therapy in brain injury is mediated by preservation of mitochondrial membrane properties. Brain Res. 2008;1221:126–133. | ||
Flaquer A, Baumbach C, Ladwig KH, et al. Mitochondrial genetic variants identified to be associated with posttraumatic stress disorder. Transl Psychiatry. 2015;5:e524. | ||
Li XM, Han F, Liu DJ, Shi YX. Single-prolonged stress induced mitochondrial-dependent apoptosis in hippocampus in the rat model of post-traumatic stress disorder. J Chem Neuroanat. 2010;40(3):248–255. | ||
Liu D, Xiao B, Han F, Wang E, Shi Y. Single-prolonged stress induces apoptosis in dorsal raphe nucleus in the rat model of posttraumatic stress disorder. BMC Psychiatry. 2012;12:211. | ||
Su YA, Wu J, Zhang L, et al. Dysregulated mitochondrial genes and networks with drug targets in postmortem brain of patients with posttraumatic stress disorder (PTSD) revealed by human mitochondria-focused cDNA microarrays. Int J Biol Sci. 2008;4(4):223–235. | ||
Wan J, Liu D, Zhang J, Shi Y, Han F. Single-prolonged stress induce different change in the cell organelle of the hippocampal cells: a study of ultrastructure. Acta Histochem. 2016;118(1):10–19. | ||
Xing G, Barry ES, Benford B, et al. Impact of repeated stress on traumatic brain injury-induced mitochondrial electron transport chain expression and behavioral responses in rats. Front Neurol. 2013;4:196. | ||
Zhang L, Li H, Hu X, et al. Mitochondria-focused gene expression profile reveals common pathways and CPT1B dysregulation in both rodent stress model and human subjects with PTSD. Transl Psychiatry. 2015;5:e580. | ||
Dave KR, Prado R, Busto R, et al. Hyperbaric oxygen therapy protects against mitochondrial dysfunction and delays onset of motor neuron disease in Wobbler mice. Neuroscience. 2003;120(1):113–120. | ||
Weber SU, Koch A, Kankeleit J, et al. Hyperbaric oxygen induces apoptosis via a mitochondrial mechanism. Apoptosis. 2009;14(1):97–107. | ||
Holbach KH, Caroli A, Wassmann H. Cerebral energy metabolism in patients with brain lesions at normo- and hyperbaric oxygen pressures. J Neurol. 1977;217:17–30. | ||
Zhang Y, Yang Y, Tang H, et al. Hyperbaric oxygen therapy ameliorates local brain metabolism, brain edema and inflammatory response in a blast-induced traumatic brain injury model in rabbits. Neurochem Res. 2014;39(5):950–960. | ||
Meng XE, Zhang Y, Li N, et al. Hyperbaric oxygen alleviates secondary brain injury after trauma through inhibition of TLR4/NF-kappaB signaling pathway. Med Sci Monit. 2016;22:284–288. | ||
Shetty AK, Mishra V, Kodali M, Hattiangady B. Blood brain barrier dysfunction and delayed neurological deficits in mild traumatic brain injury induced by blast shock waves. Front Cell Neurosci. 2014;8:232. | ||
Cevik NG, Orhan N, Yilmaz CU, et al. The effects of hyperbaric air and hyperbaric oxygen on blood-brain barrier integrity in rats. Brain Res. 2013;1531:113–121. | ||
Zhou W, Marinescu M, Veltkamp R. Only very early oxygen therapy attenuates posthemorrhagic edema formation and blood-brain barrier disruption in murine intracerebral hemorrhage. Neurocrit Care. 2015;22(1):121–132. | ||
Harch PG, Kriedt C, Van Meter KW, Sutherland RJ. Hyperbaric oxygen therapy improves spatial learning and memory in a rat model of chronic traumatic brain injury. Brain Res. 2007;1174:120–129. | ||
Tal S, Hadanny A, Berkovitz N, Sasson E, Ben-Jacob E, Efrati S. Hyperbaric oxygen may induce angiogenesis in patients suffering from prolonged post-concussion syndrome due to traumatic brain injury. Restor Neurol Neurosci. 2015;33(6):943–951. | ||
Singhal AB. Oxygen therapy in stroke: past, present, and future. Int J Stroke. 2006;1(4):191–200. | ||
Veltkamp R, Siebing DA, Sun L, et al. Hyperbaric oxygen reduces blood-brain barrier damage and edema after transient focal cerebral ischemia. Stroke. 2005;36(8):1679–1683. | ||
Yang Y, Zhang YG, Lin GA, et al. The effects of different hyperbaric oxygen manipulations in rats after traumatic brain injury. Neurosci Lett. 2014;563:38–43. | ||
Efrati S, Fishlev G, Bechor Y, et al. Hyperbaric oxygen induces late neuroplasticity in post stroke patients – randomized, prospective trial. PLoS One. 2013;8(1):e53716. | ||
Bersani FS, Wolkowitz OM, Lindqvist D, et al. Global arginine bioavailability, a marker of nitric oxide synthetic capacity, is decreased in PTSD and correlated with symptom severity and markers of inflammation. Brain Behav Immun. 2016;52:153–160. | ||
Allen BW, Demchenko IT, Piantadosi CA. Two faces of nitric oxide: implications for cellular mechanisms of oxygen toxicity. J Appl Physiol (1985). 2009;106(2):662–667. | ||
Baynosa RC, Naig AL, Murphy PS, et al. The effect of hyperbaric oxygen on nitric oxide synthase activity and expression in ischemia-reperfusion injury. J Surg Res. 2013;183(1):355–361. | ||
Bernareggi M, Radice S, Rossoni G, Oriani G, Chiesara E, Berti F. Hyperbaric oxygen increases plasma exudation in rat trachea: involvement of nitric oxide. Br J Pharmacol. 1999;126(3):794–800. | ||
Elayan IM, Axley MJ, Prasad PV, Ahlers ST, Auker CR. Effect of hyperbaric oxygen treatment on nitric oxide and oxygen free radicals in rat brain. J Neurophysiol. 2000;83(4):2022–2029. | ||
Ercin CN, Yesilova Z, Korkmaz A, Ozcan A, Oktenli C, Uygun A. The effect of iNOS inhibitors and hyperbaric oxygen treatment in a rat model of experimental colitis. Dig Dis Sci. 2009;54(1):75–79. | ||
Uusijarvi J, Eriksson K, Larsson AC, et al. Effects of hyperbaric oxygen on nitric oxide generation in humans. Nitric Oxide. 2015;44:88–97. | ||
Khan M, Dhammu TS, Matsuda F, et al. Targeting the nNOS/peroxynitrite/calpain system to confer neuroprotection and aid functional recovery in a mouse model of TBI. Brain Res. 2016;1630:159–170. | ||
Zhou JG, Fang YQ, Liu CY, Zhou YQ, Ji YF, Liu JC. [Effect of hyperbaric oxygen on the expression of nitric oxide synthase mRNA in cortex after acute traumatic cerebral injury]. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2012;28(1):38–41. Chinese. | ||
Olivera GC, Ren X, Vodnala SK, et al. Nitric oxide protects against infection-induced neuroinflammation by preserving the stability of the blood-brain barrier. PLoS Pathog. 2016;12(2):e1005442. | ||
Hasarmeh M, Itzik A, Weidenfeld J, Ovadia H. Modulation of hyperosmotic and immune-induced disruption of the blood-brain barrier by the nitric oxide system. Neuroimmunomodulation. 2016;23(1):1–7. | ||
Khan M, Dhammu TS, Sakakima H, et al. The inhibitory effect of S-nitrosoglutathione on blood-brain barrier disruption and peroxynitrite formation in a rat model of experimental stroke. J Neurochem. 2012;123(Suppl 2):86–97. | ||
Han F, Shirasaki Y, Fukunaga K. Microsphere embolism-induced endothelial nitric oxide synthase expression mediates disruption of the blood-brain barrier in rat brain. J Neurochem. 2006;99(1):97–106. | ||
Khan M, Sakakima H, Dhammu TS, et al. S-Nitrosoglutathione reduces oxidative injury and promotes mechanisms of neurorepair following traumatic brain injury in rats. J Neuroinflammation. 2011;8:78. | ||
Lavrnja I, Parabucki A, Brkic P, et al. Repetitive hyperbaric oxygenation attenuates reactive astrogliosis and suppresses expression of inflammatory mediators in the rat model of brain injury. Mediators Inflamm. 2015;2015:498405. | ||
Lin KC, Niu KC, Tsai KJ, et al. Attenuating inflammation but stimulating both angiogenesis and neurogenesis using hyperbaric oxygen in rats with traumatic brain injury. J Trauma Acute Care Surg. 2012;72(3):650–659. | ||
Lee YS, Chio CC, Chang CP, et al. Long course hyperbaric oxygen stimulates neurogenesis and attenuates inflammation after ischemic stroke. Mediators Inflamm. 2013;2013:512978. | ||
Andrews JA, Neises KD. Cells, biomarkers, and post-traumatic stress disorder: evidence for peripheral involvement in a central disease. J Neurochem. 2012;120(1):26–36. | ||
Baker DG, Nievergelt CM, O’Connor DT. Biomarkers of PTSD: neuropeptides and immune signaling. Neuropharmacology. 2012;62(2):663–673. | ||
Bauer ME, Wieck A, Lopes RP, Teixeira AL, Grassi-Oliveira R. Interplay between neuroimmunoendocrine systems during post-traumatic stress disorder: a minireview. Neuroimmunomodulation. 2010;17(3):192–195. | ||
Gill JM, Saligan L, Woods S, Page G. PTSD is associated with an excess of inflammatory immune activities. Perspect Psychiatr Care. 2009;45(4):262–277. | ||
Gola H, Engler H, Sommershof A, et al. Posttraumatic stress disorder is associated with an enhanced spontaneous production of pro-inflammatory cytokines by peripheral blood mononuclear cells. BMC Psychiatry. 2013;13:40. | ||
Lozano D, Gonzales-Portillo GS, Acosta S, et al. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat. 2015;11:97–106. | ||
O’Donovan A, Chao LL, Paulson J, et al. Altered inflammatory activity associated with reduced hippocampal volume and more severe posttraumatic stress symptoms in Gulf War veterans. Psychoneuroendocrinology. 2015;51:557–566. | ||
Plantinga L, Bremner JD, Miller AH, et al. Association between posttraumatic stress disorder and inflammation: a twin study. Brain Behav Immun. 2013;30:125–132. | ||
Wilson CB, McLaughlin LD, Nair A, Ebenezer PJ, Dange R, Francis J. Inflammation and oxidative stress are elevated in the brain, blood, and adrenal glands during the progression of post-traumatic stress disorder in a predator exposure animal model. PLoS One. 2013;8(10):e76146. | ||
Chen LF, Tian YF, Lin CH, Huang LY, Niu KC, Lin MT. Repetitive hyperbaric oxygen therapy provides better effects on brain inflammation and oxidative damage in rats with focal cerebral ischemia. J Formos Med Assoc. 2014;113(9):620–628. | ||
Geng F, Ma Y, Xing T, Zhuang X, Zhu J, Yao L. Effects of hyperbaric oxygen therapy on inflammasome signaling after traumatic brain injury. Neuroimmunomodulation. 2016;23(2):122–129. | ||
Hui J, Zhang ZJ, Zhang X, Shen Y, Gao YJ. Repetitive hyperbaric oxygen treatment attenuates complete Freund’s adjuvant-induced pain and reduces glia-mediated neuroinflammation in the spinal cord. J Pain. 2013;14(7):747–758. | ||
Lim SW, Wang CC, Wang YH, Chio CC, Niu KC, Kuo JR. Microglial activation induced by traumatic brain injury is suppressed by postinjury treatment with hyperbaric oxygen therapy. J Surg Res. 2013;184(2):1076–1084. | ||
Vlodavsky E, Palzur E, Soustiel JF. Hyperbaric oxygen therapy reduces neuroinflammation and expression of matrix metalloproteinase-9 in the rat model of traumatic brain injury. Neuropathol Appl Neurobiol. 2006;32(1):40–50. | ||
Wilson HD, Wilson JR, Fuchs PN. Hyperbaric oxygen treatment decreases inflammation and mechanical hypersensitivity in an animal model of inflammatory pain. Brain Res. 2006;1098(1):126–128. | ||
Yang L, Tang J, Chen Q, et al. Hyperbaric oxygen preconditioning attenuates neuroinflammation after intracerebral hemorrhage in rats by regulating microglia characteristics. Brain Res. 2015;1627:21–30. | ||
Chen X, Duan XS, Xu LJ, et al. Interleukin-10 mediates the neuroprotection of hyperbaric oxygen therapy against traumatic brain injury in mice. Neuroscience. 2014;266:235–243. | ||
Almzaiel AJ, Billington R, Smerdon G, Moody AJ. Hyperbaric oxygen enhances neutrophil apoptosis and their clearance by monocyte-derived macrophages. Biochem Cell Biol. 2015;93(4):405–416. | ||
Harvey LD, Yin Y, Attarwala IY, et al. Administration of DHA reduces endoplasmic reticulum stress-associated inflammation and alters microglial or macrophage activation in traumatic brain injury. ASN Neuro. 2015;7(6):1759091415618969. | ||
Lucke-Wold BP, Turner RC, Logsdon AF, et al. Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J Neurosurg. 2016;124(3):687–702. | ||
Han F, Yan S, Shi Y. Single-prolonged stress induces endoplasmic reticulum-dependent apoptosis in the hippocampus in a rat model of post-traumatic stress disorder. PLoS One. 2013;8(7):e69340. | ||
Wen L, Han F, Shi Y, Li X. Role of the endoplasmic reticulum pathway in the medial prefrontal cortex in post-traumatic stress disorder model rats. J Mol Neurosci. 2016;59(4):471–482. | ||
Xiao B, Yu B, Liu DJ, Han F, Shi YX. Single prolonged stress induces dysfunction of endoplasmic reticulum in a rat model of post-traumatic stress disorder. Mol Med Rep. 2015;12(2):2015–2020. | ||
Xie J, Han F, Shi Y. The unfolded protein response is triggered in rat neurons of the dorsal raphe nucleus after single-prolonged stress. Neurochem Res. 2014;39(4):741–747. | ||
Liu X, Yang J, Li Z, et al. Hyperbaric oxygen treatment protects against spinal cord injury by inhibiting endoplasmic reticulum stress in rats. Spine (Phila Pa 1976). 2015;40(24):E1276–E1283. | ||
Domachevsky L, Pick CG, Arieli Y, Krinsky N, Abramovich A, Eynan M. Do hyperbaric oxygen-induced seizures cause brain damage? Epilepsy Res. 2012;100(1–2):37–41. | ||
Domachevsky L, Pick CG, Peled N, Gomori JM, Abramovich A, Tempel-Brami C. MRI findings after hyperbaric oxygen-induced seizures. Epilepsy Res. 2013;105(1–2):62–68. | ||
Domachevsky L, Rachmany L, Barak Y, Rubovitch V, Abramovich A, Pick CG. Hyperbaric oxygen-induced seizures cause a transient decrement in cognitive function. Neuroscience. 2013;247:328–334. | ||
Lehm JP, Bennett MH. Predictors of middle ear barotrauma associated with hyperbaric oxygen therapy. SPUMS J. 2003;33(3):127–133. | ||
Camporesi EM. Side effects of hyperbaric oxygen therapy. Undersea Hyperb Med. 2014;41(3):253–257. | ||
Wolf EG, Prye J, Michaelson R, Brower G, Profenna L, Boneta O. Hyperbaric side effects in a traumatic brain injury randomized clinical trial. Undersea Hyperb Med. 2012;39(6):1075–1082. | ||
Norris FH, Friedman MJ, Watson PJ, Byrne CM, Diaz E, Kaniasty K. 60,000 disaster victims speak: part I. An empirical review of the empirical literature, 1981–2001. Psychiatry. 2002;65(3):207–239. | ||
Iverson KM, Hendricks AM, Kimerling R, et al. Psychiatric diagnoses and neurobehavioral symptom severity among OEF/OIF VA patients with deployment-related traumatic brain injury: a gender comparison. Womens Health Issues. 2011;21(4 Suppl):S210–S217. | ||
Eckert S, Shaw J, Koduri S, et al. Sex-dependent changes in depression and facial allodynia in the chronic period following mild TBI in the mouse. Soc Neurosci. 2015;2015:500.03/I37. | ||
Acosta SA, Tajiri N, Shinozuka K, et al. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS One. 2013;8(1):e53376. | ||
Acosta SA, Tajiri N, Shinozuka K, et al. Combination therapy of human umbilical cord blood cells and granulocyte colony stimulating factor reduces histopathological and motor impairments in an experimental model of chronic traumatic brain injury. PLoS One. 2014;9(3):e90953. | ||
Hayashi T, Kaneko Y, Yu S, et al. Quantitative analyses of matrix metalloproteinase activity after traumatic brain injury in adult rats. Brain Res. 2009;1280:172–177. | ||
Jacotte-Simancas A, Costa-Miserachs D, Coll-Andreu M, Torras-Garcia M, Borlongan CV, Portell-Cortes I. Effects of voluntary physical exercise, citicoline, and combined treatment on object recognition memory, neurogenesis, and neuroprotection after traumatic brain injury in rats. J Neurotrauma. 2015;32(10):739–751. | ||
Kaneko Y, Tajiri N, Yu S, et al. Nestin overexpression precedes caspase-3 upregulation in rats exposed to controlled cortical impact traumatic brain injury. Cell Med. 2012;4(2):55–63. | ||
Tajiri N, Acosta SA, Shahaduzzaman M, et al. Intravenous transplants of human adipose-derived stem cell protect the brain from traumatic brain injury-induced neurodegeneration and motor and cognitive impairments: cell graft biodistribution and soluble factors in young and aged rats. J Neurosci. 2014;34(1):313–326. | ||
Tajiri N, Hernandez D, Acosta S, et al. Suppressed cytokine expression immediately following traumatic brain injury in neonatal rats indicates an expeditious endogenous anti-inflammatory response. Brain Res. 2014;1559:65–71. | ||
Matar MA, Zohar J, Cohen H. Translationally relevant modeling of PTSD in rodents. Cell Tissue Res. 2013;354(1):127–139. | ||
Mu J, Ostrowski RP, Soejima Y, et al. Delayed hyperbaric oxygen therapy induces cell proliferation through stabilization of cAMP responsive element binding protein in the rat model of MCAo-induced ischemic brain injury. Neurobiol Dis. 2013;51:133–143. | ||
Golden Z, Golden CJ, Neubauer RA. Improving neuropsychological function after chronic brain injury with hyperbaric oxygen. Disabil Rehabil. 2006;28(22):1379–1386. | ||
Li Y, Dong H, Chen M, et al. Preconditioning with repeated hyperbaric oxygen induces myocardial and cerebral protection in patients undergoing coronary artery bypass graft surgery: a prospective, randomized, controlled clinical trial. J Cardiothorac Vasc Anesth. 2011;25(6):908–916. | ||
Ohguri T, Imada H, Kohshi K, et al. Effect of prophylactic hyperbaric oxygen treatment for radiation-induced brain injury after stereotactic radiosurgery of brain metastases. Int J Radiat Oncol Biol Phys. 2007;67(1):248–255. | ||
Xiong L, Zhu Z, Dong H, Hu W, Hou L, Chen S. Hyperbaric oxygen preconditioning induces neuroprotection against ischemia in transient not permanent middle cerebral artery occlusion rat model. Chin Med J (Engl). 2000;113(9):836–839. | ||
Lu PG, Feng H, Yuan SJ, et al. Effect of preconditioning with hyperbaric oxygen on neural cell apoptosis after spinal cord injury in rats. J Neurosurg Sci. 2013;57(3):253–258. | ||
Ostrowski RP, Graupner G, Titova E, et al. The hyperbaric oxygen preconditioning-induced brain protection is mediated by a reduction of early apoptosis after transient global cerebral ischemia. Neurobiol Dis. 2008;29(1):1–13. | ||
Hu SL, Hu R, Li F, et al. Hyperbaric oxygen preconditioning protects against traumatic brain injury at high altitude. Acta Neurochir Suppl. 2008;105:191–196. | ||
Peng Y, Feng SF, Wang Q, et al. Hyperbaric oxygen preconditioning ameliorates anxiety-like behavior and cognitive impairments via upregulation of thioredoxin reductases in stressed rats. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(6):1018–1025. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.