Back to Journals » Drug Design, Development and Therapy » Volume 8
Hydrogen sulfide, a potential novel drug, attenuates concanavalin A-induced hepatitis
Authors Cheng P, Chen K, Xia Y, Dai W, Wang F, Shen M, Wang C, Yang J, Zhu R, Zhang H, Li J, Zheng Y, Wang J, Zhang Y, Lu J, Zhou Y, Guo C ![]()
Received 21 April 2014
Accepted for publication 4 June 2014
Published 9 September 2014 Volume 2014:8 Pages 1277—1286
DOI https://doi.org/10.2147/DDDT.S66573
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Ping Cheng,* Kan Chen,* Yujing Xia, Weiqi Dai, Fan Wang, Miao Shen, Chengfen Wang, Jing Yang, Rong Zhu, Huawei Zhang, Jingjing Li, Yuanyuan Zheng, Junshan Wang, Yan Zhang, Jie Lu, Yingqun Zhou, Chuanyong Guo
Department of Gastroenterology, Shanghai Tenth People's Hospital, Tongji University of Medicine, Shanghai, People's Republic of China
*These authors contributed equally to this work
Background: Hydrogen sulfide (H2S) is known to exert anti-inflammatory properties. Apoptosis and autophagy play important roles in concanavalin A (Con A)-induced acute hepatitis. The purpose of this study was to explore both the effect and mechanism of H2S on Con A-induced acute hepatitis.
Methods: BALB/c mice were randomized into sham group, Con A-injection group, and 14 µmol/kg of sodium hydrosulfide (NaHS, an H2S donor) pretreatment group.
Results: Aspartate aminotransferase, alanine aminotransferase, and pathological damage were significantly ameliorated by NaHS pretreatment. NaHS pretreatment significantly reduced the levels of interleukin-6 and tumor necrosis factor-α compared with those of the Con A group. The expression of Bcl-2, Bax, Beclin-1, and LC3-2, which play important roles in the apoptosis and autophagy pathways, were also clearly affected by NaHS. Furthermore, NaHS affected the p-mTOR and p-AKT.
Conclusion: H2S attenuates Con A-induced acute hepatitis by inhibiting apoptosis and autophagy, in part, through activation of the PtdIns3K-AKT1 signaling pathway.
Keywords: NaHS, apoptosis, PtdIns3K-AKT, autophagy
Introduction
Hepatitis seriously threatens human health and daily life. It is caused by various factors such as viruses, drugs, chemicals, alcohol, genetic factors, or a patient’s own immune system. Currently, effective drugs in the treatment of hepatitis are lacking, so effective therapies must be explored to provide new applications in the clinic. Concanavalin A (Con A) can induce necrosis of hepatocytes; this causes hepatic dysfunction and also attracts and activates immune cells. Pathological studies have shown infiltration and a large number of lymphocyte accumulation of mainly the CD4+ T cells in the liver parenchyma.1,2 Therefore, Con A is often applied in the animal model of acute hepatitis.3
There are complicated mechanisms in the evolution of Con A-induced hepatitis. Recent studies show that blocking MAPK and activating AKT cell death pathways can both significantly reduce Con A-induced hepatitis.3,4 Apoptosis, named type 1 programmed cell death, may be a major cell death mechanism in Con A-induced hepatitis. The Bcl-2 family is considered to have an important role in the apoptosis pathway. In the Bcl-2 family, the representative antiapoptotic genes are Bcl-2 and Bcl-xl, and the proapoptotic genes are Bax and Bad. It has been reported that the balance between Bax and Bcl-2 proteins determines the survival status of cells after particular kinds of stimulus or damage.1,5
Autophagy, named type 2 programmed cell death, is a newly described type of programmed cell death. Autophagy is a protective cell behavior for surviving harsh environments.6,7 However, beyond a certain range, it eventually leads to cell death, with the excessive accumulation of autophagosomes.8 Damaged organelles are surrounded by isolation membrane to form autophagosomes, and these autophagosome fuse with lysosomes to form autophagy-lysosomes. It has been confirmed that many autophagy-related genes are linked with the formation of autophagy. Autophagy-related gene protein Beclin-1 and the diaphragm combine to generate the production of the Atg12-Atg5-Atg16 complex. Further, raised LC3-2 binds to the diaphragm to promote the extension of autophagy membrane. Mature autophagosomes eventually form while the Atg12-Atg5-Atg16 complex is deactivated. Therefore, restraining the expression of the Atg family can effectively block the formation of autophagosome.
Hydrogen sulfide (H2S), a third common endogenous signaling molecule after nitric oxide and carbon monoxide, is known to exert anti-inflammatory properties.9 H2S is naturally synthesized in the liver from L-cysteine by two main enzymes, cystathionine-β-synthetase and cystathionine-γ-lyase (CSE).10 DL-propargylglycine (PAG), an irreversible inhibitor of CSE, penetrates cell membranes and decreases H2S production in many rat tissues.11,12 Generally, two-thirds of H2S molecules dissociate into bisulfide ions and hydrogen ions. Therefore, sodium hydrosulfide (NaHS) can be seen as a water-soluble H2S donor.13 In this study, we explored both the effect of H2S on Con A-induced acute hepatitis and the regulation of apoptosis and autophagy.
Materials and methods
Reagents and drug administration
Con A and dimethyl sulfoxide were purchased from Sigma-Aldrich (St Louis, MO, USA) and stored at 4°C. PAG was purchased from Sigma-Aldrich, and the RNA polymerase chain reaction (PCR) kit was obtained from Takara Biotechnology (Dalian, People’s Republic of China). Fetal bovine serum and Dulbecco’s Modified Eagle’s Medium were obtained from Thermo Fisher Scientific (Waltham, MA, USA). LC3-2 and Beclin-1 were purchased from Abcam (Cambridge, UK). The antibodies used for immunoblotting and immunohistochemical staining were anti-tumor necrosis factor (TNF)-α, anti-interleukin (IL)-6, anti-mTOR, anti-p-mTOR, anti-p-AKT, and anti-AKT (Santa Cruz Biotechnology, Dallas, TX, USA). Con A was dissolved in pyrogen-free physiological saline and intravenously injected at a dose of 20 mg/kg body weight to induce hepatitis as previously described.1 NaHS was dissolved in saline and intravenously injected at a dose of 14 μmol/kg body weight. PAG was dissolved in pyrogen-free physiological saline and intraperitoneally injected at a dose of 50 mg/kg body weight.
Animals and ethics statement
Male BALB/c mice (23±2 g, 6–8 weeks old) were obtained from Shanghai SLAC Laboratory Animal Co, Ltd (Shanghai, People’s Republic of China). This study conscientiously implemented principles from the Guide for the National Science Council of the Republic of China and the Animal Care and Use Committee of The Tenth People’s Hospital of Shanghai (permit number 2011-0111).
Model establishment and experimental design
Based on previous experiments by our team, time points at 8, 12, and 24 hours were used. Con A was dissolved in pyrogen-free physiological saline at a concentration of 2.5 mg/mL as previously described. Next, 72 mice were randomly divided into four groups of 18 mice each.
Group 1 (normal control, n=18) mice were intravenously injected in the tail with physiological saline only; group 2 (model group, n=18) mice were intravenously injected in the tail with 20 mg/kg Con A; group 3 (PAG group, n=18) mice were intravenously injected in the tail with 50 mg/kg PAG 1 hour prior to Con A injection;14 group 4 (protected group, n=18) mice were intravenously injected in the tail with NaHS (14 μmol/kg) 1 hour prior to Con A injection. Eighteen mice from each group were randomly killed at 8, 12, and 24 hours. All liver tissue (median and left lobes) and serum were collected and stored at -80°C.
Biochemical analysis
After blood collection, serum was separated by centrifugation at 2,000 rpm for 10 minutes at room temperature. Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were tested using an automated chemistry analyzer (Olympus AU1000; Olympus Corporation, Tokyo, Japan).
Histopathology
When mice were killed, their liver tissues (median and left lobes) were collected, incubated in 4% paraformaldehyde for more than 24 hours, and prepared in paraffin blocks according to the traditional method. The degree of inflammation and tissue damage was observed in paraffin sections (3 μm thick) stained with hematoxylin and eosin using an optical microscope.
Measurement of plasma H2S
Plasma H2S was measured as described by Chunyu et al.15
Assay of liver H2S-synthesizing activity
H2S-synthesizing activity in liver was detected as described by Zhang et al16 and Labarca and Paigen.17
Immunohistochemistry
Immunohistochemistry using the antibodies against Beclin-1 (1:2,000), LC3-2 (1:500), p-AKT (1:1,000), p-mTOR (1:1,000), and rabbit anti-mouse was performed according to standard procedures.18 Slides were then observed by optical microscopy.
Western blot analysis
Total protein was prepared using standard procedures. Protein samples were separated by electrophoresis in a sodium dodecyl sulfate polyacrylamide gel and transferred to polyvinylidene fluoride membrane. After blocking, membranes were incubated overnight with different primary antibodies and β-actin at 4°C. After incubation with peroxidase-conjugated secondary antibodies for 1 hour at 37°C, membranes were developed with the Odyssey two-color infrared laser imaging system (LI-COR Biosciences, Lincoln, NE, USA).18
Reverse transcription-PCR and real-time PCR
The mRNA transcripts from liver tissues were detected and analyzed via quantitative reverse transcription-PCR. Total RNA was extracted using TRIzol® reagent (Thermo Fisher Scientific) as described by the manufacturer’s instructions. Reverse transcription-PCR was performed in a 7900HT Fast Real-time PCR system (Thermo Fisher Scientific) according to the manufacturer’s guidelines. The primers used in the PCR reactions are listed in Table 1.
 | Table 1 Primers used in the polymerase chain reactions |
Transmission electron microscopy
Male BALB/c mice were treated as described above, and laparotomy was performed under ketamine/xylazine anesthesia. The liver was perfused with 2 mL of 2.5% glutaraldehyde in phosphate buffered saline and was then treated as previously described.19
Statistical analysis
Statistical significance was assessed using the Student’s t-test or Tukey’s post hoc tests. All statistical analyses were performed using SPSS 17.0 (SPSS, Inc., Chicago, IL, USA), and P<0.05 was considered significant.
Results
H2S pretreatment ameliorates Con A-induced hepatitis
We performed an assay using a Con A-induced hepatitis model. The expression level of CSE mRNA in the liver was significantly increased in the Con A group compared with that in the saline group. Con A resulted in a significant increase in the plasma H2S level and the amount of H2S formed in liver compared with the saline group, and NaHS preconditioning further increased them compared with the Con A group. In addition, PAG pretreatment significantly decreased the expression level of CSE mRNA, reduced the plasma H2S level and the amounts of H2S formed in liver compared with the Con A group (P<0.05) (Figure 1A). Liver function was assessed by measuring the serum level of ALT and AST. As shown in Figure 1B, the level of ALT and AST clearly increased after Con A injection compared with the saline group at 8, 12, and 24 hours (P<0.05), and NaHS pretreatment significantly attenuated the Con A injection-induced elevation of serum ALT and AST (P<0.05). Conversely, PAG pretreatment aggravated serum ALT and AST at all three time points compared to the Con A group (P<0.05) (Figure 1B). Histopathological changes in the liver tissues from the three groups were examined after hematoxylin and eosin staining (Figure 1C). The structure of the liver tissues was completely maintained and ordered in the saline group, whereas a disordered lobular structure with some hepatocyte necrosis and polymorphonuclear cell infiltration were observed in the model group at 12 hours and 24 hours. Pretreatment with NaHS at 12 hours attenuated these pathological changes. It is worth noting that at all three time points, the administration of PAG clearly accentuated these pathological features, which included significant congestion of liver, periportal inflammatory cell infiltration, and large fragments of liver tissue necrosis.
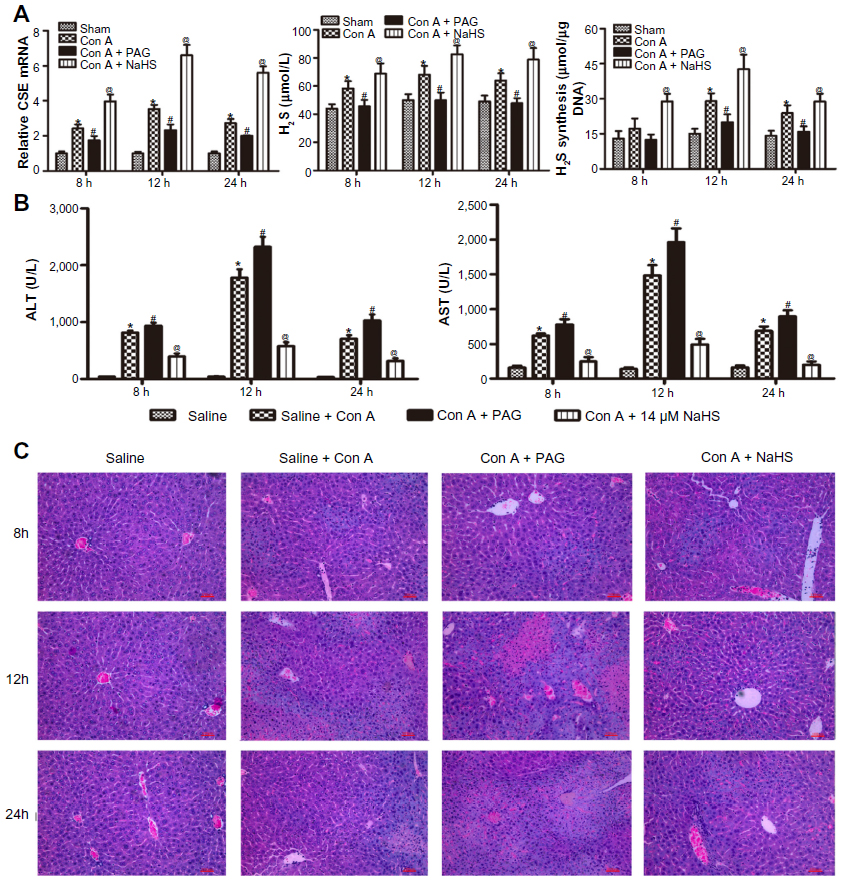 | Figure 1 H2S pretreatment ameliorates Con A-induced hepatitis. |
H2S pretreatment inhibits the release of cytokines during Con A-induced hepatitis
Con A-induced hepatitis is associated with changes in the levels of inflammatory cytokines. We demonstrated with real-time PCR and immunoblotting that the expression of TNF-α and IL-6 was significantly increased in the model group compared to that in the saline group (Figure 2A and B). In addition, the location and expression of TNF-α and IL-6 by immunohistochemical staining were stronger in the model group than in the saline group at 12 hours (P<0.05) (Figure 2C). Furthermore, NaHS pretreatment significantly attenuated the expression of TNF-α and IL-6 compared to the model group (P<0.05) (Figure 2C). In contrast, with PAG treatment, there were opposite experimental results compared to the NaHS group (P<0.05) (Figure 2C).
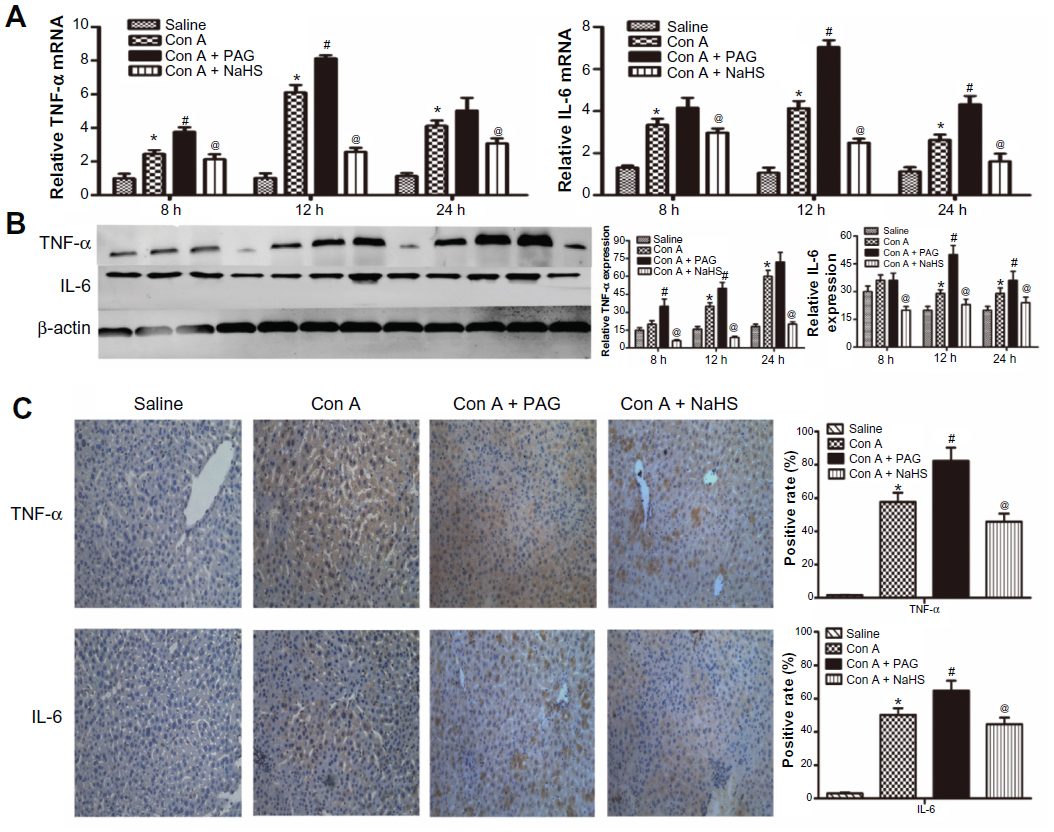 | Figure 2 H2S pretreatment inhibits the release of cytokines during Con A-induced hepatitis. |
H2S attenuates hepatocyte apoptosis in Con A-induced hepatitis in mice
We next investigated the effect of H2S on apoptosis in Con A-induced hepatitis in mice. The expression of Bcl-2 and Bax cDNAs was detected with real-time PCR, as expected. NaHS pretreatment significantly increased the expression of Bcl-2 at all three time points and reduced the expression of Bax at 12 hours and 24 hours. PAG preconditioning significantly elicited the opposite effect of NaHS (Figure 3A). NaHS also increased the expression of Bcl-2 at the protein level at 8 hours and 24 hours, and the expression of Bax was mainly reduced at all three time points with NaHS pretreatment (Figure 3B). However, PAG preconditioning did not displayed the opposite effect of NaHS, it only reduced the role of NaHS.
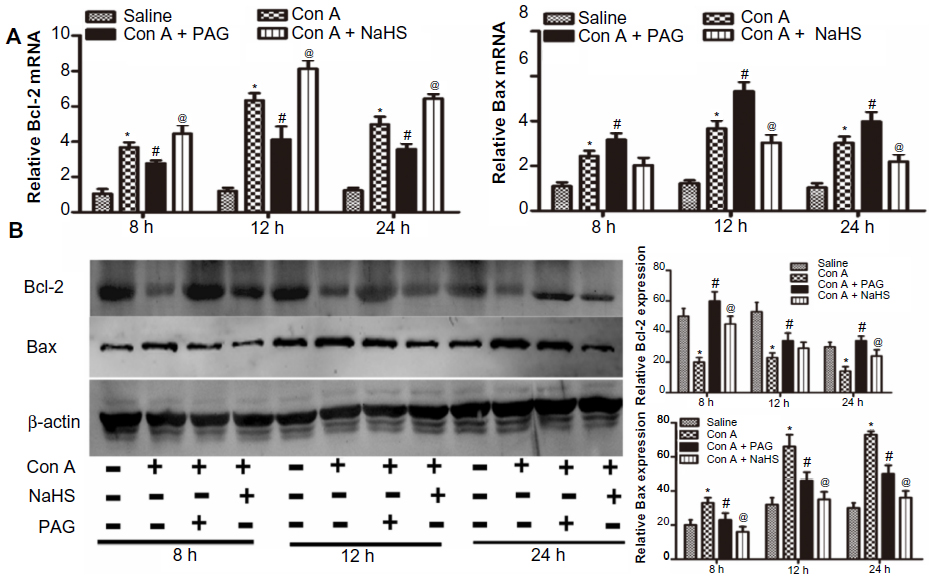 | Figure 3 H2S attenuates hepatocyte apoptosis in Con A-induced hepatitis in mice. |
H2S attenuates autophagy in Con A-induced hepatitis in mice
It is well known that Con A-induced hepatitis can involve autophagy. LC3 is an important marker of autophagy, and Beclin-1 plays an important role in autophagy. Therefore, to further assess activation of autophagy by NaHS and PAG pretreatment in the Con A model, the expression of LC3 and Beclin-1 in liver tissues were investigated with real-time PCR and immunoblotting (Figure 4A and B). These results indicate that the levels of LC3 were significantly reduced after NaHS preconditioning compared to the model group. In contrast, the influence of PAG on autophagy in Con A-induced hepatitis exhibited an increased conversion of LC3. However, Beclin-1 did not appear to have a role in the Con A model and NaHS/PAG models (Figure 4A and B). Analysis of the immunohistochemical changes in the mouse livers confirmed these results (Figure 4C). In addition, the formation of autophagosomes is a pivotal process in autophagy; hence, electron microscopy was applied to observe the ultrastructure of hepatic cells (Figure 4D). Compared with basal levels in the saline group, autophagic vacuoles were dramatically increased in the model group. However, after NaHS treatment, the liver nuclear chromatin was more homogeneous, and the integrity of the cell structure was still intact. Meanwhile, there were more autophagic vacuoles in PAG-treated murine liver tissues.
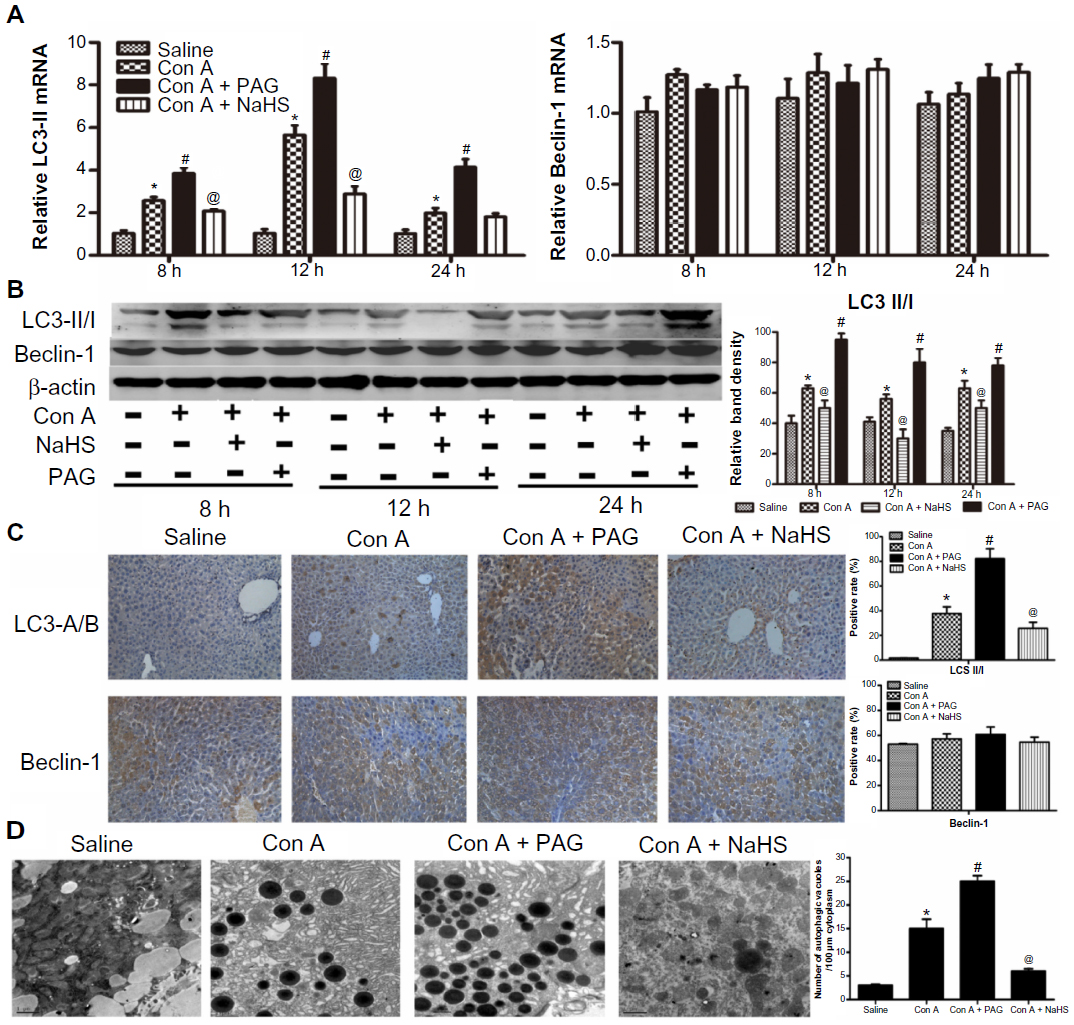 | Figure 4 H2S attenuates autophagy in Con A-induced hepatitis in mice. |
H2S activates PtdIns3K-AKT1 signaling in Con A-induced hepatitis in mice
As PtdIns3K-AKT1 signaling plays a key role in regulating inflammatory responses, to further explore the mechanism and effect of H2S, the activation of AKT and mTOR were analyzed through Western blot analysis of Con A-induced acute hepatitis in mice (Figure 5A). Con A actually enhanced p-AKT1 and p-mTOR levels in the mouse liver (P<0.05) (Figure 5A), which is further increased by NaHS pretreatment compared to the Con A model group at 12 hours (P<0.05) (Figure 5A). Pretreatment with PAG decreased the activation of AKT1 and mTOR compared to the NaHS group (P<0.05) (Figure 5A). To confirm the results of Western blot analysis, immunohistochemical changes were further detected (Figure 5B).
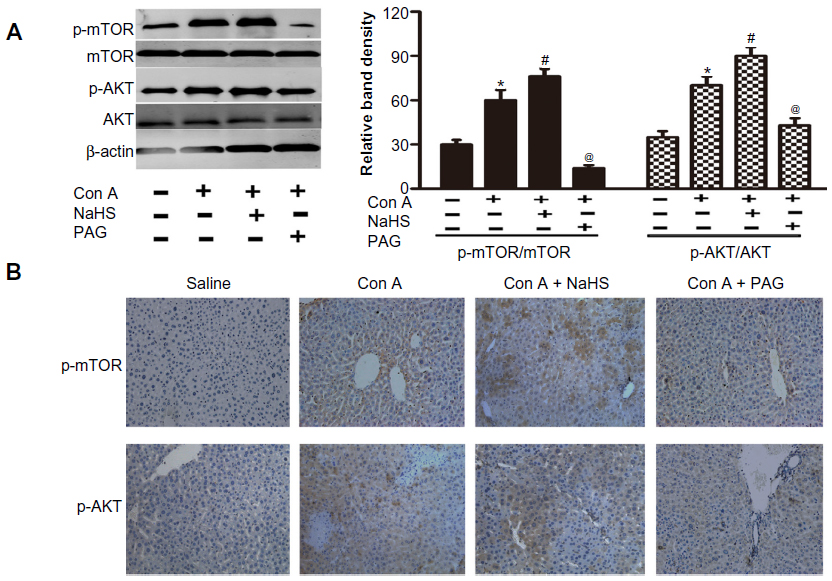 | Figure 5 H2S activates PtdIns3K-AKT1 signaling in Con A-induced hepatitis in mice. |
Discussion
Hepatitis, as a transmitted disease of high incidence, seriously threatens human health and daily life.19 Currently, effective drugs in the treatment of hepatitis are not readily available, so we must explore new therapies to provide effective applications in the clinic. As previously discussed, Con A is often applied in animal models of acute hepatitis.
The gas H2S is a new regulator of important physiologic functions. Zhang et al20 reported that H2S protects liver against ischemia/reperfusion injury, exhibiting anti-inflammatory and antiapoptotic activities. As an irreversible inhibitor of CSE, PAG decreases H2S production in many rat tissues.21 Based on the multiple physiologic functions of H2S, we speculate that it might be an effective therapy for Con A-induced hepatitis. Our results, indeed, confirmed that H2S pretreatment ameliorates Con A-induced hepatitis and inhibits the release of cytokines (Figures 1 and 2). It is worth noting that the administration of PAG, which can decrease H2S production by inhibiting CSE, clearly accentuated Con A-induced hepatitis. This confirmed the protective effect of H2S from another angle.
PtdIns3K-AKT1 is one of the most important cell survival pathways and is known to play an essential role in hepatitis. Mayoral et al22 have reported that tissue-specific COX-2-dependent prostaglandins exert efficient protection against Con A-induced hepatitis in mice through an antiapoptotic effect by activating AKT and AMP kinase. Therefore, the PtdIns3K signaling pathway may contribute to multiple endogenous protective pathways to reduce liver injury. AKT can inhibit cytochrome c release through blocking the channel formed by Bcl-2-associated X protein (Bax) and phosphorylating Bad.23 Therefore, we attempted to examine the AKT signaling pathway to illuminate how H2S pretreatment ameliorates Con A-induced hepatitis. Wang et al24 previously reported that H2S preconditioning produces liver protective effects against hepatic ischemia/reperfusion injury in mice through activation of the PtdIns3K-AKT1 pathway. We have also observed in the present study that NaHS pretreatment significantly increased p-AKT compared to the Con A group (Figure 5). Hence, we supposed that NaHS ameliorated cell death in Con A-induced hepatitis by activating the PI3K/AKT signaling pathway. It is well known that Bax is the representative proapoptotic gene, Bcl-2 is the representative antiapoptotic gene, and the balance between Bax and Bcl-2 proteins has been linked with induction of apoptosis in cell death.19 Also, our results showed that the Con A increase of Bax and decrease of Bcl-2 finally resulted in the cell death. However, with NaHS treatment, the balance between Bax and Bcl-2 trended to normal, with the upregulation of Bcl-2 and downregulation of Bax (Figure 3B). Hence, we supposed that NaHS ameliorated cell death in Con A-induced hepatitis by inhibiting the intrinsic pathway of apoptosis.
A recent paper has shown that activation of the PtdIns3K-AKT1 signaling molecules link receptor tyrosine kinases to mTOR activation (PI3K-AKT-mTOR), which is an important signal that inhibits autophagy.25 Studies have reported that Con A can directly induce autophagic cell death of hepatocytes.26,27 Con A, after endocytosis, binds to mitochondria to cause mitochondrial dysfunction and autophagy.28,29 The role of autophagy in hepatitis is worthy of further exploration, particularly whether H2S-preconditioning affects autophagy during Con A-induced hepatitis and whether the PI3K-AKT-mTOR pathway mediates it. In our study, the results showed that LC3-II (an important autophagy marker) was inhibited by H2S preconditioning in hepatocytes, but Beclin-1 was not affected (Figure 4). Con A could upregulate the levels of p-AKT1, and NaHS preconditioning could further enhance AKT1 phosphorylation compared to the Con A group in hepatocytes, while p-AKT1 was markedly decreased after using PAG (Figure 5). This indicates that PtdIns3K-AKT1 may play an important role in the autophagy processes of Con A-induced hepatitis. Chang et al30 reported that either 3-MA or small interfering RNA for BNIP3 and LC3, but neither Beclin-1 nor ATG5, partially inhibited Con A-induced cell death. Petiot et al31 also used RNA interference technology and found that Con A triggers autophagic cell death through increasing of BNIP3 protein and the generation of LC3-II, but other autophagy proteins such as Beclin-1 and ATG5 are not involved. This may explain why Beclin-1 was not affected in our experiment.
The mechanism underlying how H2S inhibits apoptosis and autophagy through activation of the PtdIns3K-AKT1 signaling during Con A-induced acute hepatitis is not clear. We intend to probe this unknown mechanism in future studies.
Acknowledgments
We thank all members of the Central Laboratory of the Tenth Hospital of Tongji University. This project was supported by the National Natural Science Foundation of China (ID: 81270515), the Shanghai Municipal Health Bureau (ID: 2011287, 2012107), and the Liver Diseases Study Fund of China Foundation for Hepatitis Prevention and Control (ID: WBN20100021, CFHPC20131011).
Disclosure
The authors report no conflicts of interest in this work.
References
Shen M, Lu J, Cheng P, et al. Ethyl pyruvate pretreatment attenuates concanavalin a-induced autoimmune hepatitis in mice. PLoS One. 2014;9(2):e87977. | |
Zhou Y, Dai W, Lin C, et al. Protective effects of necrostatin-1 against concanavalin A-induced acute hepatic injury in mice. Mediators Inflamm. 2013;2013:706156. | |
Shao X, Qian Y, Xu C, et al. The protective effect of intrasplenic transplantation of Ad-IL-18BP/IL-4 gene-modified fetal hepatocytes on Con A-induced hepatitis in mice. PLoS One. 2013;8(3):e58836. | |
Tang G, Yue Z, Talloczy Z, et al. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet. 2008;17(11):1540–1555. | |
Guo J, Zhang K, Ji Y, Jiang X, Zuo S. Effects of ethyl pyruvate on myocardial apoptosis and expression of Bcl-2 and Bax proteins after ischemia-reperfusion in rats. J Huazhong Univ Sci Technolog Med Sci. 2008;28(3):281–283. | |
Velentzas PD, Velentzas AD, Mpakou VE, et al. Detrimental effects of proteasome inhibition activity in Drosophila melanogaster: implication of ER stress, autophagy, and apoptosis. Cell Biol Toxicol. 2013;29(1):13–37. | |
Maiuri MC, Grassia G, Platt AM, Carnuccio R, Ialenti A, Maffia P. Macrophage autophagy in atherosclerosis. Mediators Inflamm. 2013; 2013:584715. | |
Zhou XJ, Zhang H. Autophagy in immunity: implications in etiology of autoimmune/autoinflammatory diseases. Autophagy. 2012;8(9):1286–1299. | |
Ekundi-Valentim E, Mesquita FP, Santos KT, et al. A comparative study on the anti-inflammatory effects of single oral doses of naproxen and its hydrogen sulfide (H2S)-releasing derivative ATB-346 in rats with carrageenan-induced synovitis. Med Gas Res. 2013;3(1):24. | |
Sen U, Munjal C, Qipshidze N, Abe O, Gargoum R, Tyagi SC. Hydrogen sulfide regulates homocysteine-mediated glomerulosclerosis. Am J Nephrol. 2010;31(5):442–455. | |
Asimakopoulou A, Panopoulos P, Chasapis CT, et al. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br J Pharmacol. 2013;169(4):922–932. | |
Renga B. Hydrogen sulfide generation in mammals: the molecular biology of cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE). Inflamm Allergy Drug Targets. 2011;10(2):85–91. | |
Wang K, Zhu D, Yu X, Sun J, Yao W. Differences in the H2S-induced quantal release of catecholamine in adrenal chromaffin cells of neonatal and adult rats. Toxicology. 2013;312:12–17. | |
Li Y, Wang ZL, He F, et al. TP-58, a novel thienopyridine derivative, protects mice from concanavalinA-induced hepatitis by suppressing inflammation. Cell Physiol Biochem. 2012;29(1–2):31–40. | |
Chunyu Z, Junbao D, Dingfang B, Hui Y, Xiuying T, Chaoshu T. The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem Biophys Res Commun. 2003;302(4):810–816. | |
Zhang J, Sio SW, Moochhala S, Bhatia M. Role of hydrogen sulfide in severe burn injury-induced inflammation in mice. Mol Med. 2010;16(9–10):417–424. | |
Labarca C, Paigen K. A simple, rapid, and sensitive DNA assay procedure. Anal Biochem. 1980;102(2):344–352. | |
Cheng P, Dai W, Wang F, et al. Ethyl pyruvate inhibits proliferation and induces apoptosis of hepatocellular carcinoma via regulation of the HMGB1-RAGE and AKT pathways. Biochem Biophys Res Commun. 2014;443(4):1162–1168. | |
Shen M, Lu J, Dai W, et al. Ethyl pyruvate ameliorates hepatic ischemia-reperfusion injury by inhibiting intrinsic pathway of apoptosis and autophagy. Mediators Inflamm. 2013;2013:461536. | |
Zhang Q, Fu H, Zhang H, et al. Hydrogen sulfide preconditioning protects rat liver against ischemia/reperfusion injury by activating Akt-GSK-3β signaling and inhibiting mitochondrial permeability transition. PLoS One. 2013;8(9):e74422. | |
Wang X, Wang Q, Guo W, Zhu YZ. Hydrogen sulfide attenuates cardiac dysfunction in a rat model of heart failure: a mechanism through cardiac mitochondrial protection. Biosci Rep. 2011;31(2):87–98. | |
Mayoral R, Mollá B, Flores JM, Boscá L, Casado M, Martín-Sanz P. Constitutive expression of cyclo-oxygenase 2 transgene in hepatocytes protects against liver injury. Biochem J. 2008;416(3):337–346. | |
White BC, Sullivan JM, DeGracia DJ, et al. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci. 2000;179(S 1–2):1–33. | |
Wang D, Ma Y, Li Z, et al. The role of AKT1 and autophagy in the protective effect of hydrogen sulphide against hepatic ischemia/ reperfusion injury in mice. Autophagy. 2012;8(6):954–962. | |
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. | |
Chang CP, Lei HY. Autophagy induction in T cell-independent acute hepatitis induced by concanavalin A in SCID/NOD mice. Int J Immunopathol Pharmacol. 2008;21(4):817–826. | |
Lei HY, Chang CP. Induction of autophagy by concanavalin A and its application in anti-tumor therapy. Autophagy. 2007;3(4):402–404. | |
Pratt J, Roy R, Annabi B. Concanavalin-A-induced autophagy biomarkers requires membrane type-1 matrix metalloproteinase intracellular signaling in glioblastoma cells. Glycobiology. 2012;22(9):1245–1255. | |
Liu Z, Li X, Ding X, Yang Y. In silico and experimental studies of concanavalin A: insights into its antiproliferative activity and apoptotic mechanism. Appl Biochem Biotechnol. 2010;162(1):134–145. | |
Chang CP, Yang MC, Liu HS, Lin YS, Lei HY. Concanavalin A induces autophagy in hepatoma cells and has a therapeutic effect in a murine in situ hepatoma model. Hepatology. 2007;45(2):286–296. | |
Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275(2):992–998. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.