Back to Journals » Drug Design, Development and Therapy » Volume 14
Hydrogen-Rich Saline Regulates Microglial Phagocytosis and Restores Behavioral Deficits Following Hypoxia-Ischemia Injury in Neonatal Mice via the Akt Pathway
Authors Ke H, Liu D, Li T, Chu X, Xin D, Han M, Wang S, Wang Z
Received 28 May 2020
Accepted for publication 13 August 2020
Published 21 September 2020 Volume 2020:14 Pages 3827—3839
DOI https://doi.org/10.2147/DDDT.S264684
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Tin Wui Wong
Hongfei Ke1 ,* Dexiang Liu2 ,* Tingting Li,1 Xili Chu,1 Danqing Xin,1 Min Han,1 Shuanglian Wang,1 Zhen Wang1
1Department of Physiology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, People’s Republic of China; 2Department of Medical Psychology and Ethics, School of Basic Medicine Sciences, Cheeloo College of Medicine, Shandong University, Jinan 250012, Shandong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhen Wang
Department of Physiology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, People’s Republic of China
Email [email protected]
Introduction: We have reported previously that hydrogen-rich saline (HS) plays a neuroprotective role in hypoxia-ischemia (HI) brain damage in newborn mice. However, the mechanisms for this neuroprotection resulting from HS remain unknown. In this study, we examined the potential for HS to exert effects upon microglial phagocytosis via involvement of the Akt signaling pathway as one of the neuroprotective mechanisms in response to neonatal HI.
Methods: The HI brain injury model was performed on postnatal day (PND) 7 (modified Vannucci model). The acute brain damage was detected at 3 days after HI exposure. The behavioral and functional screening of the pups at PND11 and PND13 and their long-term outcomes (PND35, 28-days post-HI) were evaluated sensorimotor performance and cognitive functions, respectively.
Results: The result showed that HS administration alleviated HI-induced edema, infract volume and cellular apoptosis within the cortex of neonatal mice. Accompanying these indices of neuroprotection from HS were reductions in HI-induced phagocytosis in microglia as demonstrated in vivo and in vitro, effects that were associated with increasing levels of Akt phosphorylation and improvements in neurobehavioral responses. These beneficial effects of HS were abolished in mice treated with an Akt inhibitor.
Discussion: These results demonstrate that HS treatment attenuates neurobehavioral deficits and apoptosis resulting from HI, effects which were associated with reductions in phagocytosis and appear to involve the Akt signaling pathway.
Keywords: microglia, phagocytosis, hypoxia-ischemia, HS, Akt
Introduction
Hypoxia-ischemia (HI) brain injury is a leading cause of mortality and neurological disabilities in infants and young children.1 The long-term neurological defects resulting from HI are often manifested in behavioral, social, attention, cognitive and/or functional motor deficits.2 HI brain injury involves a complex array of factors, with the main feature being a decrease in oxygen concentration and blood flow levels, eventually leading to an insufficient supply of nutrition to the brain.3 Inflammation also represents a crucial factor leading to brain damage, with microglia playing an important role in the early inflammatory responses after HI injury.4 As therapeutic interventions for these HI-induced effects are extremely limited, an increased understanding of the underlying mechanisms of this condition is sorely needed for the development of more effective measures in the prevention and treatment of neonatal HI encephalopathy.
Hydrogen is a transparent, colorless and odorless, highly flammable gas. Hydrogen possesses a selective antioxidant effect which can discriminatively remove some of the more toxic hydroxyl radicals and nitrite anions while maintaining a low level of toxicity and exerting minimal effects on other reactive oxygen species with important biological functions.5 Moreover, hydrogen can effectively exert anti-inflammatory effects through its capacity to inhibit active oxygen production, neutralize hydroxyl radicals and inhibit the release of pro-inflammatory factors. Unfortunately, hydrogen administration is problematic as use of inhaled hydrogen possesses a high risk of explosion and is difficult to control while oral ingestion of hydrogen-saturated water can result in detrimental changes in certain biological indicators.6 Therefore, researchers have employed an alternate method of hydrogen administration consisting of an injection of hydrogen-rich saline (HS). HS could promote renal function recovery after ischemia/reperfusion injury in rats.7 HS exerts a protective effect against cisplatin-induced ovarian injury by regulating oxidative stress.8 With use of this method, we have found that the neuroprotective effects of HS on HI injury in newborn mice are achieved via regulation of endoplasmic reticulum stress and autophagy mechanisms.9 Moreover, this HS treatment can produce preventative effects on neuro-inflammation and behavioral dysfunction after HI injury. These effects are linked to the promotion of M2 polarization in microglia and the regulation of complement-mediated synapse loss through activation of AMPK.10 A related issue of significance to the present report is the findings that protein kinase B (Akt) mediates diverse cellular functions, such as cellular survival, apoptosis and metabolism.11 Given this background information, in this report, we examined whether the beneficial effects of HS upon neonatal HI may involve mechanisms related to effects upon phagocytosis via the Akt signaling pathway.
Materials and Methods
HI Model
All animal experiments in this research were approved by the Animal Ethics and Welfare Committee of Shandong University (approval No. ECSBMSSDU 2018-2-059). The use of experimental animals in this project conforms to the “3R” principle of animal experiment ethics. The entire experimental process followed the “Regulations on the Administration of Laboratory Animals” formulated by the Science and Technology Commission of the People’s Republic of China, and the “Guiding Opinions on the Good Treatment of Laboratory Animals” issued by the Ministry of Science and Technology of the People’s Republic of China. The Center for Experimental Animals of Shandong University provided pregnant C57BL/6J mice (≥15 days gestation) which then delivered under standard environmental conditions. The HI brain injury model used in this experiment represented a modified version of the Rice-Vannucci model and was performed at postnatal day (PND) 7 as described previously.1 A total of 144 male mice were used in this study Under conditions of a clean surgical environment, mice were anesthetized with 2.5% isoflurane and the right carotid artery was exposed and ligated. Following a one-hour postoperative recovery period, the mice were placed in a hypoxic environment for 1.5 h, with a temperature maintained at 37 °C and an oxygen concentration of approximately 8%. Sham controls were subjected to the anesthesia and exposure of the right carotid artery. The wound was sutured after the operation, and the 75% alcohol was used to disinfect the wound. Mice from each group have post-operative procedure.
Animal Treatments
The hydrogen was dissolved in physiological saline with use of a hydrogen generator for 12 h to prepare the HS. This procedure requires that it be performed at 4 °C in a 0.4 Megapascal (MPA) environment to ensure that the hydrogen concentration is >0.6 mmol and that the preparation is fresh with each use. LY294002 (ab120243, Abcam, Cambridge, MA), a PI3-kinase inhibitor, was diluted in phosphate-buffered saline (PBS). The in vivo dose of LY29400 was based upon results from preliminary experiments using different concentrations. The animals were randomly divided into 4 groups: 1) Sham + vehicle (PBS), 2) HI + vehicle (PBS), 3) HI + HS and 4) HI + HS +LY294002. The HS-saturated saline (5 mL/kg) was injected intraperitoneally at 24 h, 48 h and 72 h after HI injury according to their corresponding group assignment. In the HI+HS+LY294002 group, LY294002 (i.p. 5 mL/kg) was injected at 30 minutes before the HS injection. The procedures for these administrations were based upon methods as established from our previous research.12 The Sham and HI groups were injected identically with the same volume of the vehicle (PBS) relative to body weight. At one hour later after the last injection, the brains were removed for next experiment.
Measurement of Brain Water Content
The brains of these mice (N=5/group) were removed at 72 h after HI injury. The cerebral hemispheres were bisected and the remaining parts, such as cerebellum, medulla oblongata and brain stem were removed. The ipsilateral hemisphere was weighed and the wet weight recorded. Both ipsilateral and contralateral hemispheres from all mice were placed in a 60 °C oven for 24 h until dried, then quickly weighed to determine their dry weight. Weight ratios were then calculated to establish the degree of cerebral edema. Brain water contents were determined as based on the following calculation:12,13
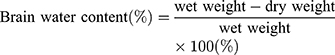
Measurement of Infarct Size
For infarct size determinations, brain tissue of the mice (N=4/group) was removed at 72 h after HI injury then placed in a −20 °C refrigerator for rapid cooling and freezing for approximately 30 min. Coronal sections were cut and divided into 4 pieces with each being approximately 1.0 mm. Brain slices were soaked in a prepared 2% concentration of TTC (Sigma-Aldrich) staining solution which was maintained in a 37 °C oven for approximately 20 min. Image-J image-processing software was used to calculate the infarction area. Quantitative calculations of cerebral infarction volumes were performed using the following calculation:12,14

Nissl Staining
The mice were anesthetized with isoflurane at 72 h after HI injury and perfused with 4% paraformaldehyde (pH=7.4). The brains (N=5/group) were harvested and fixed in 4% formaldehyde. At the beginning of this experiment, brain slices in the region containing the infarct lesion (between −1.60 and −2.00 mm from bregma) was chosen to undergo immunohistochemistry or immunofluorescence. All the slices of each group used in every independent experiment have the similar anatomical positions. The sections and incubated with 0.5% cresyl violet acetate (Sigma-Aldrich - St Louis, MO, USA) for 20 min. After washing with distilled water and dehydration with alcohol, the slide slices were treated with transparent xylene and then observed under a microscope. The surviving neurons in different regions of the brain (ipsilateral to HI) were counted in selected microscopic fields at ×200 magnification. Results are expressed as the number of surviving neuronal cells within each group relative to that of the Sham group within the different areas.10,13
Western Blot Analysis
The animals (N=3/group) were decapitated and removed the cerebral cortex ipsilateral to the HI insult at 72 h after HI injury. The tissue was thawed and homogenized in RIPA buffer containing additional protease inhibitors (100mM PMSF, #ST506-2, Beyotime, Shanghai, China) and phosphatase inhibitors (#04-906-845-001, Roche, Shanghai, China) to prevent protein degradation and ensure the stability of phosphorylated proteins and centrifuged at 13,800×g for 10 min. BCA Protein Assay Kits were used to quantity total protein concentrations. An appropriate amount of 5× SDS-PAGE protein loading buffer was added to the resultant supernatant and heated to 100 °C for 10 min to fully denature the proteins. After the sample was cooled to room temperature, it was loaded onto the SDS-PAGE gel for electrophoresis, typically using 80V constant voltage in the upper gel layer while 120 V was used when bromophenol blue entered the lower gel layer. The membrane was transferred to the PVDF membrane at a current of 300 mA for ≥1 h using a Bio-Rad standard wet membrane transfer device. After the membrane transfer was completed, there were 3 washing at 5 min each. The membranes were incubated for 1 h in 5% non-fat milk. Membranes were then incubated overnight at 4 °C with primary antibodies for AKT (1:1000, #9272S, Cell Signaling Technology, Danvers, MA, USA), p-AKT (1:1000, #9271S, Cell Signaling Technology), β-actin (1:1000, #TA-09, ZSGB, Peking, China). The secondary antibody was then added to the PVDF membrane and incubated at room temperature for 1 h. The chemiluminescence signal was generated with use of the ECL kit (Millipol, USA) and detected using the Tanon imaging system (Tanon-4600).
Immunohistochemical Imaging
At 3 days post-HI, mice from each group (N=4/group) were anesthetized and perfused with 4% paraformaldehyde (pH 7.4). Brains were collected and placed in the same fixative at 4°C for 24 h. Brain samples were sliced into 4 µm thick coronal sections. The sections between −1.60 and −2.00 mm from bregma were selected and assayed for immunohistochemistry imaging. Briefly, brain slices were incubated overnight at 4°C with antibodies directed against the ionized calcium binding adaptor molecule 1 (Iba-1) (1:100). The slices were then treated with secondary antibodies at room temperature for 30 min. Fluorescent microscopy (OLYMPUS-BX51) and the Magna Fire SP system was used to analyze microphotographs. Activated microglia scores were assigned as previously described (Table 1).15 The activated microglia was measured in three randomly selected microscopic fields at ×200 magnification. These fields initiated at the core region of the infarct (N=4 mice/group). The numbers of activated microglia within each brain were expressed as the average of three fields per section.
 |
Table 1 Semi-Quantitative Score for Iba-1 Staining |
Primary Microglia Culture
C57BL/6J (PND 1–2) neonatal mice were anesthetized with isoflurane. The brain tissues were removed and their meninges were completely stripped in pre-cooled Hank’s balanced salt solution (HBSS). Brain tissues were snipped and digested at 37°C for 30 min using trypsin containing 0.125% EDTA. The single cell suspension was obtained by slowly blowing the larger tissue mass and filtering with a 70 μm sieve. The cells suspension was centrifuged at 400 × g for 10 min to obtain precipitation, which was re-suspended with Dulbecco’s Modified Eagle Media (DMEM)/F12. Two weeks later, the cells were shaken on an orbital shaker for 2 h (250 rpm, 37°C) to obtain microglia. Microglia was cultured for 14 days before treatment. TLR agonists, such as LPS or amyloid β (Aβ), increase the phagocytic capacity of microglia and induce phosphatidylserine exposure, which devour viable neurons.16,17 In the present study, microglia was stimulated with LPS with/without HS, LY294002 for 24 h. The cells were randomly divided into four groups: 1) Sham+PBS, 2) Lipopolysaccharide (LPS, 500 ng/mL) + PBS, 3) LPS+ HS (10 μM) and 4) LPS+ HS (10 μM) and LY294002 (1 μM). LPS, from Sigma-Aldrich (L2630), were diluted by PBS.
TUNEL Staining
TUNEL staining was performed with the TUNEL kit (KeyGEN BioTECH, Jiangsu, China) instructions using the manufacturer’s instructions. In briefly, the sections (N=4/group) were incubated at 37°C in an appropriate concentration of protease k working solution for 15–20 min, and then the TdT enzyme reaction solution was added and incubated at 37°C in dark for 60 min. Streptavidin-tritc marker solution was then added to each sample and incubated at 37°C in dark for 30 min. Each section was randomly searched for 6 fields (20×), and the proportion of TUNEL-positive cells was expressed as the percentage of the total cells counted.
Phagocytosis Assay
The fluorescent zymosan A BioparticlesTM Alexa FlourTM (Invitrogen, Z23373) was performed in the phagocytosis assay of microglia in vitro. Primary microglia was used for phagocytosis assay before reaching 80% confluency. Cells were treated with or without LPS, HS, LY294002 for 22 h and then added zymosan fluorescent beads (75,000 particles/mL) for 2 h. Following blocking with 10% goat serum for 1 h, the cells were incubated with rabbit polyclonal anti-Iba1 (1:400, Wako, Japan), then with second antibody. DAPI staining showed nucleus. Images were obtained with fluorescent microscopy (OLYMPUS-BX51, Olympus Corporation, Japan). Phagocytic index (PI) or phagocytic activity was determined employing the equation:18
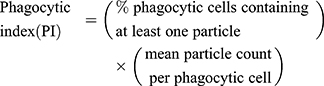
Grip Strength Test
As a means to assess the sensorimotor performance of the mice, a grip strength test was administered on the 4th (PND 11) and 6th (PND 13) days after HI injury. For this test the mice (N=11/group) were placed on a horizontal 20 × 20 cm 16-mesh wire maintained at approximately 10 cm above the surface of a tabletop and allowed 5–10 seconds to adapt to the wire mesh. The wire mesh was then slowly rotated from the horizontal position until the mouse was displaced and fell into a cushion. The angle of rotation at which the mouse fell from the wire mesh was recorded. Each mouse was tested three times and the average angle at which the mouse fell was calculated. The grip strength was then indicated by the value of the rotation angle at the time of falling from the mesh wire. The average age for a rodent to be able to grasp a horizontal screen is PND 8 with a range from PND 5–15.19
Y-Maze Test
Performance of the mice in a Y-maze was used as a means to evaluate the cognitive abilities of the mice. The test was performance at PND 35 (28 days after HI insult). Position and movement times of the mice were recorded using the SMART video tracking system, which can display movement of the mice in real time and record relevant data including time, trajectory and speed into the background database. The potential confounding effects of light and positional cues were controlled to ensure that the formation of memory was not affected by these factors.
The test consisted of a 5 min sample trial followed by a 5 min retrieval trial. Each mouse was tested once per test.20 For the training component, the main arm of the “Y” (the base of Y, zone 1) was blocked, forcing the mice to select one of the side arms (zones 2 or 3). The mice were then allowed to move freely within either of the two regions of the side arms for a 5 min period. After a 15 minute period, the partition of the closed arm was removed enabling access to all three arms. For the testing component, mice were placed at a defined starting point and the number of times the mice entered each area (defined as the limbs and body completely entering into the area) and time spent in each area within the 5 min test period were recorded. The number of arm entries and time spent in each arm was then calculated to provide an evaluation of the cognitive abilities of the mice:
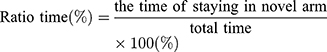

Statistical Analysis
The SPSS software program was used to perform the statistical analyses. All values presented were expressed as the mean ±standard deviation. Data were analyzed using a one-way ANOVA, following by Bonferroni corrections for multiple post hoc comparisons of means. A p <0.05 was required for results to be considered as statistically significant.
Results
HS Treatment Up-Regulates Akt Phosphorylation in the Cortex Ipsilateral to HI
As shown in Figure 1, levels of p-Akt at 72 h post-HI were decreased in the HI versus the Sham group [F(3,8)=29.835, p < 0.001; post hoc p < 0.001]. Treatment with HS increased these levels of p-Akt in the HI + HS group as compared with those in the HI group (post hoc p < 0.05). The combination of HS and LY294002 partially blocked the capacity for HS to promote Akt phosphorylation (post hoc p < 0.05).
 |
Figure 1 HS activates Akt following HI. Levels of Akt and phosphorylated Akt (p-Akt) in the ipsilateral cortex were assessed at 3 days after HI injury using Western blot. Quantification of the relative levels of p-Akt/Akt are presented. N=3/group. Values represent the mean ± SD, *p < 0.05, ***p < 0.001 according to ANOVA. |
Akt Inhibition Attenuates Neuroprotective Effects of HS on Edema and Infarct Volume Following HI
Consistent with our previous report,10 HI insult significantly increased brain water content (90.73 ± 1.04%) as compared with that observed in the Sham group (87.86 ± 1.31%) [F(3,16)=11.416, p < 0.001; post hoc p<0.001], while HS treatment significantly decreased this increase in brain water content by HI (80.29 ±0.71%, post hoc p<0.01) (Figure 2A). In addition, as shown with TTC staining, HS treatment also decreased infarct volume (23.21 ± 7.13%) [F(3,12)=34.100, p < 0.001; post hoc p<0.001] as compared with that obtained in the HI group (66.96 ± 11.24%). When HS was combined with LY294002 the protective effects of HS on edema (89.09 ±1.75%, post hoc p<0.01) and infarct volume (49.36 ± 13.81%, post hoc p<0.01) were partially blocked (Figure 2B).
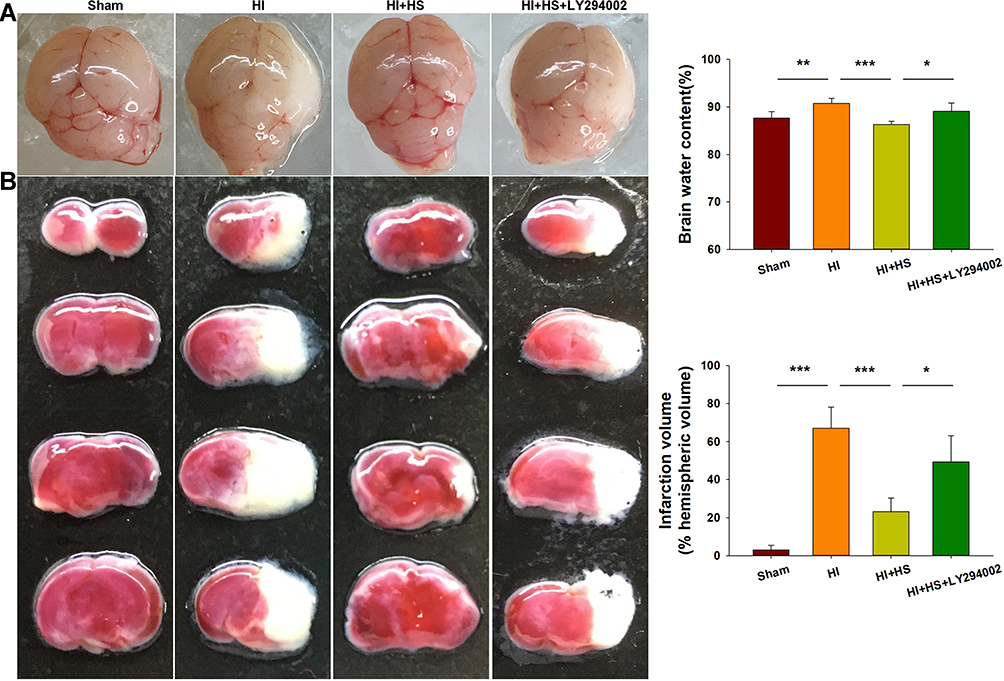 |
Figure 2 HS attenuates edema and infarct volume via the Akt pathway. (A) Representative images of brain edema at 3 days post-HI. Brain water contents within the ipsilateral hemispheres were determined using both dry and wet methods. N=5/group. (B) Brain slices were stained with TTC for quantification of infarct volumes. N=4/group. Values represent the mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001 according to ANOVA. |
Akt Inhibition Attenuates the Neuroprotective Effects of HS on Tissue Loss Following HI
Results from Nissl staining demonstrated that HI exposure significantly increased tissue loss in the prefrontal cortex [F(3,16)=35.521, p < 0.001; post hoc p<0.001], CA1 [F(3,16)=409.175, p < 0.001; post hoc p<0.001], CA3 [F(3,16)=77.240, p < 0.001; post hoc p<0.001] and DG [F(3,16)=48.952, p < 0.001; post hoc p<0.001] regions within the ipsilateral hemisphere as compared with that in the Sham group. HS treatment significantly decreased this tissue loss in these sites, prefrontal cortex (post hoc p<0.01), CA1 (post hoc p<0.05), CA3 (post hoc p<0.05) and DG (post hoc p<0.01), as compared with that observed in the HI group. The combination of HS and LY294002 partially blocked this protective effect of HS on tissue loss in the prefrontal cortex (post hoc p<0.01), CA (post hoc p<0.01), CA3 (post hoc p<0.01) and DG (post hoc p<0.05) regions (Figure 3).
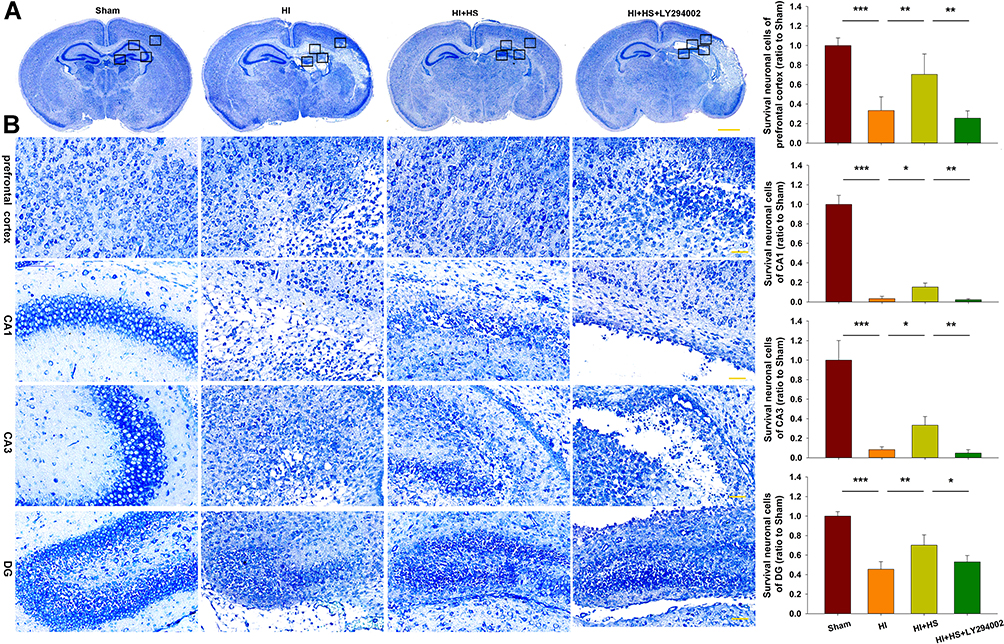 |
Figure 3 HS attenuates tissue loss via the Akt pathway. (A) Representative images of Nissl staining from each group. Scale bar = 1000 µm. (B) Magnified views of boxed regions in A showing neuronal cell loss. Scale bar = 50 µm. Results are expressed as the number of surviving neuronal cells within each group relative to that of the Sham group within the different areas. N=5/group. Values represent the mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001 according to ANOVA. |
Akt Inhibition Attenuates the Neuroprotective Effects of HS on Neuronal Apoptosis Following HI
Next, TUNEL staining was used to further assess the capacity for LY294002 to block the effects of HS on neuronal apoptosis after HI insult. HS treatment significantly decreased the number of apoptotic cells (11.06 ± 2.34%) [F(3,12)=23.537, p < 0.001; post hoc p<0.001] within the ipsilateral hemisphere as compared with that obtained in the HI group (42.15 ± 11.97%). This protective effect of HS on neuronal apoptosis was blocked by LY294002 (31.12 ± 8.44%) (post hoc p<0.05) (Figure 4).
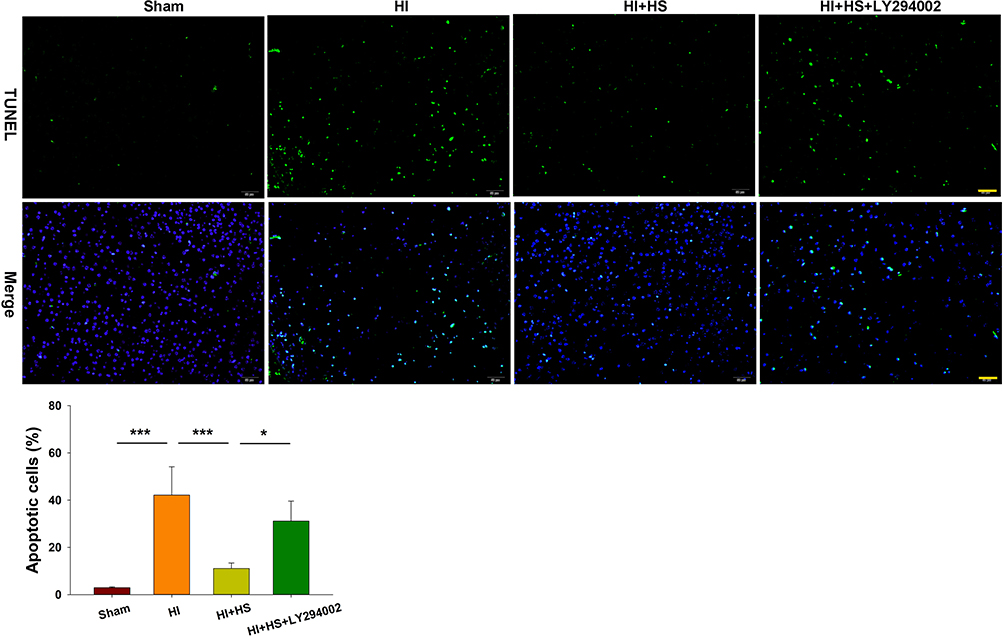 |
Figure 4 HS attenuates apoptosis via the Akt pathway. TUNEL staining (green) was used to investigate neuronal survival as determined at 3 days after HI exposure. Three randomly selected images (× 20) were captured from each section per animal. Scale bar = 50 µm. N=4/group. Values represent the mean ± SD, *p < 0.05, ***p < 0.001 according to ANOVA. |
Akt Inhibition Attenuates the Neuroprotective Effect of HS on Microglial Activation and Phagocytic Capacity Following HI
Following HI exposure, Iba-1+ cells in the ipsilateral hemisphere exhibited a rounded amoeboid-like appearance, while HS treatment restored the appearance of these cells to that of an extended and elongated morphology [F(3,12)=100.240, p < 0.001; post hoc p<0.001] (Figure 5).
 |
Figure 5 HS suppresses phagocytotic activation of microglia following HI insult via the Akt pathway. Immunofluorescent staining of Iba-1 (green) within the ipsilateral cortex was used to investigate microglial activation as determined at 3 days following HI insult. Scale bar = 50 μm. Quantification of Iba-1+ immunoreactivity score. Six randomly selected images (× 20) were captured from each section per animal. N=4/group. Values represent the mean ± SD, ***p < 0.001 according to ANOVA. |
Moreover, we tested for the phagocytotic activities of microglia with engulfment of E. coli opsonized FITC-labelled bioparticles. The addition of LPS (500 ng/mL) for 24 h increased the percent of phagocytic cells [F(3,12)=24.880, p < 0.001; post hoc p < 0.001], mean number of particles per cell [F(3,12)=15.952, p < 0.001; post hoc p < 0.01] and phagocytic index [F(3,12)=44.465, p < 0.001; post hoc p < 0.01] as compared to the control. These three parameters were all significantly decreased in response to HS treatment (post hoc p < 0.01, 0.01 and 0.001, respectively). These effects of HS on morphological changes in microglia and phagocytic capacity were all blocked by LY294002 (Figures 5 and 6).
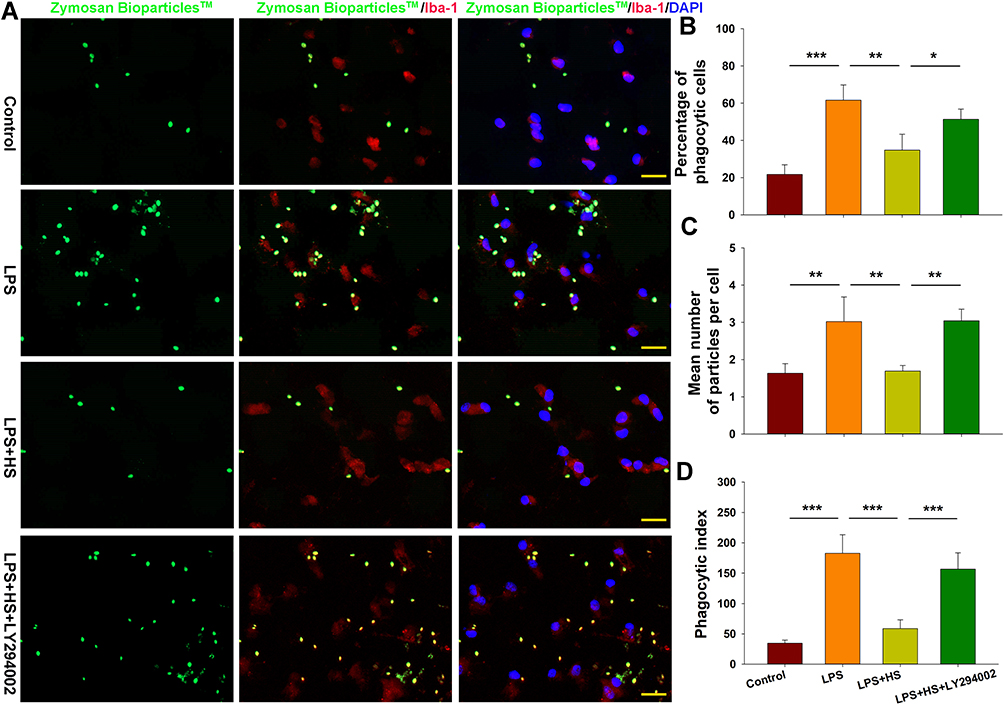 |
Figure 6 HS treatment suppresses phagocytotic activation of microglia following LPS stimulation via the Akt pathway. (A) Representative photographs of BioParticlesTM Fluorescent Particles after treatment with LPS for 24 h. (B) Quantification of percent of phagocytic cells. (C) Quantification of mean number of particles ingested per cell. (D) Quantification of phagocytic index. N=4/group. Four randomly selected images (× 20) were captured from each section. Values represent the mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001 according to ANOVA. |
Akt Inhibition Attenuates Improvements in Neurobehavioral Responses by HS Following HI
Significant reductions in grasp strength were observed in HI mice as determined on PND11 [F(3,38)=9.487, p < 0.001; post hoc p < 0.05] and PND13 [F(3,37)=17.429, p < 0.001; post hoc p < 0.001] when compared with that obtained in the Sham group. HS treatment reversed these reductions in grasp strength at PND 11 (post hoc p < 0.001) and PND 13 (post hoc p < 0.001) as compared with those obtained in the HI group (Figure 7A). LY294002 blocked this restorative effect of HS on grasp strength at PND 11 (post hoc p < 0.001) and PND 13 (post hoc p < 0.001) (Figure 7A).
 |
Figure 7 HS treatment improves neurobehavioral responses following HI via the Akt pathway. (A) Performance of mice in the grip strength test at PND11 indicating the average angle at which mice fell from the wire mesh for the Sham, HI, HI + HS and HI+HS+LY294002 groups (N = 11, 10, 10 and 11, respectively). (B) Performance of mice in the grip strength test at PND13 indicating the average angle at which mice fell from the wire mesh for the Sham, HI, HI + HS and HI+HS+LY294002 groups (N = 11, 10, 9 and 11, respectively). (C and D) Performance of the mice in the Y-maze test at PND 35 for mice in the Sham, HI, HI + HS and HI+HS+LY294002 groups (N = 8, 10, 8 and 8, respectively) indicating (C) Ratio time and (D) Ratio entry. (E) Relationship between the number of arm entries and the time spent within each arm (correlation coefficient = 0.560, p < 0.01; N = 34). Values represent the mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001 according to ANOVA. |
At PND 35, mice were tested in a Y-maze task to assess their working memory. HI exposure decreased Y-maze alternations [F(3,30)=11.896, p < 0.001; post hoc p < 0.01] as compared with that of the Sham group. HS treatment in HI mice increased their Y-maze alternations (post hoc p < 0.01) as compared with the HI group. A coadministration of LY294002 and HS reversed this effect of HS on Y-maze alternations (post hoc p < 0.01). HI exposure also decreased the total number of arm entries [F(3,30)=4.383, p < 0.05; post hoc p < 0.05] as compared to the Sham group (Figure 7B). Interestingly, HS treatment of HI mice had no effect on the total number of arm entries (post hoc p > 0.05). A significant positive correlation was present between the total number of arm entries and Y-maze alterations (correlation coefficient = 0.560, p < 0.05; N = 34) (Figure 7C–E).
Discussion
In this current study, we show that HS treatment activated Akt phosphorylation in response to HI injury in newborn mice. Suppression of this Akt activity following HI exposure attenuated HS’s neuroprotective capacity in HI mice, as indicated by increases in apoptosis, microglial activation and phagocytic activity.
Hydrogen can function as a novel, selective antioxidant capable of protecting tissue by anti-oxidation and anti-inflammation effects as shown in clinical and animal studies.5,21 Treatment of ischemic injury with hydrogen has been shown to suppress oxidative stress, proinflammatory responses and cell apoptosis in kidney, brain, liver, heart and intestines.22–26 Previous work within our laboratory has demonstrated that HS administration shifts microglia from a M1 to M2 polarization and diminishes synaptic loss after HI exposure in newborn mice through the promotion of AMPK phosphorylation.10 In line with these findings, infarct volume and apoptotic cell numbers were also decreased by HS treatment following HI exposure.
A number of pathophysiological events occur following neonatal HI, including energy failure, glutamate excitotoxicity, free radical generation, edema, and inflammation.27 Brain edema is generally classified into cytotoxic edema and vasogenic edema. Cytotoxic edema is defined as a cellular swelling wherein water moves from the interstitial to intracellular space. Vasogenic edema is characterized by the blood-brain barrier breakdown, resulting in circulating immune cells to infiltrate to the brain.28 Edema leads to further aggravate secondary brain from multiple mechanisms. HS could alleviate early brain injury via suppressing oxidative stress and brain edema after experimental subarachnoid hemorrhage in rabbits.29 Hydrogen inhalation exerted the neuroprotective effect on intracerebral hemorrhage in mice, evidence as decreasing brain edema and improved neurological outcomes.30 In accordance with the previous related reports, HS abates significantly the brain edema and brain infarct after HI exposure in the present study.
Here, we now show that the Akt pathway represents a critical component of HS’s neuroprotective ability. Akt, a downstream target of PI3K, is involved in the regulation of various neuronal functions, such as cell proliferation, apoptosis and survival.31 Akt phosphorylation can inactivate BCL-2 associated X (Bax) and suppress neuronal apoptosis after cerebral ischemia in rats and can attenuate the phosphorylation of pro-caspase-9 and caspase-9 to inhibit apoptotic activation.32,33 Results from previous studies have demonstrated the potential value of Akt activity in mediating the beneficial effects of hydrogen as shown in acute kidney injury, lung cancer, myocardial injury and traumatic brain injury.34–37 To investigate the mechanism underlying the neuroprotective effects of HS, we measured the ratio of p-Akt/Akt following HI insult. The results indicated that the ratio of p-Akt/Akt decreased significantly at 3 days after HI exposure. Administration of HS increased the ratio of p-Akt/Akt in the corresponding group compared with the control group at 3 days after HI exposure, associating with attenuating neuronal apoptosis. Taken together, these results suggest that HS attenuates HI brain damage by activating the Akt signaling pathway.
Microglia, the phagocytes of the CNS, play a role in the regulation of synaptic circuit remodeling and synaptic pruning in the healthy CNS.38 In response to brain injury, microglia are activated and display a number of remarkable characteristics associated with changes in morphology, migration and phagocytosis.39 For example, in response to ipsilateral ischemic stroke injury, microglia initially display increases in ramification, followed later by de-ramification and eventually increases in phagocytic activity.40 We observed that HI exposure induced phagocytic activity of microglia, from a mostly short and compact morphology to that of a phagocytotic shape, an effect which was further confirmed with primary microglia. And, suppressing this excessive phagocytosis with HS results in neuroprotective effects. Activated microglia via Toll-like receptor (TLR)-2 or TLR4 could result in neuronal loss by phagocytosing otherwise-viable neurons.41 TLR agonists, such as LPS or amyloid β (Aβ), increase the phagocytic capacity of microglia and induce phosphatidylserine exposure, which devour viable neurons.16,17 Blocking of microglial phagocytosis can prevent neuronal death and, in this way, exert therapeutic benefits for inflammatory brain pathologies. Based upon our current results, we propose that one effect of HI involves an activation of microglial phagocytosis which then leads to the death of otherwise-viable neurons. Confirmation of this hypothesis awaits further investigation.
However, this study had some limitations. First, the neuroprotective effect of HS was observed in many pathways, but only its role in Akt pathway was investigated in the present study. Second, although HS exerted neuroprotective effects via activating the Akt signaling pathway, the underlying mechanisms explaining how HS mediated Akt phosphorylation are still unclear. Moreover, the mechanism detailing how HS suppressed microglial phagocytosis has not been thoroughly explored either.
In conclusion, the results of the present study reveal that HS attenuates HI-induced apoptosis and phagocytosis, effects that appear to involve the Akt signaling pathway. These neuroprotective effects of HS thus contribute to a reduction in the neurobehavioral deficits resulting from HI insult in newborn mice.
Abbreviations
Akt, protein kinase B; CNS, central nervous system; DAPI, 4′,6′-diamidino-2-phenylindole dihydrochloride hydrate; DMEM, Dulbecco’s Modified Eagle Media; HBSS, Hank’s balanced salt solution; HI, hypoxia-ischemia; Iba-1, ionized calcium binding adapter molecule 1; IL, interleukin; LPS, lipopolysaccharide; MPA, Megapascal; PFA, paraformaldehyde; TTC, 2,3,5-triphenyltetrazolium chloride monohydrate.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors have no financial, personal, or other conflicts of interest to disclose for this work.
References
1. Concepcion KR, Zhang L. Corticosteroids and perinatal hypoxic-ischemic brain injury. Drug Discov Today. 2018;23(10):1718–1732. doi:10.1016/j.drudis.2018.05.019
2. Li B, Concepcion K, Meng X, Zhang L. Brain-immune interactions in perinatal hypoxic-ischemic brain injury. Prog Neurobiol. 2017;159:50–68. doi:10.1016/j.pneurobio.2017.10.006
3. Toro-Urrego N, Vesga-Jimenez DJ, Herrera MI, Luaces JP, Capani F. Neuroprotective role of hypothermia in hypoxic-ischemic brain injury: combined therapies using estrogen. Curr Neuropharmacol. 2019;17(9):874–890. doi:10.2174/1570159X17666181206101314
4. Le K, Chibaatar Daliv E, Wu S, et al. SIRT1-regulated HMGB1 release is partially involved in TLR4 signal transduction: a possible anti-neuroinflammatory mechanism of resveratrol in neonatal hypoxic-ischemic brain injury. Int Immunopharmacol. 2019;75:105779. doi:10.1016/j.intimp.2019.105779
5. Ohsawa I, Ishikawa M, Takahashi K, et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med. 2007;13(6):688–694. doi:10.1038/nm1577
6. Saitoh Y, Harata Y, Mizuhashi F, Nakajima M, Miwa N. Biological safety of neutral-pH hydrogen-enriched electrolyzed water upon mutagenicity, genotoxicity and subchronic oral toxicity. Toxicol Ind Health. 2010;26(4):203–216. doi:10.1177/0748233710362989
7. Li J, Hong Z, Liu H, et al. Hydrogen-rich saline promotes the recovery of renal function after ischemia/reperfusion injury in rats via anti-apoptosis and anti-inflammation. Front Pharmacol. 2016;7:106. doi:10.3389/fphar.2016.00106
8. Meng X, Chen H, Wang G, Yu Y, Xie K. Hydrogen-rich saline attenuates chemotherapy-induced ovarian injury via regulation of oxidative stress. Exp Ther Med. 2015;10(6):2277–2282. doi:10.3892/etm.2015.2787
9. Bai X, Liu S, Yuan L, et al. Hydrogen-rich saline mediates neuroprotection through the regulation of endoplasmic reticulum stress and autophagy under hypoxia-ischemia neonatal brain injury in mice. Brain Res. 2016;1646:410–417. doi:10.1016/j.brainres.2016.06.020
10. Chu X, Cao L, Yu Z, et al. Hydrogen-rich saline promotes microglia M2 polarization and complement-mediated synapse loss to restore behavioral deficits following hypoxia-ischemic in neonatal mice via AMPK activation. J Neuroinflammation. 2019;16(1):104. doi:10.1186/s12974-019-1488-2
11. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169(3):381–405. doi:10.1016/j.cell.2017.04.001
12. Liu S, Xin D, Wang L, et al. Therapeutic effects of L-Cysteine in newborn mice subjected to hypoxia-ischemia brain injury via the CBS/H2S system: role of oxidative stress and endoplasmic reticulum stress. Redox Biol. 2017;13:528–540. doi:10.1016/j.redox.2017.06.007
13. Xin D, Chu X, Bai X, et al. l-Cysteine suppresses hypoxia-ischemia injury in neonatal mice by reducing glial activation, promoting autophagic flux and mediating synaptic modification via H2S formation. Brain Behav Immun. 2018;73:222–234. doi:10.1016/j.bbi.2018.05.007
14. O’Donnell ME, Tran L, Lam TI, Liu XB, Anderson SE. Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab. 2004;24(9):1046–1056. doi:10.1097/01.WCB.0000130867.32663.90
15. Thei L, Rocha-Ferreira E, Peebles D, Raivich G, Hristova M. Extracellular signal-regulated kinase 2 has duality in function between neuronal and astrocyte expression following neonatal hypoxic-ischaemic cerebral injury. J Physiol. 2018;596(23):6043–6062. doi:10.1113/JP275649
16. Fricker M, Neher JJ, Zhao JW, Thery C, Tolkovsky AM, Brown GC. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J Neurosci. 2012;32(8):2657–2666. doi:10.1523/JNEUROSCI.4837-11.2012
17. Neniskyte U, Neher JJ, Brown GC. Neuronal death induced by nanomolar amyloid beta is mediated by primary phagocytosis of neurons by microglia. J Biol Chem. 2011;286(46):39904–39913. doi:10.1074/jbc.M111.267583
18. Fuentes AL, Millis L, Vapenik J, Sigola L. Lipopolysaccharide-mediated enhancement of zymosan phagocytosis by RAW 264.7 macrophages is independent of opsonins, laminarin, mannan, and complement receptor 3. J Surg Res. 2014;189(2):304–312. doi:10.1016/j.jss.2014.03.024
19. HC J. Assessment of developmental milestones in rodents. J Curr Protoc Essent Lab Tech. 2004;8.
20. Anisman H. Dissociation of disinhibitory effects of scopolamine: strain and task factors. Pharmacol Biochem Behav. 1975;3(4):613–618. doi:10.1016/0091-3057(75)90182-3
21. Hong Y, Chen S, Zhang JM. Hydrogen as a selective antioxidant: a review of clinical and experimental studies. J Int Med Res. 2010;38(6):1893–1903. doi:10.1177/147323001003800602
22. Uto K, Sakamoto S, Que W, et al. Hydrogen-rich solution attenuates cold ischemia-reperfusion injury in rat liver transplantation. BMC Gastroenterol. 2019;19(1):25. doi:10.1186/s12876-019-0939-7
23. Li H, Luo Y, Yang P, Liu J. Hydrogen as a complementary therapy against ischemic stroke: a review of the evidence. J Neurol Sci. 2019;396:240–246. doi:10.1016/j.jns.2018.11.004
24. Li L, Liu T, Liu L, et al. Effect of hydrogen-rich water on the Nrf2/ARE signaling pathway in rats with myocardial ischemia-reperfusion injury. J Bioenerg Biomembr. 2019;51(6):393–402. doi:10.1007/s10863-019-09814-7
25. Gao G, Wang W, Tadagavadi RK, et al. TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J Clin Invest. 2014;124(11):4989–5001. doi:10.1172/JCI76042
26. Shigeta T, Sakamoto S, Li XK, et al. Luminal injection of hydrogen-rich solution attenuates intestinal ischemia-reperfusion injury in rats. Transplantation. 2015;99(3):500–507. doi:10.1097/TP.0000000000000510
27. Millar LJ, Shi L, Hoerder-Suabedissen A, Molnar Z. Neonatal hypoxia ischaemia: mechanisms, models, and therapeutic challenges. Front Cell Neurosci. 2017;11:78. doi:10.3389/fncel.2017.00078
28. Kimelberg HK. Current concepts of brain edema. Review of laboratory investigations. J Neurosurg. 1995;83(6):1051–1059. doi:10.3171/jns.1995.83.6.1051
29. Zhuang Z, Zhou ML, You WC, et al. Hydrogen-rich saline alleviates early brain injury via reducing oxidative stress and brain edema following experimental subarachnoid hemorrhage in rabbits. BMC Neurosci. 2012;13:47. doi:10.1186/1471-2202-13-47
30. Manaenko A, Lekic T, Ma Q, Ostrowski RP, Zhang JH, Tang J. Hydrogen inhalation is neuroprotective and improves functional outcomes in mice after intracerebral hemorrhage. Acta Neurochir Suppl. 2011;111:179–183. doi:10.1007/978-3-7091-0693-8_30
31. Li Y, Ma X, Wang Y, Li G. miR-489 inhibits proliferation, cell cycle progression and induces apoptosis of glioma cells via targeting SPIN1-mediated PI3K/AKT pathway. Biomed Pharmacother. 2017;93:435–443. doi:10.1016/j.biopha.2017.06.058
32. Kamada H, Nito C, Endo H, Chan PH. Bad as a converging signaling molecule between survival PI3-K/Akt and death JNK in neurons after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2007;27(3):521–533. doi:10.1038/sj.jcbfm.9600367
33. Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282(5392):1318–1321. doi:10.1126/science.282.5392.1318
34. Guo SX, Fang Q, You CG, et al. Effects of hydrogen-rich saline on early acute kidney injury in severely burned rats by suppressing oxidative stress induced apoptosis and inflammation. J Transl Med. 2015;13:183. doi:10.1186/s12967-015-0548-3
35. Jiang Y, Liu G, Zhang L, et al. Therapeutic efficacy of hydrogen-rich saline alone and in combination with PI3K inhibitor in nonsmall cell lung cancer. Mol Med Rep. 2018;18(2):2182–2190. doi:10.3892/mmr.2018.9168
36. Song D, Liu X, Diao Y, et al. Hydrogen-rich solution against myocardial injury and aquaporin expression via the PI3K/Akt signaling pathway during cardiopulmonary bypass in rats. Mol Med Rep. 2018;18(2):1925–1938. doi:10.3892/mmr.2018.9198
37. Wang L, Yin Z, Wang F, et al. Hydrogen exerts neuroprotection by activation of the miR-21/PI3K/AKT/GSK-3beta pathway in an in vitro model of traumatic brain injury. J Cell Mol Med. 2020;24(7):4061–4071. doi:10.1111/jcmm.15051
38. Lehrman EK, Wilton DK, Litvina EY, et al. CD47 protects synapses from excess microglia-mediated pruning during development. Neuron. 2018;100(1):120–134e6. doi:10.1016/j.neuron.2018.09.017
39. Sun Y, Hei M, Fang Z, Tang Z, Wang B, Hu N. High-mobility group box 1 contributes to cerebral cortex injury in a neonatal hypoxic-ischemic rat model by regulating the phenotypic polarization of microglia. Front Cell Neurosci. 2019;13:506. doi:10.3389/fncel.2019.00506
40. Morrison HW, Filosa JA. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation. 2013;10:4. doi:10.1186/1742-2094-10-4
41. Neher JJ, Neniskyte U, Hornik T, Brown GC. Inhibition of UDP/P2Y6 purinergic signaling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia. 2014;62(9):1463–1475. doi:10.1002/glia.22693
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.