")
Back to Journals » Infection and Drug Resistance » Volume 17
Hybrid Sequencing-Based Genomic Analysis of Klebsiella pneumoniae from Urinary Tract Infections Among Inpatients at a Tertiary Hospital in Beijing
Authors Zhang W, Wang Y, Wang K, Li J, Liu J, Li S, Song L, Liao C, Yang X, Li P, Liu X
Received 15 November 2023
Accepted for publication 26 March 2024
Published 12 April 2024 Volume 2024:17 Pages 1447—1457
DOI https://doi.org/10.2147/IDR.S448253
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Sandip Patil
Wei Zhang,1,* Yufei Wang,1,* Kaiying Wang,2,* Jinhui Li,2 Jia Liu,1 Shulei Li,1 Lijie Song,1 Chunchen Liao,2 Xiaoli Yang,1 Peng Li,2 Xiong Liu2
1Department of Clinical Laboratory, The Third Medical Center, PLA General Hospital, Beijing, 100039, China; 2Chinese PLA Center for Disease Control and Prevention, Beijing, 100071, China
*These authors contribute equally to this work
Correspondence: Peng Li; Xiong Liu, Chinese PLA Center for Disease Control and Prevention, Beijing, 100071, China, Email [email protected]; [email protected]
Background: Urinary tract infection (UTI) associated with Klebsiella pneumoniae poses a serious threat for inpatients. This study aimed to describe the genomic characteristics of K. pneumoniae causing UTI in a tertiary-care hospital in Beijing, China.
Methods: A total of 20 K. pneumoniae strains collected from 2020 to 2021 were performed whole-genome sequencing. The Antibiotic susceptibility of 19 common antimicrobial agents was tested against all strains. The multi-locus sequence types (MLSTs) and serotypes were determined from the WGS data. De novo assemblies were used to identify resistance and virulence genes. The presence and characteristics of the plasmids were detected using hybrid assembly of long and short-read data.
Results: These K. pneumoniae strains were clustered into nine sequence types (STs) and twelve K-serotypes. All the carbapenem-resistant K. pneumoniae (CRKP) strains acquired carbapenemase blaKPC-2 (n=7). Two CRKP strains exhibited increased resistance to Polymyxin B with MIC ≥ 4 mg/L due to insertion of an IS5-like sequence in the mgrB gene, and they were also involved in a transmission event in Intensive Care Unit. Long-read assemblies identified many plasmids co-carrying multiple replicons. Acquisition of a new IncM2_1 type blaCTX-M-3 positive plasmid was observed after transfer from ICU to neurovascular surgery by comparing the two strains collected from the same patient.
Conclusion: K. pneumoniae is a significant pathogen responsible for urinary tract infections. The ST11-KL47 strain, prevalent at our hospital, exhibits a combination of high drug resistance and hypervirulence. It is imperative to enhance ongoing genomic surveillance of urinary tract infection-causing pathogens.
Keywords: Antimicrobial resistance, Klebsiella pneumoniae, urinary tract infections
Introduction
Urinary tract infection (UTI) commonly denotes a bacterial infection that originates in the region extending from the urethral opening to the perirenal fascia.1 It is one of the most prevalent types of infections in hospital, with an estimated 150 million patients worldwide affected every year.2 The diagnosis of urinary tract infection is usually based on symptoms and positive urine test or culture.3 In China between 2006 and 2016, cases of urinary tract infections accounted for 11.65% of all healthcare-related infections,4 and in the past three decades, there has been a notable surge in elderly women developing such infections.5
K. pneumoniae is a significant causative agent of UTI, which can result in high rates of illness and death.6,7 Due to the high empiric use of antibiotics for the treatment of UTI, the antibiotic resistance of K. pneumoniae increased significantly worldwide.8 In China, resistance rates to K. pneumoniae UTI isolates have risen for all tested antimicrobials, including carbapenems. The most severe cases were found in medical ICUs, where resistance rates have increased to 50–60% for amikacin, ciprofloxacin, imipenem, and ertapenem.9 Colistin is regarded as a last-resort agent to combat infections caused by multidrug-resistant bacteria, however reports of colistin-resistant K. pneumoniae are increasing in recent years.10
The isolation rate of K. pneumoniae in UTI exhibits regional variations, indicating a degree of geographical disparity. Over the period from 2009 to 2017, an extensive surveillance study at a Beijing hospital reported that K. pneumoniae accounted for 9.6% of all pathogens isolated from UTI cases,11 positioning it as the second most prevalent Gram-negative bacterium just after Escherichia coli. Comparable findings were also documented in another hospital within Beijing, a notably elevated isolation rate of K. pneumoniae was observed among elderly patients admitted through the emergency department, reaching up to 21% prevalence over the period from 2009 to 2020.12
Genomic surveillance is increasingly pivotal in tracking and managing the spread of pathogenic bacteria. As yet, no genomic research on UTIs caused by K. pneumoniae within Beijing hospitals has been published to our knowledge. In this study, we aim to characterize the genomic traits of K. pneumoniae strains isolated from UTI cases at our facility. The hospital where this study was conducted has implemented robust infection control measures, yet understanding the genomic epidemiology of K. pneumoniae circulating in the local population is crucial for refining these efforts.
Materials and Methods
Setting
This study was conducted in The Third Medical Center of People’s Liberation Army (PLA) General Hospital. It is a 900-bed tertiary general hospital with an estimated of 800 admissions per year in Beijing China.
Sample Collection and Identification
The urine culture results of hospitalized patients in all departments in our hospital from January 2020 to December 2021 were reviewed retrospectively, all samples were collected as part of routine patient care and processed according to standard operating procedures as described previously.12 In 2020, a total of 862 urine samples were collected with 228 (26.4%) yielding positive bacterial cultures, including 34 (14.9% of positives) identified as Klebsiella species. In 2021, 1929 urine samples were collected, with 371 (19.2%) showing positive results; among these, 45 (12.1% of positives) were identified as Klebsiella species.
Although a total of 79 strains were identified as Klebsiella species from urine samples collected between 2020 and 2021, only 21 of these strains were isolated from inpatients exhibiting symptoms of UTI and were subsequently subjected to whole-genome sequencing. One strain was subsequently excluded after being re-identified as Klebsiella quasipneumoniae based on the results of whole-genome sequencing, the remaining 20 strains confirmed as K. pneumoniae were subjected to further analysis.
The 20 confirmed K. pneumoniae strains were isolated from 16 inpatients across six different departments including Urology Surgery, Geriatric ward, Intensive care unit, Endocrinology ward, Neurovascular surgery and Obstetrics and gynecology ward. All K. pneumoniae strains used in this study were grown at 37°C for 18–24 h on Luria-Bertani (LB) agar plates.
Antimicrobial Susceptibility Testing
Species identification and antimicrobial susceptibility were conducted using the VITEK2 compact system (BioMerieux, France) as described previously.13 The AST-N334 card was used for antimicrobial susceptibility testing. The list of tested antimicrobial agents includes the following: Meropenem, Imipenem, Gentamicin, Amikacin, Levofloxacin, Ciprofloxacin, Nitrofurantoin, Ampicillin/Sulbactam, Piperacillin/clavulanic acid, Ticarcillin/Clavulanic acid, Cefoxitin, Cefepime, Ceftriaxone, Cefuroxime, Ceftazidime, Cefoperazone/Sulbactam, Minocycline, Trimethoprim/Sulfamethoxazole. The broth microdilution method was used to determine the minimum inhibitory concentration (MIC) values of Polymyxin B. Quality control was performed with both a colistin susceptible strain (E. coli ATCC 25922) and the colistin resistant (E. coli NCTC 13846). The susceptibility breakpoint was interpreted in accordance with the Clinical and Laboratory Standards Institute (CLSI-M100-2021).14
Whole Genome Sequencing
Whole-genome sequencing was performed as described previously.13 Briefly, genomic DNA was extracted from bacterial cultures using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. DNA concentration and purity were measured using Qubit 4.0 (Thermo Fisher Scientific). Sequencing libraries were generated using NEB Next Ultra DNA Library Prep Kit for Illumina (New England Biolabs) following the manufacturer’s recommendations. DNA libraries were constructed with 150-bp paired-end fragments and sequenced on the HiSeq 2500 sequencer at Novogene Company (Beijing, China). The nanopore sequencing library was prepared using the SQK-RAD004 rapid sequencing kit (Oxford Nanotechnology, UK) and sequenced on MinION Mk1B with R9.4 flowcell in our lab.
Bioinformatics Analysis
Filtered reads were obtained by using the FastQC v0.11.8 software,15 adapters and low-quality reads were removed and filtered out using Trimmomatic v0.39.16 Assembly of sequencing reads was carried out with Unicycler v0.4.817 in the normal mode. The assembly was annotated by Prokka v1.12.18
MLST and capsular locus determination were carried out by analyzing each genome with Kleborate v2.2.0.19 Unknown MLSTs were verified by stringMLST v0.6.3.20 Novel alleles of ST profiles were submitted to BIGSdb.21 Resistance genes, virulence genes, and plasmids were identified with Abricate v0.8 (accessible at https://github.com/tseemann/abricate) using resfinder, vfdb and plasmidfinder database respectively. Plasmid images were produced in CGView Comparison Tool v1.0.3.22
The K. pneumoniae strain 16HN-263 (NZ_CP045263.1) was selected as the best-matching chromosomal reference by the genomic distance estimation tool Mash v2.3.23 Single nucleotide polymorphisms (SNPs) were identified using Snippy v4.6.0 software (accessible at https://github.com/tseemann/snippy). The recombination regions were identified and excluded by using ClonalFrameML v1.1224 with default parameters, except that the “output_filtered” and “embranch” option were set to “true”. SNPs occurring outside the putative genomic recombination regions are identified and integrated with clinical data to infer transmission relationships among the collected strains. A maximum likelihood phylogenetic tree based on the non-recombination alignment was constructed using Raxml-ng v1.0.325 implementing with 1000 bootstrap replicates, GTR+I+G4 was selected as the best evolutionary model by using Modeltest-ng v0.1.7.26 The phylogeny was midpoint rooted and visualized using Figtree v1.4.4 (accessible at http://tree.bio.ed.ac.uk/software/Figtree/). All analyses were performed using default software parameters unless otherwise specified.
Statistical Analyses
Software R v4.1.1 and RStudio Desktop 2023.03.1 were used for statistical analysis. Categorical variables were expressed as percentages and analyzed by using χ2. p< 0.05 was considered statistically significant.
Ethical Statement
Due to the retrospective nature of the study, the need of informed consent was waived by the institutional ethics committees of The Third Medical Center of PLA General Hospital (Beijing, China). The study was also supervised by Chinese PLA Center for Disease Control and Prevention. We confirmed that all methods were performed in accordance with the relevant regulations and guidelines outlined in the Declaration of Helsinki.
Data Availability
This Whole-Genome Shotgun project has been deposited at GenBank under accession numbers from SAMN27593772 to SAMN27593792, BioProject ID: PRJNA826970.
Results
General Features of the Patients and Strains
Following a retrospective examination of urine bacterial culture results from inpatients over the course of 2020 to 2021, a selection of 20 K. pneumoniae strains (Table 1) were identified as originating from inpatients presenting with symptoms of UTI. These strains were isolated from 16 inpatients across six different wards but mainly from the Urology Surgery (35.0%, 7/20), Geriatric ward (35.0%, 7/20) and Intensive Care Unit (15.0%, 3/20). One inpatient (Pa05) had three strains, two inpatients (Pa01 and Pa08) had two strains, and the other inpatients each had one strain. The mean age of these inpatients was 67.7 years (range, 22–92 years), except for the inpatient Pa01, all other inpatients were over 45 years old. Among the 16 inpatients, 11 (68.8%) inpatients were males, and 5 (31.2%) were females. Two (Pa05 and Pa08) of the 16 inpatients were admitted to the hospital twice and the average length of stays was 30.5 days for all the 18 hospitalization. Apart from 2 inpatients (Pa01 and Pa10), all others have used catheters during hospitalization.
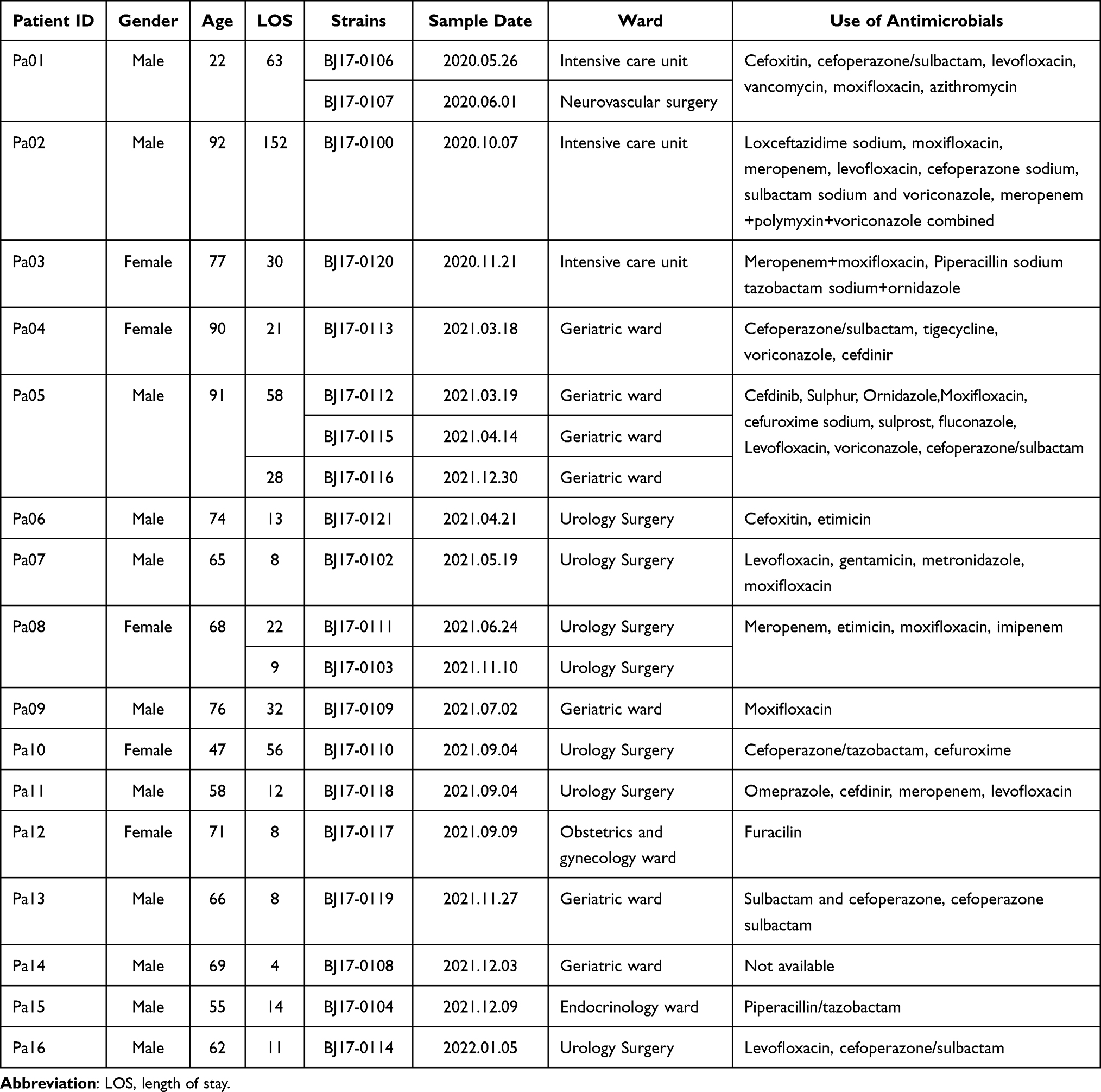 |
Table 1 Characteristics of the 16 Patients and 20 K. Pneumoniae Strains |
Antimicrobial Resistance Phenotype
The Antibiotic susceptibility of 19 common antimicrobial agents was tested against all strains (Supplementary Table 1). In terms of carbapenem antibiotics, 35.0% strains (7/20) exhibited resistance to both imipenem and meropenem with MICs ≥16 mg/L, including all the three strains isolated from Intensive Care Unit. In terms of Amino glycosides, 50.0% strains (10/20) exhibited resistance to Gentamicin, and 45.0% (9/20) strains were resistant to Amikacin. In terms of Quinolone antibiotics, 80.0% strains (16/20) were resistant to Levofloxacin, and 85.0% strains (17/20) were resistant to Ciprofloxacin. 75.0% strains (15/20) were resistant to Nitrofurantoin, which was widely used during urinary tract infection. In addition, two strains (10.0%, 2/20) isolated from Intensive Care Unit exhibited resistance to Polymyxin B with MIC ≥ 4 mg/L.
Distribution of Multilocus Sequence Typing Types and Serotypes
All reads of the 20 K. pneumoniae strains were de novo assembled (Supplementary Table 2), the average of genomic size was 5.70 Mb, and the median of GC % content was 57.0%. The multi-locus sequence types (MLSTs) and serotypes were determined from the assemblies (Table 2). Twenty K. pneumoniae strains belonged to nine sequence types, including ST11(10), ST1(1), ST23(1), ST65(1), ST147(1), ST628(1), ST1076(1) and two novel MLST types. Three strains were identified as a novel sequence type ST6902, which was closest to the known ST147 with only one mutation in the tonB-38 allele. One strain belonged to another novel sequence type ST6903, which had only one mutation in the phoE-9 allele with ST6903. The analysis revealed there exists five O-serotypes and twelve K-serotypes, and the most prevalent serogroup of O and K locus was O101 (45.0%, 9/20) and KL47 (30.0%, 6/20), respectively.
 |
Table 2 MLST, Serotypes, Virulence Genes, Replicon Types and Numbers of 20 K. Pneumoniae Genomes |
Resistance and Virulence Gene Profiles
De novo assemblies were screened for virulence (Table 2) and resistance genes (Supplementary Table 3). Seven ST11 strains carried the blaKPC-2 gene (35.0%, 7/20), which was consistent with the antibiotic susceptibility test results of imipenem and meropenem (Supplementary Table 1). Thirteen strains (65.0%, 13/20) were CTX-M positive, one ST11 strain co-carried blaCTX-M-65 and blaCTX-M-3, five ST11 strains carried blaCTX-M-65, while seven other ST strains carried blaCTX-M-14 (4), blaCTX-M-27 (2) and blaCTX-M-15 (1). Two ST11 strains isolated from Intensive Care Unit had the MICs of Polymyxin B≥ 4 mg/L, and genetic analysis further revealed an insertion of an IS5-like sequence in the mgrB gene, which were associated with colistin resistance.27
Seven ST11 strains harboured a set of virulence genes including aerobactin (iuc1), yerseniabactin (ybt9; ICEKp3) and rmpA2, six of them belonged to KL47 (85.7%, 6/7) and one belonged to KL64 (1/7, 14.3%). One strain belonged to ST65-KL2-O1/O2v2 harbored a different set of virulence genes including aerobactin (iuc1), yerseniabactin (ybt17; ICEKp10), Clb3, Iro1 and rmpA2. Six ST11-KL47-OL101 strains, including the two strain with elevated resistance to Polymyxin B, co-harbored virulence genes (iuc1, ybt9 and rmpA2) and carbapenemase gene (blaKPC-2).
Phylogenetic Analysis of Klebsiella Pneumoniae Strains
Reads mapping to K. pneumoniae reference genome NZ_CP045263.1 showed an average of 92.5% coverage among all 20 genomes (Supplementary Table 2). A reference-based whole-genome alignment was constructed, from which 103,894 SNPs were identified. Putative recombination loci were further detected and removed, the final 4,289,862bp recombination-free alignment consisted of 49,483 variable SNP sites among the K. pneumoniae strains. The pairwise disparity between these strains ranges from 3 to 17,887 SNPs (Figure 1). This recombination-free SNP variation across the whole genome was used to construct a maximum likelihood phylogenetic tree. Interestingly, we found 4 groups of strains within which the pair-wise SNPs distances were obviously less than other strains. Three groups of strains were isolated from three patients, respectively, in addition, one group of strains were isolated from two different patients whom were both admitted in the Intensive Care Unit, suggesting a transmission event.
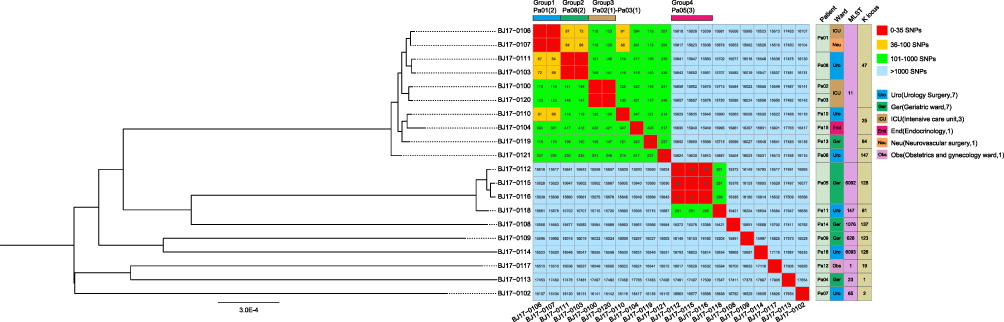 |
Figure 1 Phylogenetic analysis of Klebsiella pneumoniae strains A maximum-likelihood phylogenetic tree based on the recombination-free alignment of 20 Klebsiella pneumoniae strains was constructed, the tree was mid-point rooted. The single-nucleotide polymorphisms (SNPs) numbers between each K. pneumoniae strain, 4 groups of strains (red) were observed within which the pair-wise SNPs distances were less than 35. |
Plasmid Carriage Profile
The presence and characteristics of the plasmids were detected using hybrid assembly of long and short-read data. With exception of the ST23-KL1 strain BJ17-0113, all other K. pneumoniae strains have at least one plasmid replicon. An average of 4.3±2.7 plasmid replicons per strain was observed. Among all the strains, 11 ColRNAI_1 and 9 IncFII (pHN7A8) _1 plasmid replicons were identified, and 90.9% (10/11) and 100% (9/9) of which were found in ST11 strains, respectively. One ST147-KL81 strain BJ17-0118 had the largest plasmid carriage with 8 different plasmid replicons. Long-read assemblies revealed that multiple replicons coexist in the same plasmid. For example, IncR_1 and IncFII(pHN7A8) _1 plasmid replicons were located within the same plasmid sequence in three strains. Compared with strain BJ17-0106 (Figure 2), BJ17-0107 isolated from the same patient Pa01 had acquired a IncM2_1 plasmid (pBJ17-0107_1). However, no difference in the antimicrobial resistance phenotype were observed between strains BJ17-0106 and BJ17-0107. The plasmid pBJ17-0107_1 had high similarity (95.1%) to Enterobacter hormaechei strain C15 plasmid pC15_002 (NZ_CP042490.1) from Australia, but had acquired two additional resistance genes (aac (3)-IId and blaCTX-M-3) mediated by ISEcp1 and IS26 family transposases (Figure 2).
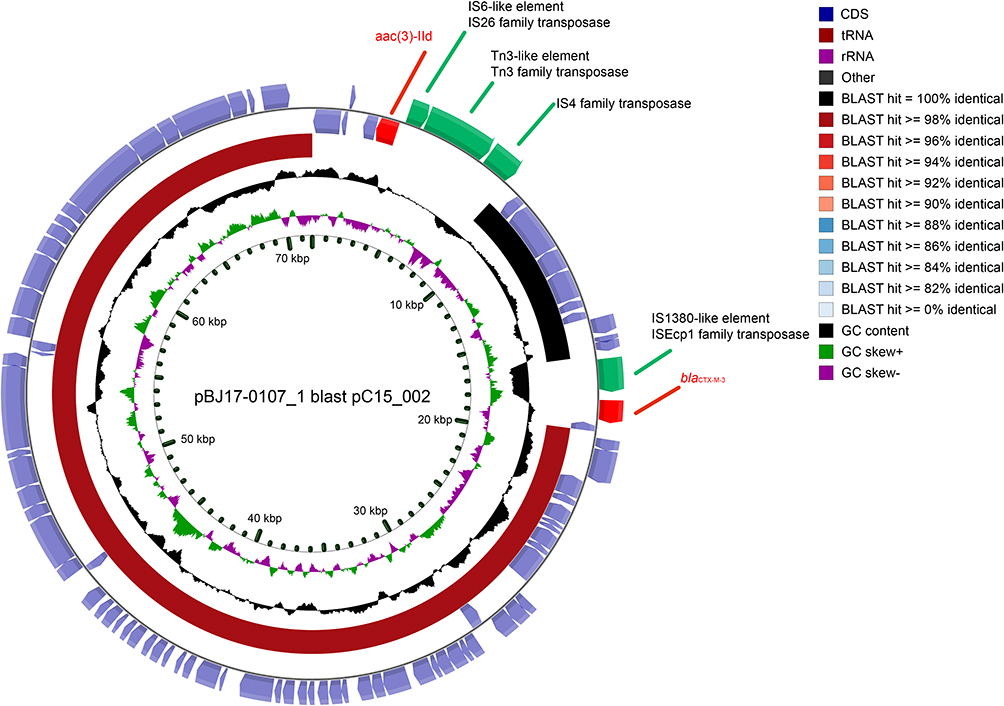 |
Figure 2 CGView Comparison between two plasmids. The newly acquired plasmid from BJ17-0107 (outer ring) had high similarity to a previously plasmid pC15_002 (inner ring) from Australia. Arrows are proportional to the lengths of the genes and oriented in the direction of transcription, two resistance genes were marked as red, four transposase were marked as yellow, other genes were marked blue. Tracks shown are (from inner to outer): GC skew (G−C/G+ (C), G+C content. |
Discussion
In this study, we focused on Urinary tract infection caused by K. pneumoniae among inpatients at a tertiary hospital in Beijing and performed a high-resolution genomic analysis using short- and long-read sequencing. Traditionally, UTIs have been regarded as a female-specific disease due to anatomical factors such as shorter urethral length, reduced distance from the anus to the urethral meatus, and the permissive nature of the vaginal and perineal environments for microbial colonization.28 We found that there were more than twice as many male patients (n=11) as female patients (n=5), in addition, the average age was nearly 70 years old, indicating that elderly men are also at high risk of UTI. We found that 87.5% (14/16) patients had used catheters during hospitalization, the use of indwelling urinary catheters is widely recognized as a major predisposing factor for urinary tract infection,29 healthy monitoring of these patients should be strengthened, especially regarding the insertion, maintenance, and timely removal of urinary catheters.
We identified that 35% strains were resistant to imipenem and meropenem, all of which belonged to ST1130 and carried the blaKPC-2 gene. Whereas in a study conducted in Portuguese, most carbapenem-resistant strains detected were found to harbor the blaKPC-3 gene within the ST14 lineage highlighting the geographical differences.31 Two carbapenem-resistant strains isolated from ICU showed increased MICs to Polymyxin B due to insertion of an IS5-like sequence in the mgrB gene which encoding a negative regulator of the PhoQ/PhoP signaling system.27 Polymyxin B is one of the few agents that retain activity against Carbapenem-resistant K. pneumoniae,32 raising the great concern of more strict infection control in ICU.
A total of 45% (9/20) of the strains were identified as hypervirulent K. pneumoniae, based on the presence of virulence genes, which was significant higher (p<0.01) than that found in a similar study in Changsha, China, which report a rate 24.8% (30/121).33 Although hypervirulent strains in both hospitals predominantly belonged to ST11, with prevalence rates of 77.8% and 56.7% respectively, there was a clear disparity in the distribution of K serotypes. Among our 9 hypervirulent strains, 3 were classified as K1, K2, and K64 respectively, while the remaining 6 strains were all belong to the K47 serotype (66.7%, 6/9). In contrast, at the hospital in Changsha, a notably different pattern emerged where 17 strains were identified as K64 (56.7%), 5 were K2, 1 was K1, and 7 other K serotypes strains, no K47 strains were identified. There is mounting evidence suggesting that KL64 has evolved from KL47 via genomic recombination events.34 The current study will provide a important basis for an in-depth examination of the adaptive genomic changes that take place during the progression from KL47 to KL64.
The utilization of WGS data permitted us to gain further insight into potential transmissions of these strains. In all six wards, two or more patients were found in three specific wards: the Intensive Care Unit (ICU), Geriatrics ward and Urology Surgery. Notably, only found within the ICU, two patients (Pa01 and Pa02) carried strains of the same sequence type (ST) and serotype, with a difference of 5 single nucleotide polymorphism (SNP) sites between the strains isolated from two distinct patients. The overlapping admission and hospitalization date strongly supported a transmission event or a shared source of infection between these two patients in ICU. However, in the Geriatrics ward, where 5 patients (Pa04, Pa05, Pa09, Pa13 and Pa14) were collected, and the Urology surgery with 6 patients (Pa06, Pa07, Pa08, Pa10, Pa11 and Pa16), the STs, serotypes, and genomic SNP variations between strains from different patients showed significant diversity, indicating that their sources of infection were likely distinct from one another. Therefore, we inferred that the ICU was potentially one of the critical areas for nosocomial transmission within the hospital, and hence, heightened infection control measures are warranted in this unit to mitigate the risk of cross-infection and outbreaks.
The hybrid assembly of long and short-read data helped to found a plasmid acquisition event during the within-host evolution of UTI.35 Two strains isolated from the same patient with six days apart showed only 3 SNP differences; nevertheless, the plasmid carriage was different between them. The latter isolated strain had acquired a new IncM2_1 aac(3)-IId and blaCTX-M-3 positive (71,730 bp) plasmid after transfer to Neurovascular surgery from ICU, which highly suggesting the dynamics of plasmid in K. pneumoniae and the important influence of environment.36–38 Most of ST11 strains co-harbored aerobactin (iuc1), yerseniabactin (ybt) and rmpA2 genes and were KPC-2 positive, showing both carbapenem-resistant and hypervirulent, which are anticipated to pose a significant threat to human health. Future research should concentrate on devising intervention strategies to prevent the dissemination and transmission of K. pneumoniae ST11 strains in healthcare settings.39
Our study has a main limitation; specifically, the number of isolates and patients is limited. Our objective was to characterize the genomic features and possible transmission of K. pneumoniae causing UTI in a tertiary-care hospital. Therefore, among the 79 Klebsiella species strains collected, we only selected the 20 strains from 16 patients who presented with urinary tract infection symptoms. Measures were implemented to control nosocomial infection of multidrug-resistant bacteria in the hospital. The limited isolation of K. pneumoniae strains over a two-year period indicates the efficacy of these measures in preventing and controlling nosocomial infections.
Conclusion
K. pneumoniae is a notable causative agent of urinary tract infections (UTIs), with the ST11-KL47 lineage being notably prevalent at our hospital, characterized by a worrying dual phenotype of heightened drug resistance and hypervirulence. Strengthening the ongoing genomic surveillance of pathogens responsible for UTIs is of utmost importance to effectively monitor and manage the emergence and spread of such multi-drug resistant and highly virulent strains.
Data Confidentiality Statement
In this study, patient data confidentiality was stringently upheld. Due to the retrospective nature of the research, the institutional ethics committees of the Third Medical Center, PLA General Hospital (Beijing, China) waived the requirement for informed consent from individual patients. All methods were conducted in compliance with the relevant regulations and guidelines outlined in the Declaration of Helsinki. We have implemented strict protocols to protect patient confidentiality throughout the process, including during the analysis and dissemination of the genomic data obtained from the bacterial isolates causing urinary tract infections in the studied population.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from National Key Research and Development Program of China (Grant No. 2023YFC2604400), the National Science and Technology Major Project (Grant No. 2018ZX10305410) and Chinese National Natural Science Foundation (Grant No. 31900151).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Foxman B. The epidemiology of urinary tract infection. Nat Rev Urol. 2010;7(12):653–660. doi:10.1038/nrurol.2010.190
2. McLellan LK, Hunstad DA. Urinary tract infection: pathogenesis and outlook. Trends Mol Med. 2016;22(11):946–957. doi:10.1016/j.molmed.2016.09.003
3. Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol. 2015;13(5):269–284. doi:10.1038/nrmicro3432
4. Wang J, Liu F, Tartari E, et al. The prevalence of healthcare-associated infections in mainland china: a systematic review and meta-analysis. Infect Control Hosp Epidemiol. 2018;39(6):701–709. doi:10.1017/ice.2018.60
5. Yuan S, Shi Y, Li M, Hu X, Bai R. Trends in incidence of urinary tract infection in mainland China from 1990 to 2019. Int J Gen Med. 2021;14:1413–1420. doi:10.2147/IJGM.S305358
6. Krawczyk B, Wysocka M, Michalik M, Gołębiewska J. Urinary tract infections caused by K. pneumoniae in kidney transplant recipients – epidemiology, virulence and antibiotic resistance. Front Cell Infect Microbiol. 2022;12:1–14. doi:10.3389/fcimb.2022.861374
7. Perween N, Rai S, Nandwani S, Kumar SK. Retrospective analysis of urinary tract infection in the pediatric population at a tertiary care centre. Cureus. 2022;14(5). doi:10.7759/cureus.24796
8. Chapelle C, Gaborit B, Dumont R, Dinh A, Vallée M. Treatment of UTIs due to Klebsiella pneumoniae carbapenemase-producers: how to use new antibiotic drugs? a narrative review. Antibiotics. 2021;10(11):1332. doi:10.3390/antibiotics10111332
9. Zhang H, Zhang G, Yang Y, et al. Antimicrobial resistance comparison of Klebsiella pneumoniae pathogens isolated from intra-abdominal and urinary tract infections in different organs, hospital departments and regions of China between 2014 and 2017. J Microbiol Immunol Infect. 2021;54(4):639–648. doi:10.1016/j.jmii.2020.03.009
10. Liu X, Wu Y, Zhu Y, et al. Emergence of colistin-resistant hypervirulent Klebsiella pneumoniae (CoR-HvKp) in China. Emerg Microbes Infect. 2022;11(1):648–661. doi:10.1080/22221751.2022.2036078
11. Xie Z, Jian J, Chen L. Analysis of antimicrobial susceptibility in bacterial pathogens associated with urinary tract infections from Beijing teaching hospital in China, 2009-2017. Can J Infect Dis Med Microbiol. 2023;2023. doi:10.1155/2023/4360342
12. Huang L, Huang C, Yan Y, Sun L, Li H. Urinary tract infection etiological profiles and antibiotic resistance patterns varied among different age categories: a retrospective study from a tertiary general hospital during a 12-year period. Front Microbiol. 2022;12:1–10. doi:10.3389/fmicb.2021.813145
13. Liu X, Wang K, Chen J, et al. Clonal spread of carbapenem-resistant Klebsiella pneumoniae sequence type 11 in Chinese pediatric patients. Microbiol Spectr. 2022;10(6). doi:10.1128/spectrum.01919-22.
14. Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing. CLSI M100-2021. Wayne, PA: Clinical and Laboratory Standards Institute; 2021.
15. Andrews S FastQC: a quality control tool for high throughput sequence data. Available from http//www.bioinformatics.babraham.ac.uk/projects/fastqc/.
16. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi:10.1093/bioinformatics/btu170
17. Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13(6):1–22. doi:10.1371/journal.pcbi.1005595
18. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi:10.1093/bioinformatics/btu153
19. Lam MMC, Wick RR, Watts SC, et al. A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat Commun. 2021;12(1). doi:10.1038/s41467-021-24448-3.
20. Gupta A, Jordan IK, Rishishwar L. stringMLST: a fast k-mer based tool for multilocus sequence typing. Bioinformatics. 2017;33(1):119–121. doi:10.1093/bioinformatics/btw586
21. Jolley KA, Maiden MCJ. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinf. 2010;11(1). doi:10.1186/1471-2105-11-595
22. Grant JR, Stothard P. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008;36(Web Server):W181–W184. doi:10.1093/nar/gkn179
23. Ondov BD, Treangen TJ, Melsted P, et al. Mash: fast genome and metagenome distance estimation using minhash. Genome Biol. 2016;17(1). doi:10.1186/s13059-016-0997-x.
24. Didelot X, Wilson DJ. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput Biol. 2015;11(5):e1004041. doi:10.1371/journal.pcbi.1004041
25. Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35(21):4453–4455. doi:10.1093/bioinformatics/btz305
26. Darriba D, Posada D, Kozlov AM. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol. 2020;37(1):291–294. doi:10.1093/molbev/msz189
27. Cannatelli A, D’Andrea MM, Giani T, et al. In vivo emergence of colistin resistance in Klebsiella pneumoniae producing KPC-type carbapenemases mediated by insertional inactivation of the PhoQ/PhoP mgrB regulator. Antimicrob Agents Chemother. 2013;57(11):5521–5526. doi:10.1128/AAC.01480-13
28. Cristea OM, Avrămescu CS, Bălășoiu M, et al. Urinary tract infection with Klebsiella pneumoniae in patients with chronic kidney disease. Curr Heal Sci J. 2017;43:137–148.
29. Quan J, Dai H, Liao W, et al. Etiology and prevalence of ESBLs in adult community-onset urinary tract infections in East China: a prospective multicenter study. J Infect. 2021;83(2):175–181. doi:10.1016/j.jinf.2021.06.004
30. Qi Y, Wei Z, Ji S, et al. ST11, the dominant clone of KPC-producing Klebsiella pneumoniae in China. J Antimicrob Chemother. 2011;66(2):307–312. doi:10.1093/jac/dkq431
31. Caneiras C, Lito L, Melo-Cristino J, Duarte A. Community-and hospital-acquired Klebsiella pneumoniae urinary tract infections in Portugal: virulence and antibiotic resistance. Microorganisms. 2019;7(5):1–14. doi:10.3390/microorganisms7050138
32. Tian GB, Doi Y, Shen J, et al. MCR-1-producing Klebsiella pneumoniae outbreak in China. Lancet Infect Dis. 2017;17(6):577. doi:10.1016/S1473-3099(17)30266-9
33. Li J, Tang M, Liu Z, et al. Molecular and clinical characterization of hypervirulent Klebsiella pneumoniae isolates from individuals with urinary tract infections. Front Cell Infect Microbiol. 2022;12:925440.
34. Chen T, Wang Y, Zhou Y, et al. Recombination drives evolution of carbapenem-resistant Klebsiella pneumoniae Sequence Type 11 KL47 to KL64 in China. Microbiol Spectr. 2023;11(1):e0110722.
35. Van Dorp L, Wang Q, Shaw LP, et al. Rapid phenotypic evolution in multidrug-resistant Klebsiella pneumoniae hospital outbreak strains. Microb Genomics. 2019;5(4):1–11. doi:10.1099/mgen.0.000263
36. San Millan A. Evolution of plasmid-mediated antibiotic resistance in the clinical context. Trends Microbiol. 2018;26(12):978–985. doi:10.1016/j.tim.2018.06.007
37. Wyres KL, Lam MMC, Holt KE. Population genomics of Klebsiella pneumoniae. Nat Rev Microbiol. 2020;18(6):344–359. doi:10.1038/s41579-019-0315-1
38. Wyres KL, Nguyen TNT, Lam MMC, et al. Genomic surveillance for hypervirulence and multi-drug resistance in invasive Klebsiella pneumoniae from south and Southeast Asia. Genome Med. 2020;12(1):1–16. doi:10.1186/s13073-019-0706-y
39. Martin MJ, Corey BW, Sannio F, et al. Anatomy of an extensively drug-resistant Klebsiella pneumoniae outbreak in Tuscany, Italy. Proc Natl Acad Sci U S A. 2021;118(48). doi:10.1073/pnas.2110227118.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.