")
Back to Journals » Infection and Drug Resistance » Volume 13
Hybrid Genome Assembly and Annotation of a Pandrug-Resistant Klebsiella pneumoniae Strain Using Nanopore and Illumina Sequencing
Authors Ruan Z , Wu J, Chen H, Draz MS, Xu J, He F
Received 28 November 2019
Accepted for publication 11 January 2020
Published 21 January 2020 Volume 2020:13 Pages 199—206
DOI https://doi.org/10.2147/IDR.S240404
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Zhi Ruan, 1 Jianyong Wu, 2 Hangfei Chen, 1 Mohamed S Draz, 3, 4 Juan Xu, 5 Fang He 6
1Department of Clinical Laboratory, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, Hangzhou 310016, People’s Republic of China; 2Department of Clinical Laboratory, The Fourth Affiliated Hospital, Zhejiang University School of Medicine, Yiwu 322000, People’s Republic of China; 3Department of Medicine, Harvard Medical School, Boston, MA 02115, USA; 4Department of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA; 5Institute of Hygiene, Zhejiang Academy of Medical Sciences, Hangzhou 310013, People’s Republic of China; 6Department of Clinical Laboratory, Zhejiang Provincial People’s Hospital, People’s Hospital of Hangzhou Medical College, Hangzhou 310014, People’s Republic of China
Correspondence: Fang He Tel/Fax +86 571 88215596
Email [email protected]
Background: The prevalence of multidrug-resistant Klebsiella pneumoniae is increasingly being implicated worldwide in a variety of infections with high mortalities. Here, we report the complete genome sequence of K. pneumoniae strain KP58, a pandrug-resistant K. pneumoniae strain that exhibits high levels of resistance to colistin and tigecycline in China.
Methods: The K. pneumoniae strain KP58 was recovered from a urine sample of a female patient hospitalized in a tertiary hospital in Hangzhou, China. Antimicrobial susceptibility testing was performed and the minimum inhibitory concentrations (MICs) were determined. Whole-genome sequencing was performed using Illumina and Oxford nanopore sequencing technologies. Genomic features, antimicrobial resistance genes and virulence genes were comprehensively analysed by various bioinformatics approaches. In addition, genomic epidemiological and phylogenetic analyses of K. pneumoniae KP58 and closely related isolates were performed using the core genome multilocus sequence typing (cgMLST) analysis in BacWGSTdb, an online bacterial whole-genome sequence typing and source tracking database.
Results: K. pneumoniae KP58 was resistant to all antimicrobial agents tested, including tigecycline and colistin. Combining the two sequencing technologies allowed a high-quality complete genome sequence of K. pneumoniae KP58 comprising one circular chromosome and five circular plasmids to be obtained. This strain harbours a variety of acquired antimicrobial resistance and virulence determinants. It also carried an ISKpn26-like insertion in the disrupted mgrB gene, which confers colistin resistance. The tigecycline resistance was associated with overexpression of the AcrAB efflux system. The closest relative of K. pneumoniae KP58 was another clinical isolate recovered from Hangzhou that differed by only 10 cgMLST loci.
Conclusion: The dataset presented in this study provides essential insights into the evolution of antimicrobial-resistant K. pneumoniae in hospital settings and assists in the development of effective control strategies. Appropriate surveillance and control measures are essential to prevent its further dissemination.
Keywords: Klebsiella pneumoniae, whole-genome sequencing, nanopore, hybrid assembly, pandrug resistance
Introduction
Klebsiella pneumoniae was first isolated in 1882 by a German microbiologist who aided in determining the bacterial cause of pneumonia.1 K. pneumoniae has gained notoriety as an opportunistic pathogen that is commonly associated with a wide range of nosocomial infections (e.g., bloodstream infections, urinary tract infections, and liver abscesses).2,3 In addition, the prevalence of multidrug-resistant K. pneumoniae has significantly narrowed the therapeutic options for the treatment of these infections, posing a challenging threat to public health.4,5 Carbapenem-resistant K. pneumoniae is considered as an urgent threat to human health as reported by the Centers for Disease Control and Prevention (CDC), USA, in 2013,6 necessitating the development of new antimicrobials. K. pneumoniae has evolved (developed or acquired) and maintained resistance to all antimicrobials introduced for treatment due to its extraordinary ability to adapt its genome through mutations and horizontal gene transfer of antimicrobial resistance determinants.7 Interestingly, recent studies have shown that multidrug-resistant K. pneumoniae clones have a mobile pool of antimicrobial resistance and virulence genes capable of causing life-threatening infections in healthy individuals.8,9 Thus, there is a pressing need for detailed molecular studies that provide comprehensive insights into the evolutionary processes of this species and help in monitoring and controlling drug-resistant infections.
Whole-genome sequencing provides the ideal means for the in-depth study of the mechanisms that promote the emergence, evolution and spread of antimicrobial resistance; tracking pathogen populations with unprecedented sensitivity for genetic variation; and greatly improved resolution for studies in population dynamics. The cost of whole-genome sequencing has steadily decreased as a result of advances in second-generation, short-read sequencing platforms (e.g., Illumina, 454, Ion Torrent and SOLiD), enabling a variety of bacterial genomic studies.10 A challenge in assembling genomes using sequencing techniques that generate short reads of <1 kb in length (e.g., Illumina) is their inability to deal with repetitive regions, which results in fragmented genome assemblies. Long-read techniques (e.g., Oxford Nanopore and PacBio) generate long reads of >10 kb in length, enabling the correct structural resolution of complex genome regions. However, although the gaps in the assembled genome can be closed by using long-read sequencing and make it easier to read through the repetitive regions, bacterial genome assemblies made using long reads alone are still inadequate due to a high rate of errors in the raw sequence reads and the generation of artefacts.11,12 Thus, the combined use of short- and long-read sequencing technologies would allow for high-quality assemblies and has emerged as a standard approach for the characterization of bacterial genomes.
In this study, we report a hybrid assembly, generated using Oxford Nanopore and Illumina reads, of a pandrug-resistant K. pneumoniae isolate that exhibits high levels of colistin and tigecycline resistance. Genomic epidemiological analysis was performed to gain insight into the basis of the prevalent K. pneumoniae lineage in China.
Materials and Methods
Bacterial Strain
The K. pneumoniae strain KP58 was cultured from a urine sample of an 89-year-old female patient hospitalised with symptoms of pneumonia and fever. During the hospitalization, the patient received multiple broad-spectrum antibiotics including cefoperazone/sulbactam, imipenem, tigecycline and colistin. The purified K. pneumoniae isolate KP58 was grown in Mueller-Hinton broth (Oxoid, Hampshire, United Kingdom) at 37°C for 24 h. The species identification and antimicrobial susceptibility of K. pneumoniae KP58 were determined with an automated VITEK®2 AST-GN16 system (bioMérieux, Marcy-l’Étoile, France) or the broth microdilution method using antibiotic panels with the following antimicrobial agents: ceftriaxone, cefepime, cefoxitin, aztreonam, piperacillin/tazobactam, imipenem, meropenem, gentamicin, tobramycin, amikacin, trimethoprim/sulphamethoxazole, ciprofloxacin, levofloxacin, nitrofurantoin, tigecycline and colistin. The minimum inhibitory concentrations (MICs) were interpreted according to the CLSI 2019 guidelines, except for tigecycline and colistin, which were interpreted using the EUCAST 2019 guidelines due to the absence of CLSI breakpoints. The MICs of the 16 tested antibacterial agents are shown in Table S1.
Whole-Genome Sequencing
Two separate genomic DNA libraries were prepared according to the requirements of the Illumina and Oxford Nanopore systems. A combination of long-read Nanopore MinION and short-read Illumina NovaSeq 6000 platforms was used to generate the complete genome sequence of K. pneumoniae KP58. For Illumina sequencing, the extracted genomic DNA was fragmented by sonication using a Covaris M220 sonicator (Covaris, Woburn, MA, USA). The sheared DNA fragments were then used to prepare a shotgun paired-end library with an average insert size of 350 bp using a TruSeq DNA Sample Prep kit (Illumina, San Diego, CA, USA). The library was sequenced on an Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) using the 150-bp paired-end sequencing mode. The two paired FASTQ files were base called from the Illumina raw sequence read data. The quality of the raw sequence reads was assessed using FastQC v0.11.8, and Trimmomatic v0.39 was used to trim sequencing adapters, reads with a quality score <20 over a sliding window size of 4 bp, and reads with a sequence length <50 bp.13 After trimming the adaptors and filtering low-quality reads, the clean sequence data were used for further bioinformatics analyses.
For Nanopore sequencing, a MinION sequencing library was prepared using the Nanopore Ligation Sequencing Kit (SQK-LSK109; Oxford Nanopore, Oxford, UK). The library was sequenced with an R9.4.1 MinION flow cell (FlO-MIN106) for a 24 h run using MinKNOW v2.0 with the default settings. FAST5 files containing raw Nanopore signal data were base called and converted to FASTQ format in real-time using Guppy v3.3.0, after which Porechop v0.2.4 was used to trim barcode and adapter sequences.14 Filtlong v0.2.0, a quality filtering tool for Nanopore reads, was used to remove sequences shorter than 3000 bases and with mean quality scores of less than 12 to facilitate assembly.15
Genome Assembly and Annotation
The hybrid genome assembly was performed using Unicycler v0.4.7, which allows for both Illumina reads (short, accurate) and Nanopore reads (long, less accurate) to be used in the conservative mode.16 The highly accurate Illumina reads were aligned against the Nanopore reads as a reference to correct random sequencing errors and finally generate a genome assembly of high quality. Eventually, the assembled sequence was polished by aligning the Illumina paired-end reads with the BWA-MEM algorithm using Pilon v1.2.3 for several rounds to improve the quality of genome assembly.17 Multiple rounds of error correction were performed until no more errors could be fixed. The quality of the assembled genome was assessed using CheckM v1.0.18.18
The complete genome sequence of K. pneumoniae KP58 was annotated using the NCBI Prokaryotic Genomes Annotation Pipeline (PGAP).19 The programs tRNAscan-SE and RNAmmer were used for tRNA and rRNA gene prediction.20,21 Antimicrobial resistance genes, virulence genes, and plasmid replicons were identified using ABRicate V0.8.7 in tandem with ResFinder 3.2, CARD 2017, VFDB 2019, and PlasmidFinder 2.1 databases by a minimum sequence homology of 90% and a minimum length of 60% to respective database entries. The integrons and gene cassettes within the genomes were identified according to the INTEGRALL database.22 Kaptive v0.6.1 was used to predict the capsule and lipopolysaccharide serotype of K. pneumoniae.23–27 The circular maps of the chromosome and plasmids of K. pneumoniae KP58 were created using CGView Server, and multiple plasmid comparisons were performed using BLAST Ring Image Generator (BRIG).28,29
Phylogenetic Analysis
The genome sequence of K. pneumoniae KP58 was compared with publicly available genome sequences of ST11 K. pneumoniae isolates in China retrieved from the NCBI GenBank database. Bacterial whole-genome sequence typing and source tracking analysis, implementing both core genome multilocus sequence typing (cgMLST) and core genome single nucleotide polymorphism (cgSNP) strategies, were performed using the BacWGSTdb server.30,31 A publicly accessible cgMLST scheme for K. pneumoniae comprising 2358 target genes was used to characterize the cgMLST allelic profile of K. pneumoniae ST11 isolates. A K. pneumoniae ST11 strain, JM45 (NCBI Reference sequence: CP006656), was selected as a reference genome for phylogenetic analysis. The pan genome of the ST11 K. pneumoniae isolates originating from China was defined with Roary using the default BLAST identity threshold (95%).32 SNPs in the core genome were extracted using Snippy v4.4.5. The removal of recombination regions from SNP alignments was performed using ClonalFrameML v1.12, and the output was used to construct a phylogenetic tree with 1000 bootstraps under the general time reversible (GTR) model with RAxML v8.2.12.33,34 The resulting alignment was also used to determine the pairwise SNP distances using snp-dist v0.6.3. GrapeTree was used to produce and visualize a minimum spanning tree (MST) based on the comparison of cgMLST allelic profiles of ST11 K. pneumoniae isolates.35
Accession Number
The raw Illumina and Nanopore sequencing reads of K. pneumoniae KP58 were deposited in the NCBI Sequence Read Archive (SRA) database under the accession number SRP223138. The complete genome sequence of K. pneumoniae KP58 was deposited in the NCBI GenBank database under the accession number CP041373-CP041378.
Ethical Approval
The K. pneumoniae strain KP58 was generated as part of routine hospital laboratory procedures. This study was performed in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Zhejiang Provincial People’s Hospital, Hangzhou, China. Written informed consent was obtained from the patient, which included publication of the case details.
Results and Discussion
Illumina sequencing generated 6,085,230 raw sequence reads with an average read length of 150 bp that encompassed 912,784,500 bp sequenced bases. The quality of the Illumina sequencing reads was assessed with the Phred quality scores (Q score). The results showed that the quality scores of the raw Illumina reads (average Q30) were stable across all reads and that most reads had a Q score of Q40, indicating the bases are of high quality (Figure S1). Nanopore sequencing generated 76,129 sequence reads with an average read length of 9806 bp that encompassed 746.5 Mbp of sequence data. The read library N50 value was 19,440 bp, with the longest read being 125,146 kb. The sequence data were further subsampled based on a quality score of 9 and a read size greater than 3000 bp, generating 368.8 Mbp of data for assembly. Altogether, these analyses confirmed the quality of the sequencing data and its adequacy for downstream analyses.
The complete genome sequence of K. pneumoniae KP58 comprises a circular chromosome (5,483,867 bp) and five plasmids (197,415 bp, 134,972 bp, 87,095 bp, 11,970 bp, and 5596 bp) (Figure S2). The NCBI Prokaryotic Genome Annotation Pipeline (PGAP) server annotated 5913 protein-coding genes and identified 85 tRNA genes and 25 rRNA operons. An assessment of the quality of the genome showed that it exhibited a completeness of 98.62% and contamination and strain heterogeneity values of 0.36% and 0%, respectively, indicating the high quality of the assembled genome. Consistent with the antimicrobial susceptibility data (Table S1), KP58 harbours a variety of acquired antimicrobial resistance determinants conferring resistance to multiple antimicrobial agents, including aadA2, rmtB, blaCTX-M-65, blaKPC-2, blaTEM-1B, blaSHV-12, blaSHV-182, qnrS1, fosA, catA2, sul2, tet(A), and dfrA14, which confer resistance to β-lactams, fluoroquinolones, fosfomycin, phenicols, sulphonamides, tetracyclines, and trimethoprim. In addition, analysis of the quinolone resistance-determining region (QRDR) identified alterations of the target genes, including mutations in GyrA (S83I and D87G) and ParC (S80I). The expression levels of tigecycline resistance-related efflux pump genes (i.e., acrA, acrB, ramA, marA, soxS, and acrAB) were determined by qRT-PCR, which indicated that the tigecycline resistance of K. pneumoniae KP58 was associated with overexpression of the AcrAB efflux system. No plasmid-born tigecycline resistance gene tet(X) was found. Although the isolate was negative for mcr genes, it carried an ISKpn26-like element in the disrupted mgrB gene, an insertion that is known to confer colistin resistance. Notably, the possession of three potential virulence determinants, including those responsible for aerobactin, hypermucoviscosity and yersiniabactin, was also identified. K. pneumoniae KP58 was identified as sequence type (ST11), a widely disseminated multi-drug resistant clonal lineage of K. pneumoniae responsible for severe infections worldwide. The KL type (polysaccharide capsule and lipopolysaccharide O antigen) of K. pneumoniae KP58 was predicted as KL64 using Kaptive. We also identified the replicons of four plasmids belonging to incompatibility (Inc) group F [IncFIB, IncR, and IncFII] and ColRNAI.
Vertically inherited core genome SNPs and core genome MLST analysis were performed to assess the phylogenetic relationship between K. pneumoniae KP58 and a total of 416 ST11 K. pneumoniae strains recovered from China. The analysis of non-recombinant SNPs increases the accuracy of the phylogenetic reconstruction and allows calculation of the substitution rate without the effect of recombination. The phylogenetic tree had a high diversity, with the most geographically related isolates being grouped into the same clade, which differed by <10 SNPs (Figure 1). The majority of these isolates were recovered from Sichuan and Hangzhou, which carried the highest proportion of the known antimicrobial resistance determinants. Our results indicated that the most closely related strain to K. pneumoniae KP58 is L39_2 (NCBI Accession number: CP033954), another ST11 strain collected from a human stool sample in Hangzhou, with these strains differing from each other by only 10 cgMLST loci or 9 SNPs (Figure 2, Table S2).
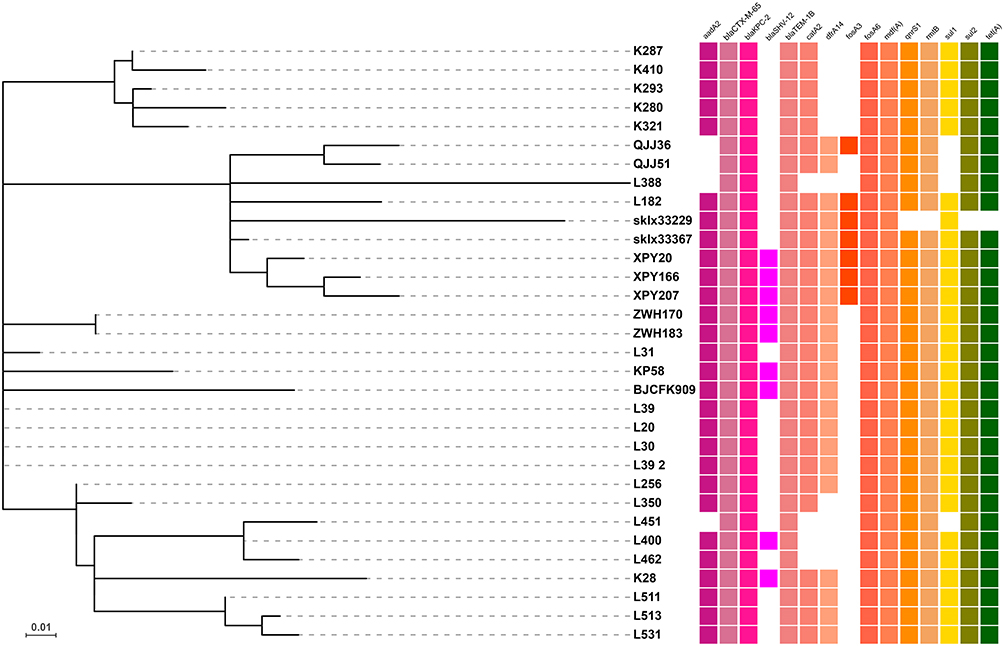 |
Figure 1 Recombination-filtered core genome phylogeny and the distribution of antimicrobial resistance genes for ST11 K. pneumoniae isolates recovered from Hangzhou, China. The cell in different colour indicates the presence of the gene while the blank cell indicates the absence of the gene. |
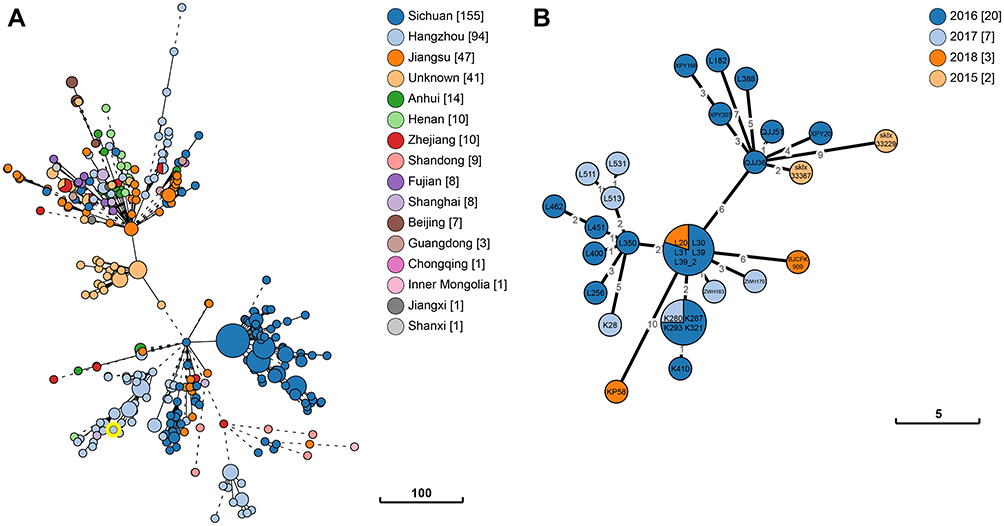 |
Figure 2 Phylogenetic relationship between K. pneumoniae KP58 and closely related K. pneumoniae strains presently retrieved from BacWGSTdb. Clonal relationships between different isolates are depicted by the line length connecting each circle. The numbers given in square brackets indicate the number of isolates from each country. (A) The minimum spanning tree of all ST11 K. pneumoniae strains recovered from China. (B) The minimum spanning tree of the closely related strains with K. pneumoniae KP58 (differing by <10 cgMLST loci). |
Among the plasmids of K. pneumoniae KP58, the IncR/IncFII plasmid (134,972 bp), which carries the carbapenem-resistance gene blaKPC-2, shared 99% identity and 75% query coverage with plasmid pKPC2_020037 from the KPC-2-producing K. pneumoniae strain WCHKP020037 isolated in Chengdu, China. The backbone region of pKP58-2 was found to contain genes for replication and plasmid stability but lacked genes for conjugation. The genetic context of blaKPC-2 on plasmid pKP58-2 is organized of Tn3-tnpA, Tn3-tnpR, ISKpn27, Tn3-ΔblaTEM-1, blaKPC-2, ΔISKpn6, korC, klcA, ΔrepB and ΔTn1721, which is almost identical to three plasmids: p3_L39 (GenBank accession number: CP033956), p2b2 (GenBank accession number: CP034125), and pKPC2_020037 (GenBank accession number: CP036372). Another IncFII-type plasmid (87,095 bp) harboured the antimicrobial resistance genes qnrS1, tet(A), catA2, sul2, and dfrA14, which are known to confer resistance to fluoroquinolone, tetracycline, phenicol, sulphonamide, and trimethoprim, respectively. In addition, a set of virulence genes comprising iucABCD, iutA, rmpA, and rmpA2 were all located on an IncFIB/IncHI1B plasmid (197,415 bp), whereas no resistance gene was detected in the same plasmid. A comparison of this virulence plasmid, named pKP58-1, and other similar plasmids deposited in the NCBI GenBank database revealed its identity with three previously reported virulence plasmids, including pK2044 (99% identity and 93% coverage), pLVPK (99% identity and 89% coverage), and pVir-CR-HvKP4 (99% identity and 78% coverage). In addition, pKP58-1 was smaller in size than the other two plasmids, pK2044 and pLVPK, but was larger than pVir-CR-HvKP4 (178,154 bp) in K. pneumoniae (Figure 3).
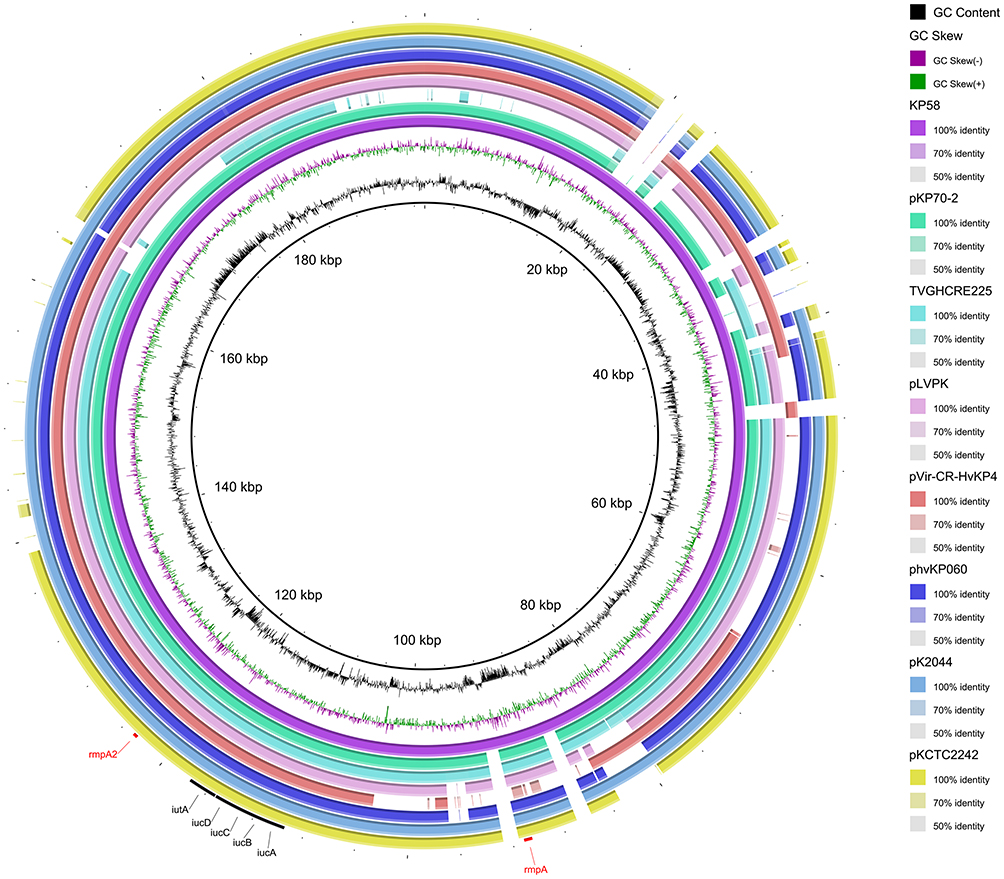 |
Figure 3 Genetic comparison of eight virulence-encoding plasmids harboured by different K. pneumoniae strains. Virulence factors, such as aerobactin (iucABCD, iutA), and mucoid phenotype (rmpA, rmpA2) are annotated. |
In summary, our study presents the hybrid genome assembly and annotation of a pandrug-resistant K. pneumoniae strain in China. These data unravelled a rich repertoire of antimicrobial resistance and virulence determinants in K. pneumoniae and may provide insight into the development of strategies for prevention, diagnosis and treatment of K. pneumoniae infections.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81702042), Natural Science Foundation of Zhejiang Province (LQ19H200003) and the Zhejiang Provincial Medical and Health Science and Technology Plan (2018KY344 and 2019KY311).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Friedlaender C. Ueber die Schizomyceten bei der acuten fibrösen Pneumonie. Archiv für pathologische Anatomie und Physiologie und für klinische Medicin. 1882;87(2):319–324.
2. Bengoechea JA, Sa Pessoa J. Klebsiella pneumoniae infection biology: living to counteract host defences. FEMS Microbiol Rev. 2019;43(2):123–144. doi:10.1093/femsre/fuy043
3. Russotto V, Cortegiani A, Fasciana T, et al. What healthcare workers should know about environmental bacterial contamination in the intensive care unit. Biomed Res Int. 2017;2017:6905450. doi:10.1155/2017/6905450
4. Geraci DM, Bonura C, Giuffre M, et al. Is the monoclonal spread of the ST258, KPC-3-producing clone being replaced in southern Italy by the dissemination of multiple clones of carbapenem-nonsusceptible, KPC-3-producing Klebsiella pneumoniae? Clin Microbiol Infect. 2015;21(3):e15–17. doi:10.1016/j.cmi.2014.08.022
5. Abdelaziz MO, Bonura C, Aleo A, Fasciana T, Mammina C. NDM-1- and OXA-163-producing Klebsiella pneumoniae isolates in Cairo, Egypt, 2012. J Glob Antimicrob Resist. 2013;1(4):213–215. doi:10.1016/j.jgar.2013.06.003
6. WHO. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Geneva: World Health Organization; 2017. Available from https://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/en.
7. Partridge SR, Kwong SM, Firth N, Jensen SO. Mobile genetic elements associated with antimicrobial resistance. Clin Microbiol Rev. 2018;31(4):e00088–17.
8. Holt KE, Wertheim H, Zadoks RN, et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc Natl Acad Sci U S A. 2015;112(27):E3574–3581. doi:10.1073/pnas.1501049112
9. Fasciana T, Gentile B, Aquilina M, et al. Co-existence of virulence factors and antibiotic resistance in new Klebsiella pneumoniae clones emerging in south of Italy. BMC Infect Dis. 2019;19(1):928. doi:10.1186/s12879-019-4565-3
10. Boolchandani M, D’Souza AW, Dantas G. Sequencing-based methods and resources to study antimicrobial resistance. Nat Rev Genet. 2019;20(6):356–370. doi:10.1038/s41576-019-0108-4
11. Senol Cali D, Kim JS, Ghose S, Alkan C, Mutlu O. Nanopore sequencing technology and tools for genome assembly: computational analysis of the current state, bottlenecks and future directions. Brief Bioinform. 2019;20(4):1542–1559. doi:10.1093/bib/bby017
12. Magi A, Semeraro R, Mingrino A, Giusti B, D’Aurizio R. Nanopore sequencing data analysis: state of the art, applications and challenges. Brief Bioinform. 2018;19(6):1256–1272. doi:10.1093/bib/bbx062
13. Andrews S. FastQC: a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
14. Wick RR. Porechop: an adapter trimmer for Oxford Nanopore reads. https://github.com/rrwick/Porechop.
15. Breese MR, Liu Y. NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics. 2013;29(4):494–496. doi:10.1093/bioinformatics/bts731
16. Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13(6):e1005595.
17. Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963. doi:10.1371/journal.pone.0112963
18. Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–1055. doi:10.1101/gr.186072.114
19. Tatusova T, DiCuccio M, Badretdin A, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44(14):6614–6624. doi:10.1093/nar/gkw569
20. Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–3108. doi:10.1093/nar/gkm160
21. Lowe TM, Chan PP. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44(W1):W54–57. doi:10.1093/nar/gkw413
22. Moura A, Soares M, Pereira C, Leitao N, Henriques I, Correia A. INTEGRALL: a database and search engine for integrons, integrases and gene cassettes. Bioinformatics. 2009;25(8):1096–1098. doi:10.1093/bioinformatics/btp105
23. Jia B, Raphenya AR, Alcock B, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45(D1):D566–D573. doi:10.1093/nar/gkw1004
24. Zankari E, Hasman H, Cosentino S, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67(11):2640–2644. doi:10.1093/jac/dks261
25. Carattoli A, Zankari E, Garcia-Fernandez A, et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58(7):3895–3903. doi:10.1128/AAC.02412-14
26. Wick RR, Heinz E, Holt KE, Wyres KL. Kaptive web: user-friendly capsule and lipopolysaccharide serotype prediction for Klebsiella Genomes. J Clin Microbiol. 2018;56(6):e00197–18. doi:10.1128/JCM.00197-18
27. Liu B, Zheng D, Jin Q, Chen L, Yang J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019;47(D1):D687–D692. doi:10.1093/nar/gky1080
28. Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi:10.1186/1471-2164-12-402
29. Grant JR, Stothard P. The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008;36(WebServer issue):W181–184. doi:10.1093/nar/gkn179
30. Ruan Z, Feng Y. BacWGSTdb, a database for genotyping and source tracking bacterial pathogens. Nucleic Acids Res. 2016;44(D1):D682–687. doi:10.1093/nar/gkv1004
31. Ruan Z, Yu Y, Feng Y. The global dissemination of bacterial infections necessitates the study of reverse genomic epidemiology. Brief Bioinform. 2019. doi:10.1093/bib/bbz010
32. Page AJ, Cummins CA, Hunt M, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31(22):3691–3693. doi:10.1093/bioinformatics/btv421
33. Didelot X, Wilson DJ. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput Biol. 2015;11(2):e1004041. doi:10.1371/journal.pcbi.1004041
34. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi:10.1093/bioinformatics/btu033
35. Zhou Z, Alikhan NF, Sergeant MJ, et al. GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018;28(9):1395–1404. doi:10.1101/gr.232397.117
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.