")
Back to Journals » International Journal of General Medicine » Volume 9
Human serum albumin homeostasis: a new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements
Received 19 December 2015
Accepted for publication 12 April 2016
Published 15 July 2016 Volume 2016:9 Pages 229—255
DOI https://doi.org/10.2147/IJGM.S102819
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
David G Levitt,1,* Michael D Levitt2,*
1Department of Integrative Biology and Physiology, University of Minnesota, 2Research Service, Veterans Affairs Medical Center, Minneapolis, MN, USA
*These authors contributed equally to this work
Abstract: Serum albumin concentration (CP) is a remarkably strong prognostic indicator of morbidity and mortality in both sick and seemingly healthy subjects. Surprisingly, the specifics of the pathophysiology underlying the relationship between CP and ill-health are poorly understood. This review provides a summary that is not previously available in the literature, concerning how synthesis, catabolism, and renal and gastrointestinal clearance of albumin interact to bring about albumin homeostasis, with a focus on the clinical factors that influence this homeostasis. In normal humans, the albumin turnover time of about 25 days reflects a liver albumin synthesis rate of about 10.5 g/day balanced by renal (≈6%), gastrointestinal (≈10%), and catabolic (≈84%) clearances. The acute development of hypoalbuminemia with sepsis or trauma results from increased albumin capillary permeability leading to redistribution of albumin from the vascular to interstitial space. The best understood mechanism of chronic hypoalbuminemia is the decreased albumin synthesis observed in liver disease. Decreased albumin production also accounts for hypoalbuminemia observed with a low-protein and normal caloric diet. However, a calorie- and protein-deficient diet does not reduce albumin synthesis and is not associated with hypoalbuminemia, and CP is not a useful marker of malnutrition. In most disease states other than liver disease, albumin synthesis is normal or increased, and hypoalbuminemia reflects an enhanced rate of albumin turnover resulting either from an increased rate of catabolism (a poorly understood phenomenon) or enhanced loss of albumin into the urine (nephrosis) or intestine (protein-losing enteropathy). The latter may occur with subtle intestinal pathology and hence may be more prevalent than commonly appreciated. Clinically, reduced CP appears to be a result rather than a cause of ill-health, and therapy designed to increase CP has limited benefit. The ubiquitous occurrence of hypoalbuminemia in disease states limits the diagnostic utility of the CP measurement.
Keywords: albumin, enteropathy, nephrosis, cirrhosis, malnutrition, clearance, synthesis
Introduction
Albumin, the major serum protein, has multiple important physiological functions including maintenance of colloidal osmotic pressure, binding of a wide variety of compounds, and provision of the bulk of plasma antioxidant activity. Thus, a correlation between serum albumin concentration (CP) and health might be expected. In fact, there is an astonishingly strong correlation between CP and mortality risk. For example, post-surgical mortality in 54,000 Veterans Administration (VA) Hospital patients increased quasi-logarithmically from <1% to 29% as the patients’ preoperative CP declined from 4.6 g/dL to < 2.1 g/dL.1 In a wide variety of diseases, CP is the serum analyte that best predicts a poor outcome.2,3 Serum albumin also predicts mortality in seemingly healthy individuals as evidenced by a study in which CP was measured in 1.7 million life insurance applicants.4 Over the ensuing 12 years, as shown in Figure 1, a clear-cut association was observed between mortality and CP concentration for all ages and sex of applicants.4 However, this persuasive evidence of the importance of albumin to health is counterbalanced by the paradoxical observation that subjects with the rare hereditary condition of analbuminemia (CP <0.1 g/dL) are relatively asymptomatic and have a normal life expectancy.5,6 This article provides a comprehensive review of the physiology and biochemistry of albumin, and the concluding section discusses the clinical aspects of serum albumin including the seemingly paradoxical observations concerning the relationship of serum albumin to health.
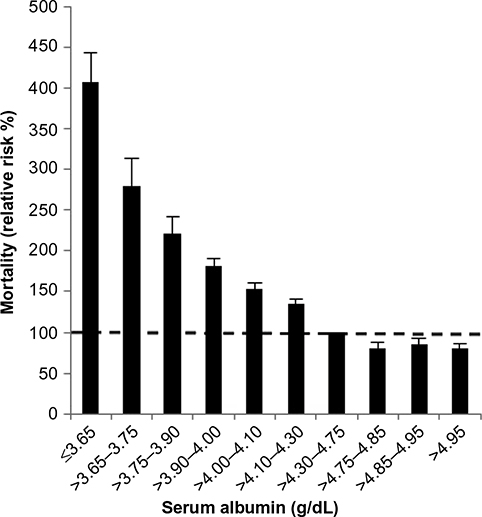 | Figure 1 Relationship of serum albumin concentration to relative risk of mortality for 50- to 69-year-old male life insurance applicants. Notes: The dashed line indicates a relative risk of 1.0. The means and 95% confidence limits of relative mortality (medium follow-up of 12 years) are shown for groups of subjects with the mean serum albumin concentrations listed . Adapted from Fulks M, Stout RL, Dolan VF. Albumin and all-cause mortality risk in insurance applicants. J Insur Med. 2010;42(1):11–17. Copyright © 2010 Journal of Insurance Medicine.4 |
Physiological functions of albumin
Colloidal osmotic pressure
The molecular weight of albumin (66.5 kD) is less than half that of gamma globulin (160 kD); hence, the osmotic activity of albumin per gram would be predicated to be about 2.3 times that of gamma globulin. In addition, the high negative charge of albumin does not bind but holds sodium ions in its field (Gibbs–Donnan effect), further increasing the intrinsic osmotic activity of albumin by about 50%. The net result is that albumin provides about 80% of the colloidal osmotic activity of normal plasma, and as CP declines, increases in globulins are insufficient to maintain normal colloidal osmotic pressure.
Binding function
Despite its strong negative charge, albumin binds a vast array of both positively and negatively charged compounds including hydrophobic organic anions such as bilirubin and long-chain fatty acids and divalent (but not monovalent) cations such as calcium and magnesium.7 Examples of other biologically important compounds bound by albumin are a wide variety of drugs, bile acids, copper, zinc, and even compounds with specific serum binders such as vitamin D and thyroxin. Albumin binding reduces the free concentration of compounds, thus limiting their biologic activity, distribution, and rate of clearance. For some compounds, such as unconjugated bilirubin, this binding is so avid that controversy exists as to how the liver is able to clear this compound at the observed rate of about 5 mL/minute.8
Antioxidant activity
Albumin provides >50% of the total antioxidant of normal plasma.9 This activity has been attributed to the abundant reduced sulfhydryl groups of albumin that have been shown to scavenge a variety of oxygen-free radicals including hypochlorous acid and nitric oxide. In addition to this inherent antioxidant activity, albumin also binds unconjugated bilirubin, a potent antioxidant. This antioxidant activity is commonly assumed to be the mechanism responsible for the strong inverse correlation between plasma unconjugated bilirubin concentration and the morbidity and mortality of many disease states.
Albumin homeostasis
In the steady state, CP is determined by the balance between albumin synthesis versus the sum of albumin removal by catabolic (C), renal (Re), and gastrointestinal (GI) processes:
|
|
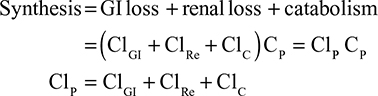
The plasma clearance (ClP) is the sum of the GI (ClGI), urinary (ClRe), and catabolic plasma clearance (ClC). The “catabolic” component (ClC) is defined as the poorly characterized fraction of total clearance that cannot be accounted for by renal or GI clearance. In most diseases, negligible amounts of intact albumin are found in urine or stool, and therefore, the GI and renal clearance components are also associated with catabolic processes that are distinct from “catabolic” component in Equation 1. Each of the components in Equation 1 will be discussed in detail in the following sections. Since albumin has a relatively long turnover time of about 25 days (see “Albumin synthesis and clearance”), this steady state Equation 1 is a valid description of CP only for the situation in which CP is relatively constant for several weeks. For example, acute changes in albumin capillary permeability produce short-term reductions in CP that are not described by Equation 1 and will be discussed in the “Acute, non-steady state shifts in albumin spaces” section.
Despite extensive study, the mechanisms involved in albumin homeostasis are not fully understood. Maximal CP levels are observed in healthy subjects, that is, there is no condition other than dehydration that might be termed “hyperalbuminemia.” This normal serum concentration is maintained by albumin synthesis/removal rates of about 10.5 g/day (Table 1). Since normal renal and GI losses are less than 6% (see “Renal albumin clearance”) and 10% (see “Gastrointestinal albumin clearance”) of synthesis, respectively, at least 84% or 8.82 g/day of clearance is accounted for by the catabolic component (ClC) (see “Albumin catabolism”). Albumin synthesis rate is thought to be regulated primarily by alterations in colloidal osmotic pressure rather than albumin concentration, per se, based primarily on measurements of albumin synthesis in the rat liver perfused with varying colloid osmotic pressure and albumin solutions.10,11
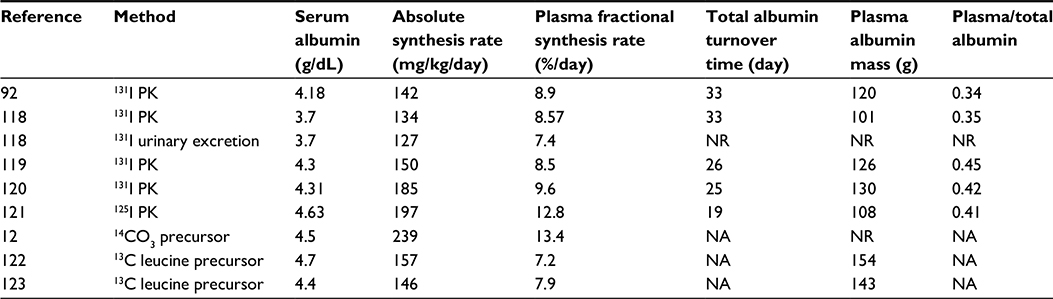 | Table 1 Selected normal values for serum albumin, absolute and fractional plasma synthesis rates, and plasma and total body distribution of albumin Note: 131I PK or 125I PK is steady-state tracer kinetics. Abbreviations: NA, not available by this technique, NR, not reported. |
An oft-stated concept is that the healthy liver can increase albumin synthesis by two- to three-fold in response to increased albumin turnover. Although such increases in synthesis have occasionally been observed in disease states,12 we are unaware of data in normal humans to support this concept. A study of a single healthy volunteer who underwent daily plasmapheresis (500 mL of blood removed, with red cells reinfused) for 30 days was the only controlled study of the response of the healthy liver to increased albumin turnover.13 An average of 9.7 g/day of albumin was removed by plasmapheresis (about 75% of the subject’s baseline rate of albumin turnover). The rate of albumin synthesis was determined from the rate of disappearance of labeled albumin before initiating plasmapheresis and then over the last 15 days of the plasmapheresis regimen when a steady state had been established presumably. The baseline albumin synthesis rate of 13.6 g/day increased to only 17.1 g/day during plasmapheresis, that is, the liver responded with only a 25% increase in albumin synthesis rather than the 75% increase needed to maintain normal albumin homeostasis. As a result, the subject’s CP declined from 4.45 g/dL at baseline to 3.64 g/dL after 4 weeks of plasmapheresis. Thus, in this one individual, the normal human liver did not respond to increased albumin turnover with a vigorous increase in synthesis, and what constitutes a “normal” response to a falling CP remains to be determined. On another occasion, the same subject underwent infusion of 10 g of albumin daily for 30 days. Albumin synthesis (measured over the last 15 days) decreased by only about 1 g/day, and the CP concentration increased from 4.45 to 5.54 g/dL. Although oncotic pressure measurements were not made in these experiments, albumin plus gamma globulin account for the vast majority of plasma colloidal osmotic pressure. Since serum gamma globulin declined by about 40% during plasmapheresis and increased by about 8% during albumin infusion, there should have been appreciable declines from normal in colloidal osmotic pressure during the course of plasmapheresis and increases during the albumin infusion. The failure to preserve the normal CP concentration in these experiments suggests that the healthy liver was relatively unresponsive to colloidal osmotic pressure changes. Similarly, the synthesis of albumin in hypoalbuminemic patients is seldom maximal (ie, two to three times normal); hence, it appears that the osmostat that is purported to regulate albumin synthesis and maintain normal albumin homeostasis is readily reset to permit hypoosmolality.
Serum albumin routinely declines in almost every disease state in which it has been studied; hence, albumin has been termed to be a negative acute phase reactant. Excessive loss of albumin into the urine or gut has been demonstrated in only a small minority of patients with hypoalbuminemia (nephrosis and protein-losing enteropathy [PLE]), thus the low CP usually results from decreased albumin production and/or increased albumin catabolism. Pinpointing the cause of hypoalbuminemia has clinical implications since manipulations to increase albumin production such as nutritional support would be indicated for reduced synthesis. In contrast, treatment of the underlying condition is required to decrease albumin catabolism. Parsing out the relative roles of defective synthesis versus increased catabolism in hypoalbuminemia is a complicated process that has received little discussion.
In order to determine whether a steady-state decrease in CP results from decreased synthesis versus increased clearance, it is necessary to measure the albumin synthesis rate. As can be seen from Equation 1, if, for example, the albumin synthesis rate is decreased 50% and clearance is unchanged, then CP will also be reduced by 50%. Another expression that is used to describe the kinetics is the “plasma fractional synthesis rate,” which is the synthesis rate divided by the total plasma albumin (see Equation 4). Since the synthesis rate = (clearance) * CP (Equation 1), if the only pathological change is a decrease in synthesis, with the clearance and plasma volume remaining normal, then CP should be reduced by the same fraction as the synthesis rate and the plasma fractional clearance (=synthesis/[CP VP]) will remain normal and unchanged. Table 2 lists various pathologic conditions, the steady state values of CP, the absolute albumin synthesis rate, the plasma fractional synthesis rate, and the total plasma albumin – all expressed as a fraction of the normal value. If the pathologic defect was simply a change in synthesis, with no change in clearance or volume of distribution, then the absolute synthesis and CP should be changed by an identical fraction, and all the other parameters in the table should be unchanged (ie, =1, the normal value). This is not the case for most of the conditions listed in Table 1, indicating that the pathology is not just a change in albumin synthesis and both synthesis and clearance may be altered. For example, a 25% decrement in synthesis with a 50% reduction in CP indicates that both decreased synthesis and increased clearance are responsible for half of the reduction in the CP.
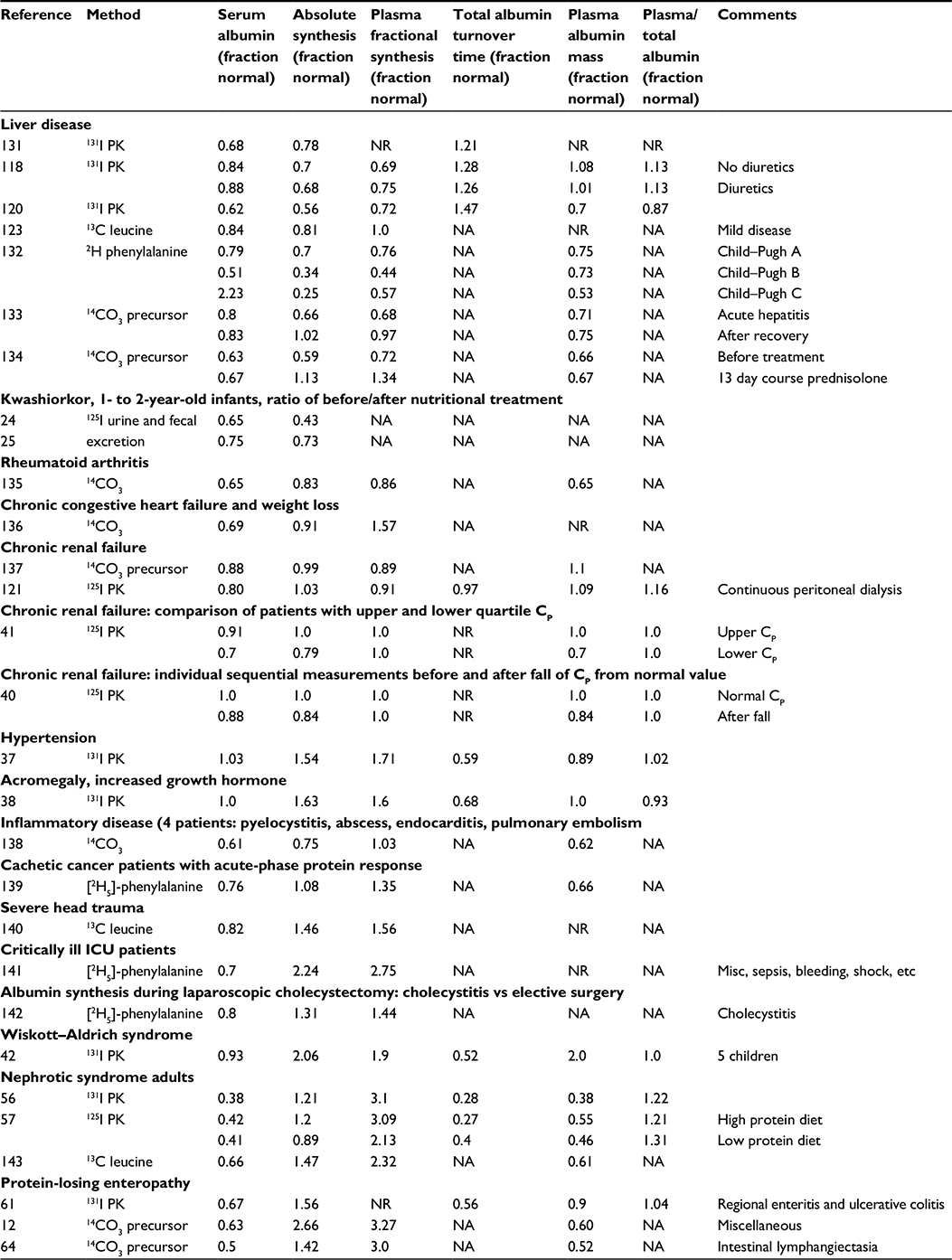 | Table 2 Human pathological albumin fractional turnover, absolute synthesis rates, and albumin plasma and total body distribution Notes: All the results are presented as the fraction of the reported normal control values. 131I PK and 125I PK, pharmacokinteic study using tracer 131I or 125I labeled albumin. Abbreviations: CP, serum albumin concentration; misc, miscellaneous; NA, not available by this technique, NR, not reported. |
However, the aforementioned analysis does not take into account that both synthesis and catabolism should respond homeostatically to a falling CP. For example, albumin synthesis has been observed to increase by at least twofold in PLE (Table 2) and the fractional catabolic rate decreases as CP falls (see “Albumin catabolism”). It can be argued that a failure of synthesis to double in conditions with severe hypoalbuminemia indicates that a defective synthetic response is contributing to the low CP. Similarly, hypoalbuminemia is normally accompanied by a decline in albumin clearance, and maintenance of a normal rather than decreased clearance could be considered to be a contributory factor in hypoalbuminemia. The permutations of synthesis, clearance, and CP in various disease states are explored in the subsequent sections of this review.
Albumin synthesis and clearance
Measurement techniques
Two fundamentally different approaches have been used to measure human albumin synthesis and clearance. The first approach is based on the measurements of the plasma clearance (ClP) of a bolus injection of a tracer of albumin. In the steady state, ClP × CP must be equal to the albumin synthesis rate (Equation 1). Since albumin has a half time of about 17 days, this approach requires measurements of tracer albumin over 2–3 weeks during which it is assumed that there is a steady state. The second approach uses labeled albumin precursors to directly measure the liver albumin synthesis rate over a period of a few hours. This approach does not provide data on albumin clearance. The technical demands of these methodologies account for the surprisingly limited available information concerning albumin synthesis, most of which is summarized in the tables and figures presented in this review. The steady-state pharmacokinetic analysis (described in detail in the Supplementary materials) provides two fundamental pharmacokinetic parameters: the steady-state albumin clearance (Clss) and volume of distribution (Vss), which can be related to the albumin synthesis rate and the total body albumin:
|
|
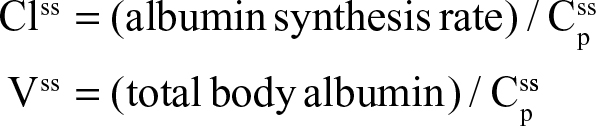
where Cpss is the steady-state CP concentration. The “total turnover rate” and “turnover time” can also be related to Clss and Vss:
|
|
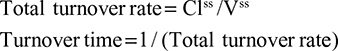
Although this approach has the experimental disadvantage that it requires the assumption of a steady state over 10–15 days to accurately determine Clss and Vss, it is the only technique that directly assesses total albumin clearance.
The alternative approach that directly measures albumin synthesis utilizes an entirely different set of assumptions. This technique involves the infusion of a labelled albumin precursor with measurement of the appearance of labelled albumin in the plasma over the ensuing 1–5 hours. The major experimental advantage of this method is that long-term measurements are not required and the theoretical advantage is that there is no steady-state assumption. Thus, this technique can be used to measure synthetic rates over short periods, such as after a meal or in patients with a fluctuating clinical course. A number of different precursors have been used, and various techniques are summarized in the Supplementary materials. This approach, which provides a direct measurement of the fraction of intravascular (ie, plasma) albumin synthesized per hour (the “plasma fractional synthesis rate”), has now become the standard measure of albumin synthesis. Fractional synthesis is related to the plasma albumin clearance (ClP, Equation 1) and plasma volume (Vp) by the relation:
|
|

The albumin synthesis rate is determined from Equation 4 with the plasma volume (VP) measured by some technique such as 125I albumin distribution. Since this technique does not provide any direct information about total body albumin, it cannot be used to measure total albumin turnover.
Synthesis of albumin in healthy subjects
Table 1 summarizes most of the reported measurements of normal human albumin catabolism or synthesis as well as the plasma and total body albumin, using the aforementioned techniques. Total body albumin is about 280 g, roughly 3% of the total body protein of 10 kg. About 40% of albumin is intravascular (Table 1) with the remaining 60% distributed in the interstitial albumin space of various organs (primarily muscle, adipose tissue, connective tissue, and skin) with an average interstitial concentration of about 60%–70% of that of plasma.14,15 The absolute synthesis rate is about 150 mg/kg/day or 10.5 g/day for a 70 kg human. Thus, roughly 8.5% of plasma albumin and 4% of the total body albumin are synthesized each day, corresponding to a total body albumin turnover time (Equation 3) of about 25 days or a half-time of 17.3 days. Compared to other circulating proteins, this turnover time is shorter than that of hemoglobin (life span 120 days), similar to that of gamma globulin, and much longer than that of many serum enzymes (lipase, amylase, etc) that have half-times of several hours. While albumin comprises only about 3% of total body protein, albumin turnover (150 mg/kg/day) represents about 19% of the total recommended dietary protein allowance of 800 mg/kg/day,16 indicating that albumin turnover is far more rapid than the average body protein.
Table 3 summarizes the reported measurements of various manipulations on the rate of albumin synthesis by healthy subjects. These observations utilize the acute precursor albumin synthesis technique discussed earlier, which should provide accurate assessment of the synthesis over 1–2 hour periods. The first three entries describe the result of a 14- to 28-day low-protein, isocaloric diet. Given that normal albumin synthesis rate is about 10.5 g/day of protein, it is not surprising that albumin synthesis fell by 20%–65% in response to administration of a very low protein (eg, 10 g/day), isocaloric diet (Table 2). It is important to note that the caloric intake was maintained at normal values in these studies. As discussed in “Albumin synthesis and protein malnutrition”, there is a strong evidence that when both protein and calories are restricted (ie, starvation), CP and, presumably, albumin synthesis remain close to normal because the breakdown of body protein to provide energy releases sufficient amino acids to maintain normal albumin synthesis.
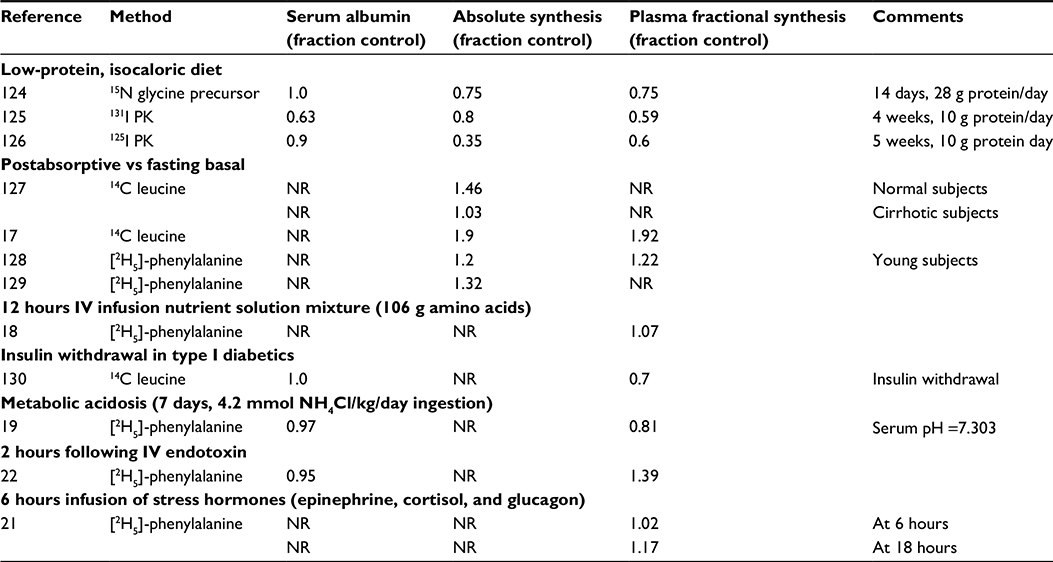 | Table 3 Acute changes in albumin synthesis rates in normal human subjects Note: Values are expressed relative to control. Abbreviations: IV, intravenous; NR, not reported. |
The next four entries in Table 3 indicate that there is a 20%–90% increase in the rate of albumin synthesis following a meal. As discussed by De Feo et al,17 this increase in albumin synthesis represents about 28% of the total increase in protein synthesis observed following a meal. Since albumin comprises only about 3% of total body protein, albumin synthesis is more responsive to oral administration of nutrients than is the average body protein, suggesting that albumin may serve as an important, but limited, storage site for body protein. The stimulus for this post-cibal increase in albumin is poorly understood. It is not simply the result of increased amino acid availability since the intravenous infusion of amino acids has no significant effect on albumin synthesis18 (Table 3).
The induction of mild acidosis (pH =7.3) in normal subjects by a 7-day NH4Cl infusion produces a 20% decrease in albumin synthesis19 (Table 3). This may be clinically significant because hypoalbuminemia is a common correlate of chronic renal disease, and acidosis has been suggested to be a possible causative factor.20 The last two entries in Table 3 indicate that infusion of several stress-related hormones (epinephrine + cortisol + glucagon)21 or endotoxin22 acutely increased albumin synthesis of healthy subjects.
Albumin synthesis and protein malnutrition
There was a general consensus in the older literature that hypoalbuminemia was an indicator of malnutrition1,20 and CP, along with other hepatically synthesized proteins such as pre-albumin, is used to evaluate nutritional status.23 However, it has now been convincingly established that CP poorly correlates with nutritional status and should not be regarded as a good marker of malnutrition.20 The classical example of a strong correlation between hypoalbuminemia and malnutrition is the kwashiorkor syndrome produced by a low protein, normal calorie diet in infants and children. As shown in Table 2, low CP in these children is associated with a low albumin synthesis rate, both of which are reversed after 1–3 months of nutritional therapy.24,25 This is consistent with the 20%–65% reduction in CP following a 14- to 28-day feeding of a low protein, isocaloric diet (Table 3). However, these examples represent special cases in which there is a marked reduction of protein intake in the presence of normal caloric intake and are not representative of general malnutrition. As reviewed by Friedman and Fadem,20 CP does not fall appreciably during a 6-month near-starvation diet or in cases of chronic anorexia nervosa. Presumably, during caloric starvation, protein released from tissue turnover, for example, muscle breakdown, is sufficient to maintain a normal CP.
As has been emphasized in the “Introduction” section, hypoalbuminemia is strongly correlated with surgical mortality1 and is one of the strongest indicators of a high risk for post-surgical morbidity.26 These correlations led to the prediction that improvements in nutrition that raise the albumin level would improve the adverse prognosis associated with hypoalbuminemia. However, this prediction could not be confirmed in a number of controlled studies designed to test the relationship between nutritional supplementation, CP and clinical status. For example, preoperative parenteral nutrition (PN) in hypoalbuminemic subjects resulted in no significant improvement in surgical mortality.26–29 Similarly, PN in critically ill patients did not alter mortality rates.29 Hypoalbuminemia in chronically ill patients does not seem to be altered by nutritional modifications. For example, 21 days of total PN in oncology patients30 or 3 months of dietary protein supplementation in hepatic encephalopathy patients31 had no effect on CP. Because of these and other results, CP is no longer considered a good marker of malnutrition, and a 2012 consensus statement of the Academy of Nutrition and Dietetics about the identification of adult malnutrition concluded that CP does not “consistently or predictably change with weight loss, calorie restriction, or nitrogen balance.”32
Albumin synthesis in hepatic diseases
Table 2 summarizes the available measurements of CP versus albumin synthesis rates in various human pathological conditions. By far the best characterized cause of hypoalbuminemia is liver disease. The first seven entries list the average reported values in patients with chronic and acute liver disease. Figure 2 shows a plot of the CP versus liver synthesis rate for the individual subjects in these reports. CP varies over a relatively small range, for example, only 7.8% of CP values were less than half the normal value (ie, <2.25 mg/dL). The solid line in Figure 2 is the least squares fit to the data. It can be seen that the reduction in CP concentration is roughly proportional to the reduction in liver synthesis rates, indicating that plasma albumin clearance (ClP) remains normal in liver disease (Equation 1). As discussed in “Gastrointestinal albumin clearance”, this normal ClP could represent a decrease in ClC balanced by an increase in GI clearance (ClGI).
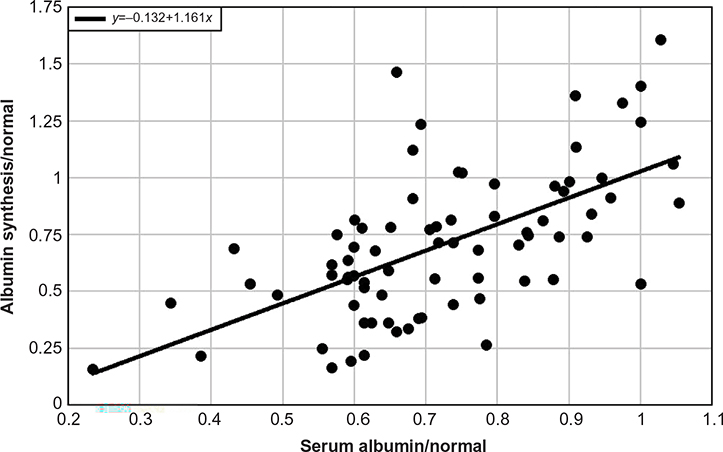 | Figure 2 Plot of albumin synthesis versus serum albumin (both normalized to the normal values) for subjects with acute and chronic liver disease. |
A measure of the relationship between liver excretory function and CP in liver disease is provided by a plot of Cp versus the clearance of exogenously administered compounds that are removed solely and rapidly by the liver. Figure 3 shows a summary plot of a total of 114 measurements of CP versus the clearance of antipyrine,33–35 indocyanine green,34 and aminopyrine36 in subjects with chronic liver disease. The solid line in Figure 3 indicates the predicted relationship if the decline in albumin synthesis (and the corresponding CP) was proportional to the decrease in clearance of these markers. Almost all the measurements fall below this line, indicating that liver clearance was decreased to a greater extent than is CP. Several different factors possibly contribute to this result: 1) the synthesis of albumin is inherently less inhibited by disrupted liver biochemistry than are drug detoxification reactions; 2) low CP specifically stimulates liver albumin synthesis which, therefore, is decreased less than the overall liver function; and 3) albumin ClC (ClC in Equation 1) is decreased when CP is decreased (see “Albumin catabolism”). Finally, CP is preserved to a greater extent than the excretory function of the cirrhotic liver.
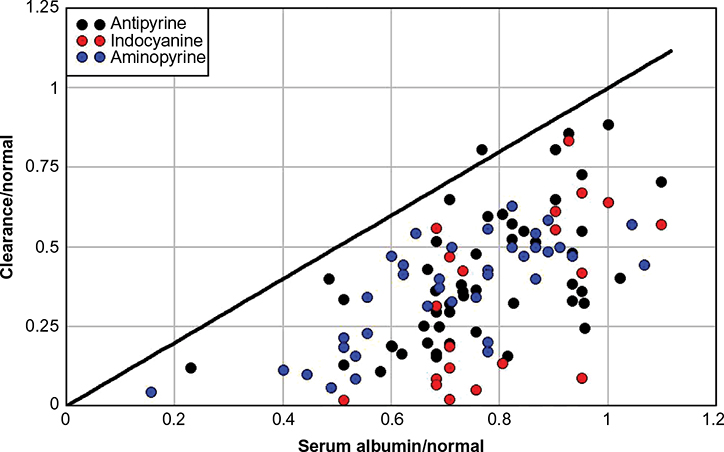 | Figure 3 Plot of the clearance of antipyrine (black), indocyanine green (red), or aminopyrine (blue) versus serum albumin (normalized to the normal value). Notes: The solid line is the expected relationship if clearance is proportional to serum albumin. Data from references 33–36. |
Albumin synthesis in extrahepatic diseases
As emphasized in the “Introduction,” relatively small decreases in CP are strongly correlated with prognosis in a wide variety of extra hepatic diseases. Table 2 lists the averages of measurements of albumin synthesis and CP for three chronic diseases not obviously associated with liver disease (rheumatoid arthritis, congestive heart failure, and chronic renal failure). In each case, the reduction in CP was greater to some extent than the corresponding reduction in albumin synthesis, suggesting that one or more of the clearance (Cl) terms in Equation 1 are increased. However, the disparities between Cp and synthesis were relatively small, and given the large variability of the data, conclusions concerning increased albumin catabolism are speculative. Friedman and Fadem20 have extensively reviewed the hypoalbuminemia of chronic renal failure and have concluded that although it is a “clinical index of illness,” its specific cause is unknown.
Table 2 describes the pharmacokinetics for hypertensive37 and acromegalic38 subjects, both of whom have a normal CP and, surprisingly, a significantly increased synthesis rate of about 1.6 times normal. The increased synthesis in the acromegalics may be a function of the hepatomegaly in these subjects38 and not of growth hormone itself which, in a short-term study, does not alter albumin synthesis.39 Both the conditions also have an increased albumin capillary permeability and, therefore, increased rates of recirculation between the blood and interstitial space, and Parving et al37 suggest that this might be associated with an increased catabolic rate. Independent of the cause of the increased synthesis, the finding of an increased synthesis and normal Cp can only be explained by an increased ClP (Equation 1).
In the older literature, the most common explanation for the hypoalbuminemia seen in various human diseases was that cytokines such as tumor necrosis factor or interleukin 6 inhibited hepatic synthesis of albumin.2 Although this hypothesis is supported by a large number of animal studies, there is only limited evidence for this in humans. Table 2 lists the measurements of CP and albumin synthesis for five conditions that may be in this “acute-phase” category: 1) patients with miscellaneous “inflammatory” diseases; 2) cachectic cancer patients with acute-phase protein response (ie, increased C-reactive protein); 3) severe head trauma patients; 4) critically ill intensive care unit patients; and 5) cholecystitis patients during laparoscopic surgery. The albumin synthesis rate was not reduced to the extent of the Cp in any of these conditions, and in most situations synthesis was actually increased above normal. Taken at face value, these data indicate that an increase in albumin clearance rather than decreased albumin synthesis is the dominant factor inducing hypoalbuminemia in these varied conditions. However, the decreased CP in these diseases may be a result of an acutely increased rate of albumin loss from the vascular space, and this decrease in CP might then stimulate the observed increased liver synthesis. For these reasons and because of the large variability of both CP and albumin synthesis in the heterogeneous pathologies listed in Table 2, it is difficult to establish a definite correlation between CP and albumin synthesis.
The strongest evidence that decreased synthesis may be the primary pathologic factor for the decrease in CP is provided by the studies by Kaysen et al40,41 in renal failure patients on dialysis. Comparing groups of these patients with high CP (upper quartile) versus those with low CP (lower quartile), the fractional decrease in CP is nearly identical to the decrease in absolute synthesis41 (Table 2), consistent with decreased synthesis as the primary cause of the low CP, with no significant change in plasma albumin clearance (see discussion of Equation 1 in “Albumin homeostasis”). More convincing support for this was provided by sequential measurements of albumin synthesis in individual patients before and after a fall in CP.40 Again, the decrease in CP could be quantitatively explained by a decrease in synthesis with no significant change in either albumin clearance or the volume of distribution (Table 2). If the pathology is a pure decrease in synthesis, then the fractional synthesis rate should be unchanged, and as shown in Table 2, this is the case for these two studies. Furthermore, the fall in CP was strongly correlated with the increases in serum inflammatory markers interleukin-6, C-reactive protein, and α1 acid glycoprotein. Thus, at least in renal failure patients, there is evidence that CP is determined primarily by the rate of liver albumin synthesis, and this synthesis is decreased in inflammatory states.
The inclusion of patients with Wiskott–Aldrich syndrome (X-linked eczema thrombocytopenia immunodeficiency syndrome) in Table 2 illustrates the rare example of a condition with a high albumin turnover (twice normal) with a normal CP and no obvious reason for the increased turnover. Since GI albumin loss was only slightly increased and there was no albuminuria, it was concluded that the increased turnover was a result of “hypercatabolism.”42 Although this disease might provide a clue about the relatively poorly understood factors involved in the catabolism of albumin (see “Albumin catabolism”), this observation has not been followed up, and there is no obvious connection between the genetic defect in Wiskott–Aldrich and albumin catabolism.43
The last six entries in Table 2 are for conditions in which the low CP is associated with a documented increase in renal (nephrotic syndrome) or GI (regional enteritis and ulcerative colitis or PLE) clearance. The increased albumin loss is partially offset by a fractional increase in albumin synthesis varying from 1.47 in nephrotic syndrome to a high of 2.66 times normal in PLE. These results will be discussed in more detail in the following sections on urinary and GI clearance.
Renal albumin clearance
Albumin excretion in the urine, which is easily quantitated, is normally <20 mg of albumin/day44,45 (<0.2% of the normal daily 10.5 gm albumin turnover). However, urinary loss underestimates the potential of the kidney to clear albumin since the proximal tubule cells take up filtered protein, which is hydrolyzed in the lysosomes with the amino acids returned to the plasma.46–48 This tubular uptake is an active process involving the specific albumin-binding proteins megalin and cubilin48 and is blocked in cubulin-deficient mice.49 Since all filtered albumin is excreted or catabolized, kidney is the one organ in which albumin catabolism could be quantitated, assuming the glomerular filtration of albumin is known. This filtration rate is a function of the glomerular sieving coefficient of albumin, which cannot be directly measured in humans. The most recent micropuncture measurements in rats suggest that the sieving coefficient is about 0.0005 of that of a freely filtered compound such as creatinine.48,50,51 Extrapolating this seemingly small sieving coefficient to humans (glomerular filtration rate =120 mL/min, CP =4.5 g/dL) yields an albumin clearance of 3.9 g/day. This value, which represents 37% of the total human albumin clearance of 10.5 g/day, is likely to be an overestimate in that the proximal tubule sample is readily contaminated with serum protein during micropuncture.50 A much lower estimate is obtained if it is assumed that there is no proximal tubule albumin uptake in cubilin-deficient mice, that is, all the filtered albumin appears in the urine. Extrapolating from the sixfold increase in urine albumin observed in cubilin-deficient mice to humans yields an estimate of 0.18 gm/day, only 1.7% of total albumin turnover.50 Another estimate is based on the measurements of urine albumin in patients with Fanconi syndrome, which represents a human “knock-out” of renal tubular protein reabsorption. If all the filtered albumin appears in the urine in these subjects, the glomerular sieving coefficient52 is 7×10–5 corresponding to a total renal albumin clearance of about 0.6 g/day or 5.7% of total albumin synthesis. A maximal possible value of ClRe is provided by the measurement of total albumin clearance in a subject with analbuminemia. As discussed in the “Albumin catabolism” section, the total clearance falls as CP falls because the ClC is decreased. Tracer measurements using 131I-labeled albumin in one analbuminemic subject indicate that the total albumin clearance is reduced by a factor of about 6%–16% of normal.5 Assuming that the ClC is reduced to zero in this subject (an overestimate) and the ClGI is normal (≈10%), the renal clearance must be <6% of the total normal albumin synthesis rate. In summary, in normal adults, the kidneys contribute probably no more than about 6% of the total albumin turnover, and this contribution is almost entirely “catabolic” with no significant intact albumin being secreted in the urine.
The best studied condition in which there is an enhanced albumin turnover is the “nephrotic syndrome” which is defined as any condition in which there is >3.5 g/day of urinary protein excreted.53 Since the bulk of this protein is albumin, this minimum protein loss represents an appreciable fraction, 3.5/10.5 or 30% of the normal rate of albumin turnover, and this loss does not account for the increased renal catabolism as a result of increased delivery of filtered albumin to the proximal tubule. In addition, the GI albumin clearance in nephrotic syndrome patients varies from negligible in about half of the patients54 up to values of about 3.5 g/day.55 Thus, nephrotic syndrome should induce a severe strain on albumin synthetic capacity of the body if the CP were to remain normal.
Two studies have reported both the urine albumin excretion and the total steady state albumin synthesis in a series of nephrotic syndrome patients with varied diagnoses and no obvious liver disease56,57 (Table 2). The data for the 42 subjects in these two studies are plotted in Figures 4 and 5. Figure 4 shows a plot of urinary albumin excretion versus either the albumin synthesis rate normalized for the control rate (red circles) or the fraction of total synthesis represented by urinary excretion (black circles). As might be expected, albumin synthesis increased with increasing loss of albumin in the urine, that is, as the urine albumin excretion rate increased to a maximal value of 0.24 g/kg/day (16.8 g/day/70 kg), total albumin synthesis approximately doubled, increasing by about 0.16 g/kg/day (red points) and the total urine albumin excretion increased to about 78% of the total synthesis rate (black points). Figure 5 shows a plot of the same two variables versus CP. It is usually assumed that the major factor controlling albumin synthesis is a decrease in CP (or, equivalently, colloid osmotic pressure).2 If this were the case, the rate of synthesis should be inversely proportional to CP. However, as seen in Figure 5 (red points), the rate of synthesis is relatively independent of CP. Thus, synthesis is responsive to the loss of albumin in the urine (Figure 5), but this responsiveness does not appear to be mediated by a falling CP concentration. One possible explanation is that the rate of synthesis is maximally turned on at all the CP values in Figure 5 and cannot be further increased as CP falls. However, the synthesis increases by an average factor of only 1.2 for these nephrotic syndrome patients, much less than the average increase of 2.66 observed in patients with PLE (Table 2), indicating that much larger increases should be possible. Perhaps these nephrotic syndrome patients have some underlying disease or other factor that limits the potential increase in liver albumin synthesis. Another explanation would be that the excretion of albumin in the urine creates a drain of body protein, whereas most of the protein lost into the gut is hydrolyzed to amino acids and reabsorbed so that there is no nutritional deficiency.
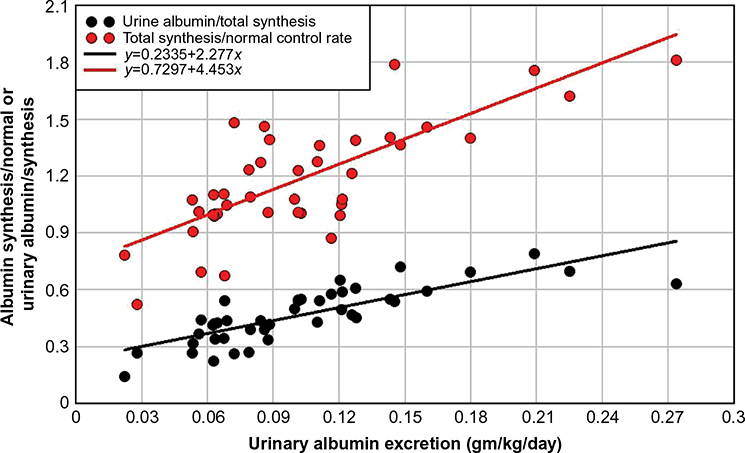 | Figure 4 Plot of either the total albumin synthesis rate relative to normal (red) or the urinary albumin excretion relative to total synthesis (black) versus the urinary albumin excretion in a series of nephrotic syndrome patients with varied diagnoses and no obvious liver disease. Note: Experimental data from references 56 and 57. |
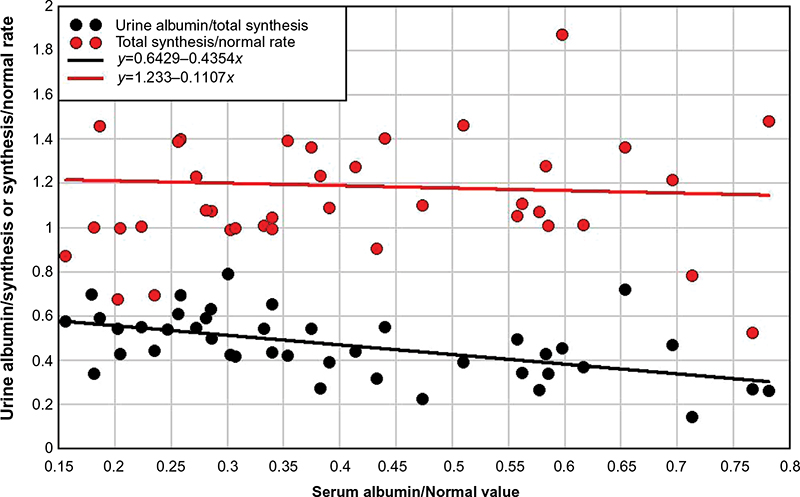 | Figure 5 Plot of either the total albumin synthesis rate relative to normal (red) or the urinary albumin excretion relative to total synthesis (black) versus the serum albumin relative to normal. Note: Experimental data from references 56 and 57. |
There is a puzzling observation related to the effect of changes in dietary protein in nephrotic patients. Kaysen et al have shown that although increasing dietary protein increases albumin synthesis in both rats58 and humans,59 it also increases albuminuria by a corresponding greater amount, so that CP actually decreases with the increased dietary protein. The authors discuss various possible explanations for this paradoxical result, with no definite conclusions.
Gastrointestinal albumin clearance
PLE describes a diverse group of diseases that have an increased GI albumin clearance in common.60 These conditions range from those that 1) specifically alter intestinal mucosal histology (Crohn’s disease, celiac disease, or Menetrier’s disease) or 2) induce anatomical changes in the mucosa as part of a systemic disease (Karposi’s sarcoma, sarcoidosis, or toxic shock syndrome) or 3) alter intestinal epithelium permeability while the epithelium remains grossly intact (increased lymphatic pressure in congestive heart failure, portal hypertension, or intestinal lymphangiectasia).60 Various techniques have been used to determine the rate of GI loss or clearance (ClGI, Equation 1) of systemic albumin. The first suggestion that hypoalbuminemia could result from increased ClGI was based on the measurements of the fraction of an intravenous dose of 131I-polyvinylpyrolidine (PVP) that appeared in a 4-day stool collection.61 Patients with regional enteritis or ulcerative colitis excreted about 5% of the intravenous 131I-PVP dose in their stool, roughly 10 times the normal value. Unfortunately, 131I-PVP measurements provided only qualitative information since PVP has a heterogeneous size distribution (mean molecular weight about 40 kD), and the gut clearance is not representative of albumin (molecular weight of 68 kD).61
The gold standard method to quantitate intestinal protein loss involves measurement of the 4-day fecal 51Cr excretion following intravenous infusion of 51Cr-albumin as described by Waldmann.62 Since Cr is neither absorbed nor secreted by the GI tract, stool Cr provides a quantitative measure of the 51Cr that leaks into the gut attached to albumin. Measurements are conventionally carried out on a 4-day stool collection.62 Since serum 51Cr disappears with a relatively fast and individually variable half time of about 5 days,63 and the stool collection is for 4 days, fecal clearance measurements are only semiquantitative unless corrected for the falling 51Cr-albumin serum concentration (which is not routinely performed). The mean 4-day fecal excretion for normal controls, about 0.23% of injected 51Cr (upper limit of normal =0.7%), rises markedly in various conditions with values as high as 10% (40-fold elevation over normal) observed in patients with intestinal lymphangiectasia.
In a Herculean study, Waldmann et al63 measured the 51Cr-albumin GI loss in 180 subjects and 50 controls (normal CP and no evidence of GI disease), and 130 subjects were “selected because they had significant hypoalbuminemia that could not be explained on the basis of either liver disease or proteinuria”. The hypoalbuminemic patients had various conditions – some with obvious GI pathology such as intestinal lymphangiectasia, Whipple’s disease, sprue, and so on and some for which the intestinal involvement was more indirect such as cardiac disease, amyloidosis, and cystic fibrosis. Figure 6 shows a plot of CP versus 51Cr lost in the GI tract (% of dose in feces/4 days) for the controls and all of the hypoalbuminemic subjects. Despite the semiquantitative nature of the fecal loss measurements and the likelihood that Cp could be influenced by factors other than fecal albumin loss, a remarkably good correlation was observed between 51Cr-albumin GI loss and CP. Nearly all patients with a CP <2.5 g/dL lost >6% of the dose/4 days in the stool (26 times normal). In a subset of 70 hypoalbuminemic subjects, 125I-albumin clearance measurements showed that the 40% with stool 51Cr in the normal range (<0.7%) had normal rates of plasma albumin fractional synthesis (=VP ClP; see Equation 4). Of the remaining 60% who had increased stool 51Cr, 81% had an increased rate of plasma albumin fractional synthesis.
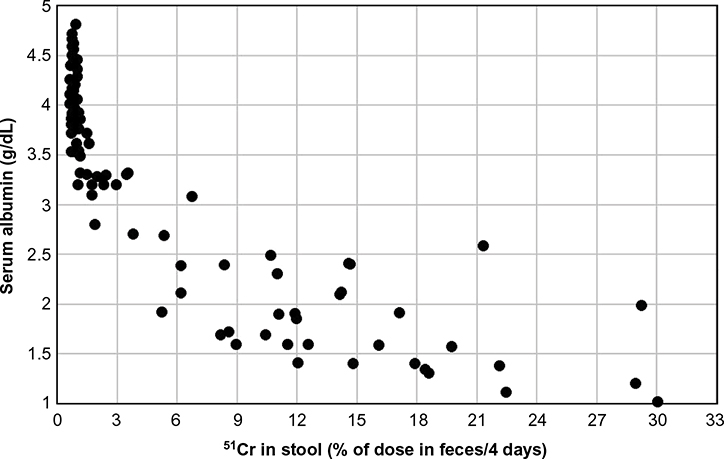 | Figure 6 Plot of serum albumin versus the percent of an intravenous dose of 51Cr-labelled albumin collected over 4 days of stool sampling in 50 control subjects and 130 patients with hypoalbuminemia and no obvious liver or renal disease. Note: Experimental data from reference 63. |
In a subset of 31 of the subjects with increased GI excretion (all with hypoalbuminemia), the ClGI was further quantitated by relating the measurements of the 51Cr collected in daily stool samples over 9 days to the daily measurements of the serum 51Cr-albumin. In normal controls, the average clearance was 13 mL of plasma/day, about 6% of the total normal albumin turnover rate. Figure 7 shows the remarkable correlation (slope of regression line 1.02) between the % of plasma albumin degraded (or synthesized) per day (=VP ClP) versus the % of plasma albumin cleared in the GI tract per day (=VP ClGI). This result demonstrates that the increased rate of plasma fractional clearance was entirely accounted for by the increases in GI excretion (ClGI) with the other two clearance terms in Equation 1 (ClRe and ClC) remaining normal and unchanged. In addition, the excellent correlation also speaks for the accuracy of both the measures of clearance. Unfortunately, individual subject data were not provided in this study. It would be of interest to know the specific diagnoses of these 31 patients. Also, since the values of Cp were not reported, it is not possible to directly determine the absolute albumin synthesis rates. A rough estimate of the synthesis rate is provided from the following calculation. The subjects with an intermediate rate of ClGI (eg, 15%/day) synthesize 25% of the total plasma albumin/day (Figure 7) and have CP of about 2 g/dL (Figure 6) so that the total synthesis rate (=0.25×CP×VP) is 14.3 g/day (assuming a normal VP of 28.5 dL), 36% greater than normal. This estimate is in rough agreement with the average values of the measurements of Steinfeld et al61 and Wochner et al64 (Table 2).
 | Figure 7 Plot of the percent of plasma albumin that is synthesized/day versus the percent of plasma albumin that is excreted into the GI tract/day in subjects with increased GI albumin clearance and hypoalbuminemia. Note: Experimental data from reference 63. Abbreviation: GI, gastrointestinal. |
The technique used to diagnose PLE clinically is the α1-antitrypsin (αAT) clearance measurement first described by Bernier et al.65 The αAT plasma clearance is determined from the αAT in a 3-day stool collection and the plasma αAT. The upper limit of normal is 24 mL/day,60 which corresponds to about 0.9% of plasma albumin/day, 1.1 gm%/day, or 10% of the total albumin synthesis rate. The validity of αAT clearance as a measure of ClGI is based on the standard assumptions that this “50 kd glycoprotein similar in size to albumin … is neither actively absorbed nor secreted in the intestine …[and] is also resistant to luminal proteolysis”.60 Although αAT clearance is linearly correlated with simultaneous measurements of 51Cr-albumin clearance, the correlation is weak (r=0.45) and the specificity and sensitivity of αAT for the diagnosis of PLE (assuming 51Cr clearance as the standard) is only 58% and 80%, respectively.66 Possible reasons for the inaccuracy of the αAT clearance include varying rates of αAT biliary secretion, intestinal proteolysis of αAT, and the presence of several protein complexes of αAT in feces that differ from the serum form.66
It is generally assumed that PLE can result from elevations in central venous pressure secondary to heart failure or constrictive pericarditis.60 The classic observation is that of Davidson et al67 who described four patients (three with chronic constrictive pericarditis and one with a large atrial septal defect) who had hypoalbuminemia, increased albumin synthesis, and PLE as measured with 131I-PVP stool collections. In each patient, these values returned to normal after surgical correction of the cardiac defect, with the stool 131I-PVP decreasing by nearly 10-fold. Most of the current literature on PLE in congestive heart failure is focused on children undergoing the palliative Fontan procedure, the current standard treatment for a congenital single ventricle. This procedure, which involves diverting venous blood directly to the pulmonary artery, results in a marked increase in central venous pressure and, in 3%–18% of subjects, is associated with PLE.68 The pathophysiology of the PLE in these children is uncertain because some patients with severely elevated venous pressure never develop PLE, and it has been suggested that the enteropathy may be produced by the chronic low cardiac output rather than the increased venous pressure.68
A number of studies have measured ClGI in patients with cirrhosis and portal hypertension. The results indicate that most cirrhotics do not have PLE. However, a small fraction of severely hypoalbuminemic cirrhotics had significantly increased ClGI,69–71 which normalized after portosystemic shunting72,73 or liver transplantation.74
Albumin catabolism
In the steady state, the albumin synthesis rate must be balanced by the renal, GI, and catabolic components defined in Equation 1. Since the normal renal and GI loss are <6% (section “Renal albumin clearance”) and 10% (section “Gastrointestinal albumin clearance”) of synthesis, respectively, at least 84% or 8.82 g/day must be removed by non-renal and non-GI albumin catabolism, referred to here as the ClC. There is a surprising lack of information concerning the specifics of this ClC. Studies using “residualizing” albumin labels that remain trapped in the lysosome after degradation suggest that about 60% of this albumin catabolism occurs in skin and muscle, apparently primarily in the fibroblasts of these organs.2,75
The most important new observation in albumin pharmacokinetics is the recognition of the importance of the major histocompatibility complex-related Fc receptor of immunoglobulin G (IgG; FcRn) in the control of albumin catabolism. FcRn had been previously shown to bind to IgG at acidic pH, diverting it from lysosomal degradation and prolonging IgG lifespan.76 It has now been shown that FcRn has the same action for albumin. FcRn knockout mice have an albumin catabolic rate twice that of normal mice and a 40% lower Cp along with a synthesis rate 20% greater than normal, presumably in response to the lower CP.77,78 It is not known whether the body is able, for example, to upregulate FcRn as a way to increase CP in hypoalbuminemia.78
It has been known for 50 years that the total plasma clearance (ClP, Equation 1) is not a constant, but decreases as CP decreases.79 The most clear-cut illustration of this phenomenon is the tracer albumin half time of about 100 days observed in an analbuminemic subject,5 six times normal and corresponding to a ClP of only 17% of normal. Since, as discussed in “Renal albumin” and “Gastrointestinal albumin clearance”, the ClRe plus ClGI can account for up to 16% of the total clearance and they should be constant, independent of CP; this observation implies that the ClC component of ClP fell to near zero in this analbuminemic subject. Recently, this result was confirmed by the finding that the half time of a tracer dose of human albumin in a humanized FcRn mouse model with a genetic absence of albumin is about six times greater than that in mice with normal albumin.80 This new observation that FcRn rescues albumin from lysosomal degradation provides a biochemical explanation of the mechanism of the decreased albumin clearance in hypoalbuminemic subjects. Since the amount of FcRn is limited, only a finite amount of albumin can be rescued, and as CP falls, the fraction of albumin rescued increases and the ClC decreases. This has several clinical implications: 1) It should provide a mechanism for maintaining normal CP, decreasing ClC when CP falls and increasing ClC when CP rises. 2) This decreased ClC would explain why CP falls only to about half normal in severe liver disease with decreased synthesis rates (Figure 3) while, in contrast, decreased ClC would not be able to compensate for the increased ClRe in nephrotic syndrome, and therefore, CP could fall to lower values (Figure 5).
Another relatively new observation is that circulating albumin, like hemoglobin, becomes increasingly glycosylated (glycosalated albumin [GA]) with time at a rate dependent on the plasma glucose, with normal levels of GA of about 13%, increasing two- to three-fold in diabetics.81–83 It is known that some forms of glycosylated albumin are rapidly cleared from the circulation by the liver,84 raising the question of whether the albumin clearance of naturally occurring GA also increases, that is, the older albumin is cleared faster. If this does occur, the process must have a relatively small effect since the single exponential decay of tracer albumin over a period ranging from 6 to 30 days5 implies that albumin clearance is a simple first-order process and does not significantly increase as albumin ages. Another argument against a more rapid clearance of GA is that there is a linear relationship between the values of GA versus glycosylated hemoglobin (HbA1C) in diabetics with varying glucose control. If the GA form was more rapidly cleared, one would predict that, as serum glucose increased, the increase in serum GA would be less than the increase in HbA1C. Glycoslyated albumin might be a clinically useful measure of blood glucose in conditions such as inflammation and renal failure that are known to reduce red cell lifetime and make HbA1C less reliable.
Measurements of serum GA (corrected for serum glucose using HbA1C) might provide a simple quantitative measure of albumin lifetime (and, therefore, synthesis rates), with the % GA being proportional to lifetime. One example where this prediction seems to be confirmed is in the measurements of the effect of thyroid hormone on GA. Patients with hypothyroidism and thyrotoxicosis had GA levels 16% greater and 15% lower, respectively, than control subjects (there was no significant difference in HbA1C values).85 This is in rough agreement with the 24% decrease in the steady-state measurement of 131I-albumin synthesis observed in hypothyroid subjects.86
Acute, non-steady-state shifts in albumin spaces
In 1896, Starling87 described measurements of fluid transfer between vascular and extravascular spaces and proposed that this fluid movement was the result of a balance between the hydrostatic and colloid osmotic pressure difference, an idea that has led to the now classical Starling equation description of ultrafiltration and vascular fluid balance. Capillaries do have finite albumin permeability, and the lymph drainage of the interstitial space has a critical role in maintaining the albumin concentration difference across the capillary wall. Surprisingly, given its relative simplicity, some important controversies remain about this classical relationship. It has still not been settled whether the normal capillary permeability is the result of movement via endocytotic vesicles or via transport through fixed anatomic tight junctions.88 There is also a disagreement about whether the colloid osmotic pressure driving force is determined by the interstitial albumin (classical Starling equation), or the local albumin concentration just across the glycocalyx that is present on the vascular surface of the capillary.89,90 There is general agreement that normally the albumin moves unidirectionally from the blood to the tissue (either via bulk fluid filtration or endocytosis) and that there is negligible diffusion of albumin from the tissue to the blood.15 This one-way loss of albumin from the blood to the tissue is balanced by the lymphatic return. Since the rate of return of albumin varies for different tissues because of varying interstitial volumes and lymphatic drainage rates, a quantitative description of the return rate requires two to three exponential terms.91,92
It is well established that the CP can fall rapidly, within hours, following acute sepsis or trauma.93 Since albumin metabolism normally has a half-time of 17 days (see “Albumin synthesis and clearance”), such a rapid decrease must represent a rapid loss from the vascular space, presumably related to an increase in capillary filtration through an enlarged pore system.37 The total capillary permeability is quantitatively described by the “transcapillary escape rate” (TER) that is estimated from the initial (1 hour) fall in concentration of a tracer dose of albumin before there is any significant back flux from the tissue. Since the albumin movement across the capillary is unidirectional, this TER represents the net rate of loss of albumin to the tissue and is balanced by a nearly equal rate of lymphatic return to the plasma since the rate of tissue catabolism is relatively small compared to this rate. The normal TER is about 5%/hour93 (half-time of 13 hours). Fleck et al93 determined that, within 3–7 hours following cardiopulmonary bypass surgery, CP fell by about 22% and TER increased by about twofold, respectively, from the presurgery values. They also found that patients in septic shock had a CP of about half normal and a TER twofold or more and, within 5 days following the shock, the TER returned to near normal.93 Similarly, Ballmer et al94 showed that, within 2 days following the onset of an acute infectious disease, CP had fallen to about 62% of normal and TER increased by about twofold.
An increase in the capillary leak will not necessarily result in a large fall in CP. For example, if this increased filtration rate is accompanied by a corresponding increase in the lymphatic return and no change in interstitial volumes, CP could fall initially by at most about 18% because the normal interstitial albumin is 70% of CP.14 The rapid fall in Cp to about half its normal value in septic shock suggests that in addition to increased capillary leak there must also be an increase in the extravascular albumin volume of distribution.
Whatever the mechanism responsible for the acute changes in CP, after a period of about 25 days, a new steady state should be reestablished and, from Equation 1, CP should return to its normal value if the synthesis and clearances are unchanged. A chronic long-term change in CP cannot be produced just by changes in, for example, TER or extravascular albumin spaces. However, increased capillary permeability could produce chronic reductions in CP if it was associated with increased tissue ClC, and it has been suggested that the albumin tissue catabolism is a function of the number of times the albumin is recycled from the vascular to the interstitial space.37
Clinical implications of serum albumin (CP) measurements
Although physicians seemingly believe that measurement of CP provides valuable clinical information, there has been a lack of discussion as to what constitutes the normal reference range for this protein. Commonly 3.5 g/dL is considered to be the lower limit of normal, presumably because laboratories have found that roughly 2.5% of values in “healthy subjects” fall below this concentration. However, mortality data on 1.6 million apparently healthy life insurance applicants4 suggest that the “normal” CP should be set much higher than 3.5 g/dL. For example, the statistically determined lower limit of normal for 50- to 69-year-old males (includes 97.5% of all values) was 3.9 g/dL. However, as shown in Figure 1, there was an increasing relative mortality over a 12-year follow-up period as the applicant’s initial CP concentration declined below 4.3 g/dL. For example, groups of subjects with CP ranges of 4.1–4.3 g/dL, 4.0–4.1 g/dL, and 3.9–4.0 g/dL had relative mortalities of 135%, 153%, and 182%, respectively. Assuming that the reference range for a laboratory test should exclude values known to be associated with high relative mortality, the case can be made that the lower limit of normal for CP is not 3.5 g/dL but 4.3 g/dL (75% of all observations). The increased mortality associated with CP well above the commonly accepted 3.5 g/dL value is on the same order as in conditions commonly assumed to have very poor health outcomes. For example, the 220% relative mortality of subjects with CP of 3.75–3.90 g/dL is greater than the 190% relative mortality of extremely obese males with a body mass index of 42.5 kg/m2.95 Given the ability of CP to predict mortality in seemingly healthy subjects, the simple measurement of CP could possibly be used to triage subjects undergoing wellness examinations, with extensive testing reserved for the 25% of subjects with albumins <4.3 g/dL.
Figure 8 shows a plot of the 223 individual measurements of CP obtained in a single day at our VA Hospital relative to the lower limits of normal derived from the life insurance data. Only about 13% and 3%, respectively, of all outpatient and inpatient CP measurements were >4.3 g/dL, the lower limit of values associated with normal mortality. Thus, at least in the VA Hospital setting, a patient whom the physician believes might benefit from the measurement of his or her CP is so overwhelmingly likely to have a low value that a subnormal value can be assumed a priori. The clinically important question is what, if any, useful prognostic/diagnostic/therapeutic information is derived from knowledge of the extent of the reduction of a patient’s CP.
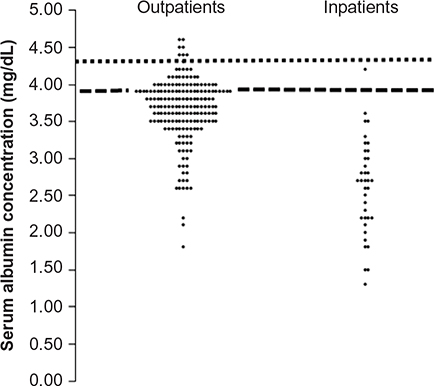 | Figure 8 Distribution of all serum albumin concentrations measured in a single day at the Minneapolis Veterans Administration Hospital for outpatients and inpatients. Note: The heavy dashed line is the lower limit of normal (lowest 2.5% of values) and the short dashed line is the average value observed in a group of 50- to 69-year-old life insurance applicants. |
Surgeons typically focus far more attention on the prognostic aspects of albumin measurements than do internists. This attention is prompted by 1) the striking increases in surgical complications associated with hypoalbuminemia, which are roughly five- to ten-fold greater for mortality and two- to three-fold greater for morbidity when patients’ serum albumins are <3 g/dL versus >4 g/dL1 and 2) the strong desire of surgeons to avoid post-surgical mortality and morbidity complications that are closely monitored for the individual surgeon. As a result, surgeons utilize CP as an important prognostic factor in determining the risk/benefit ratio for even relatively minor surgical interventions. Nonemergent surgery in hypoalbuminemic patients may be permanently shelved or temporarily postponed while attempts are made to improve the nutritional status of the subject. The stated goal of this nutritional therapy is not to raise the CP (which has been shown not to reduce surgical complications) but to improve the patient’s putatively depleted general “nutritional status” for which the low albumin is considered to be a surrogate marker. However, as discussed in “Albumin synthesis and protein malnutrition”, the concept that low albumin commonly serves as a good marker of overall nutritional status is flawed, and there is limited evidence that surgical complications can be reduced via a period of enteral or PN if the pathology causing the low albumin remains untreated.
Serum albumin concentration is a powerful predictor of a poor prognosis in nonsurgical as well as surgical patients. The pathological process in these conditions can be inflammatory, degenerative, or malignant as evidenced by statistically significant associations between albumin and a poor outcome in community-acquired pneumonia,96 chronic obstructive pulmonary disease,97 pancreatitis,98 inflammatory bowel disease,99 coronary100 and peripheral vascular101 disease, Alzheimer’s disease,102 multiple forms of malignancy,103 and all intensive care admissions.104 Even patients with psychiatric conditions such as schizophrenia105 and depression106 tend to have low CP. Although publication bias is probably playing a role in these observations, only very rare publication reported that albumin did not provide statistically significant prognostic information such as a paper showing that CP did not correlate with distant metastases for prostatic cancer.107
Despite the correlation between clinical outcomes and CP, this determination seldom plays a major role in the clinical approach to nonsurgical patients. Although there are multiple scoring systems to predict outcome of diseases, only one commonly used system formally utilizes the CP concentration – the Child–Pugh score used to predict the surgical mortality and need for liver transplantation in patients with cirrhosis. Albumin is the most predictive of the five factors that are used to calculate the Child–Pugh score, with albumin concentrations of >3.5, 2.8–3.5, and <2.8 g/dL adding 1, 2, or 3 points to the overall score, higher scores indicating worse prognoses.108 Thus, all albumin concentrations >3.5 g/dL are assumed to have the same prognostic value, which seems unlikely given the strong predictive power for mortality of values >3.5 g/dL in seemingly healthy subjects (Figure 1). Given the importance of the liver in albumin synthesis, it is surprising that albumin does not play a role in the calculation of the Model for end-stage liver disease (MELD) score, which has largely supplanted the Child–Pugh score as a prognostic indicator for liver failure. While worsening hypoalbuminemia is associated with poorer outcomes in cirrhosis, presumably albumin was found not to have prognostic value independent of the prothombin time, bilirubin, and creatinine when the MELD score was being formulated.
There are two possible limiting explanations for the strong correlation between CP and poor health outcomes. The first is that CP is essential for normal health, and even small decreases in concentration (eg, 15%) compromise albumin’s health-promoting functions. The second is that low CP is an indicator of the existence of a myriad of pathologies, some of which are so subtle that the subject appears “healthy” while harboring a lethal illness. Several observations strongly argue against the first explanation. First, congenitally analbuminemic subjects who have barely detectable CP experience a normal life expectancy6 and analbuminemic rats have normal reproductive ability.109 Second, a number of randomized studies have shown that infusion of exogenous albumin has little or no benefit on the medical outcome of hypoalbuminemic patients and may even be harmful, both in adults110–113 and children.114,115 Thus, albumin appears to be a surrogate marker for disease mortality and morbidity.
In contrast to the abundance of information concerning albumin concentrations in various disease states, there is very limited understanding of the pathophysiological mechanisms responsible for hypoalbuminemia. This article has attempted to summarize the enormous scientific literature concerning the synthesis and clearance of albumin so as to be able to discuss, albeit not fully explain, the pathophysiology of hypoalbuminemia. In the steady state, hypoalbuminemia necessarily reflects the alteration of the normal balance between albumin synthesis and albumin clearance. As discussed, accurate quantitation of albumin turnover (which in the steady state equals albumin synthesis) requires measurement of the rate that labelled albumin disappears from the serum over a several-week period during which time the subject’s albumin must remain in a steady state. The complexities of this measurement have limited its application as evidenced by the relatively few measurements that have been made in noncirrhotic conditions (Table 2).
As shown in Table 2, there is a good deal of scatter in the results of studies designed to measure the synthesis/clearance of albumin during assumed steady-state conditions in patients with hypoalbuminemia secondary to various disease states. However, there is a general tendency for synthesis to be reduced in patients with liver disease and isolated protein malnutrition and to be modestly increased in all other conditions, indicating increased catabolism in the latter groups of patients. This finding tends to refute the common belief that subtle protein malnutrition (which should reduce synthesis) is the primary cause of hypoalbuminemia in most disease states. However, it is an over-simplification to conclude that the hypoalbuminemia can be entirely attributed to decreased synthesis in liver disease and increased clearance in other conditions. For example, albumin clearance appeared to be roughly normal in the patients with liver disease although the ClC of albumin normally declines as CP concentration falls. Although speculative, loss of albumin into the intestinal tract in portal hypertension could possibly account for the maintenance of higher than expected albumin clearance in these patients and hence played a role in their hypoalbuminemia. Similarly, it is not clear to what extent failure of liver albumin synthesis to respond “normally” to the albumin drain resulting from the increased clearance in nonhepatic diseases aggravated the hypoalbuminemia in these subjects.
While the mechanism responsible for the increased albumin turnover in a wide variety of unrelated disease states is poorly understood, it has usually been assumed to result from an increase in the ClC term in Equation 1. An alternative explanation is provided by the intriguing study of Waldmann et al,63 in which both protein loss into the gut (ClGI, Equation 1) and total fractional albumin turnover (ClP, Equation 1) were measured in 130 subjects selected because they had “significant hypoalbuminemia that could not be explained on the basis of either liver disease or proteinuria.” Many of these patients had diseases not commonly considered to have appreciable intestinal pathology including neoplastic diseases, connective tissue diseases, heart failure, chronic pancreatitis, and so on. The remarkable findings of this study were that 1) when patients had no excess loss of protein into the gut, fractional turnover (ClP) of albumin was always normal, and 2) when there was increased albumin loss into the gut, this loss quantitatively accounted for the increased rate of albumin turnover, that is, ClP = ClGI (r=0.98). Thus, the absence or presence of protein loss into the gut was the sole determinant of normal versus abnormally rapid albumin turnover. These observations could be strengthened via the relatively simple assessment of fecal 1-alphtrypsin in hypoalbuminemic subjects who do not have obvious GI or liver disease. If increased GI protein loss is commonly observed, it would be of interest to determine how gut permeability to protein is altered in the vast array of conditions in which hypoalbuminemia is seemingly associated with increased albumin turnover.
While it is clear that physicians are interested in the CP of their patients (223 measurements in 1 day at a VA Hospital, Figure 8), it is less clear as to how this measurement alters diagnosis or treatment. Hypoalbuminemia ranging from minor to severe is observed in virtually every illness and thus this finding provides very little assistance in differential diagnosis. Occasionally, the CP can assist in determining whether borderline total serum calcium measurements are abnormal since the free calcium concentration is influenced by the albumin concentration. If it were easy to measure albumin synthesis/turnover, information might be obtained on whether liver disease was playing a role in the problem, since hepatic synthesis is elevated in most conditions other than liver disease. Unfortunately, measurement of synthesis is arduous and liver disease is more easily diagnosed via routine liver function studies. Given the 17-day half-life of albumin, it could be argued that the finding of a low CP is indicative of disease of several weeks duration. However, albumin can fall acutely in conditions where capillary protein permeability is increased. While some physicians understand that a low CP is indicative of severe disease or a poor outcome, this information usually does not translate into alterations in treatment. In the few clinical situations where albumin infusion has been shown to be useful – expansion of blood volume in acute renal failure, after peritoneal dialysis, or in the treatment of spontaneous bacterial peritonitis – albumin infusion is employed independent of the CP concentration. Assuming the above analysis to be correct, it appears that the care of patients would not suffer if far fewer albumin measurements were obtained.
Lastly, there is the perplexing question of how congenitally analbuminemic subjects survive with normal life expectancy and minimal morbidity despite the virtual absence of albumin, the major serum protein that putatively has many important functions including osmotic, binding, and antioxidant properties. The osmotic effect of the absence of CP has been studied in congenitally analbuminemic subjects. While albumin is responsible for about 80%–85% of the colloidal osmotic pressure of normal plasma, in analbuminemics there is a compensatory increase in globulins and other plasma proteins that raises the colloidal osmotic pressure to about 50% of normal. The finding of minimal or no edema in analbuminemic subjects indicates that 50% of the normal colloidal osmotic pressure is sufficient to limit edema formation and that factors other than colloidal osmotic pressure such as central venous pressure and venous valve competence are able to compensate. Given that albumin is freely permeable across the liver sinusoids, albumin exerts no osmotic effect on trans-sinusoidal fluid movement; hence, no tendency to ascites would be predicted in analbuminemia.8 The high-affinity albumin binding, usually regarded as an essential function,7 can also be compensated for in analbuminemics. In particular, the albumin binding of bilirubin is usually regarded as a critical factor in preventing kernicterus in newborns.116 Bilirubin binding has been carefully investigated in a rat analbuminemic model where it was shown that lipoprotein–bilirubin binding could compensate for the albumin deficiency.117 Analbuminemics provide inescapable evidence that either the functions of albumin are not critical to health or that these functions can be subsumed by other serum proteins.
Disclosure
The authors report no conflicts of interest in this work.
References
Gibbs J, Cull W, Henderson W, Daley J, Hur K, Khuri SF. Preoperative serum albumin level as a predictor of operative mortality and morbidity: results from the National VA Surgical Risk Study. Arch Surg. 1999;134(1):36–42. | ||
Peters T. All About Albumin, Biochemistry, Genetics and Medical Applications. San Diego: Academic Press; 1996. | ||
Nicholson JP, Wolmarans MR, Park GR. The role of albumin in critical illness. Br J Anaesth. 2000;85(4):599–610. | ||
Fulks M, Stout RL, Dolan VF. Albumin and all-cause mortality risk in insurance applicants. J Insur Med. 2010;42(1):11–17. | ||
Bennhold H, Kallee E. Comparative studies on the half-life of I 131-labeled albumins and nonradioactive human serum albumin in a case of analbuminemia. J Clin Invest. 1959;38(5):863–872. | ||
Watkins S, Madison J, Galliano M, Minchiotti L, Putnam FW. Analbuminemia: three cases resulting from different point mutations in the albumin gene. Proc Natl Acad Sci U S A. 1994;91(20):9417–9421. | ||
Fasano M, Curry S, Terreno E, et al. The extraordinary ligand binding properties of human serum albumin. IUBMB Life. 2005;57(12):787–796. | ||
Levitt DG, Levitt MD. Quantitative assessment of the multiple processes responsible for bilirubin homeostasis in health and disease. Clin Exp Gastroenterol. 2014;7:307–328. | ||
Taverna M, Marie AL, Mira JP, Guidet B. Specific antioxidant properties of human serum albumin. Ann Intensive Care. 2013;3(1):4. | ||
Dich J, Hansen SE, Thieden HI. Effect of albumin concentration and colloid osmotic pressure on albumin synthesis in the perfused rat liver. Acta Physiol Scand. 1973;89(3):352–358. | ||
Tracht ME, Tallal L, Tracht DG. Intrinsic hepatic control of plasma albumin concentration. Life Sci. 1967;6(24):2621–2628. | ||
Schomerus H, Mayer G. Synthesis rates of albumin and fibrinogen in patients with protein-losing enteropathy and in a patient recovering from protein malnutrition. Digestion. 1975;13(4):201–208. | ||
Andersen SB, Rossing N. Metabolism of albumin and gamma G-globulin during albumin infusions and during plasmapheresis. Scand J Clin Lab Invest. 1967;20(2):183–184. | ||
Levitt DG. The pharmacokinetics of the interstitial space in humans. BMC Clin Pharmacol. 2003;3:3. | ||
Lassen NA, Parving HH, Rossing N. Editorial: filtration as the main mechanism of overall transcapillary protein escape from the plasma. Microvasc Res. 1974;7(3):i–iv. | ||
Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids (Macronutrients): A Report of the Panel on Macronutrients, and the Standing Committee on the Scientific Evaluation of Dietary Reference Intakes. Washington, DC: National Academies Press; 2005. | ||
De Feo P, Horber FF, Haymond MW. Meal stimulation of albumin synthesis: a significant contributor to whole body protein synthesis in humans. Am J Physiol. 1992;263(4 Pt 1):E794–E799. | ||
Ballmer PE, McNurlan MA, Essen P, Anderson SE, Garlick PJ. Albumin synthesis rates measured with [2H5ring]phenylalanine are not responsive to short-term intravenous nutrients in healthy humans. J Nutr. 1995;125(3):512–519. | ||
Ballmer PE, McNurlan MA, Hulter HN, Anderson SE, Garlick PJ, Krapf R. Chronic metabolic acidosis decreases albumin synthesis and induces negative nitrogen balance in humans. J Clin Invest. 1995;95(1):39–45. | ||
Friedman AN, Fadem SZ. Reassessment of albumin as a nutritional marker in kidney disease. J Am Soc Nephrol. 2010;21(2):223–230. | ||
McNurlan MA, Sandgren A, Hunter K, Essen P, Garlick PJ, Wernerman J. Protein synthesis rates of skeletal muscle, lymphocytes, and albumin with stress hormone infusion in healthy man. Metabolism. 1996;45(11):1388–1394. | ||
Barle H, Januszkiewicz A, Hallstrom L, et al. Albumin synthesis in humans increases immediately following the administration of endotoxin. Clin Sci (Lond). 2002;103(5):525–531. | ||
Fuhrman MP, Charney P, Mueller CM. Hepatic proteins and nutrition assessment. J Am Diet Assoc. 2004;104(8):1258–1264. | ||
Cohen S, Hansen JD. Metabolism of albumin and gamma-globulin in kwashiorkor. Clin Sci. 1962;23:351–359. | ||
Picou D, Waterlow JC. The effect of malnutrition on the metabolism of plasma albumin. Clin Sci. 1962;22:459–468. | ||
Buzby GP, Williford WO, Peterson OL, et al. A randomized clinical trial of total parenteral nutrition in malnourished surgical patients: the rationale and impact of previous clinical trials and pilot study on protocol design. Am J Clin Nutr. 1988;47(2 Suppl):357–365. | ||
Verdery RB. Low weight and nutritional deficiency. J Am Geriatr Soc. 1992;40(5):536–537. | ||
Von Meyenfeldt MF, Meijerink WJ, Rouflart MM, Builmaassen MT, Soeters PB. Perioperative nutritional support: a randomised clinical trial. Clin Nutr. 1992;11(4):180–186. | ||
Heyland DK, MacDonald S, Keefe L, Drover JW. Total parenteral nutrition in the critically ill patient: a meta-analysis. JAMA. 1998;280(23):2013–2019. | ||
Gray GE, Meguid MM. Can total parenteral nutrition reverse hypoalbuminemia in oncology patients? Nutrition. 1990;6(3):225–228. | ||
Marchesini G, Dioguardi FS, Bianchi GP, et al. Long-term oral branched-chain amino acid treatment in chronic hepatic encephalopathy. A randomized double-blind casein-controlled trial. The Italian Multicenter Study Group. J Hepatol. 1990;11(1):92–101. | ||
White JV, Guenter P, Jensen G, et al. Consensus statement of the Academy of Nutrition and Dietetics/American Society for Parenteral and Enteral Nutrition: characteristics recommended for the identification and documentation of adult malnutrition (undernutrition). J Acad Nutr Diet. 2012;112(5):730–738. | ||
Andreasen PB, Ranek L, Statland BE, Tygstrup N. Clearance of antipyrine-dependence of quantitative liver function. Eur J Clin Invest. 1974;4(2):129–134. | ||
Branch RA, James JA, Read AE. The clearance of antipyrine and indocyanine green in normal subjects and in patients with chronic liver disease. Clin Pharmacol Ther. 1976;20(1):81–89. | ||
Kawasaki S, Sugiyama Y, Iga T, et al. Hepatic clearances of antipyrine, indocyanine green, and galactose in normal subjects and in patients with chronic liver diseases. Clin Pharmacol Ther. 1988;44(2):217–224. | ||
Hepner GW, Vesell ES. Quantitative assessment of hepatic function by breath analysis after oral administration of (14C)aminopyrine. Ann Intern Med. 1975;83(5):632–638. | ||
Parving HH, Rossing N, Jensen HA. Increased metabolic turnover rate and transcapillary escape rate of albumin in essential hypertension. Circ Res. 1974;35(4):544–552. | ||
Rossing N, Parving HH, Korsgaard O. Metabolism and transcapillary escape rate of albumin in acromegaly. Scand J Clin Lab Invest. 1974;33(1):39–44. | ||
Zachwieja JJ, Bier DM, Yarasheski KE. Growth hormone administration in older adults: effects on albumin synthesis. Am J Physiol. 1994;266(6 Pt 1):E840–E844. | ||
Kaysen GA, Dubin JA, Muller HG, et al. Inflammation and reduced albumin synthesis associated with stable decline in serum albumin in hemodialysis patients. Kidney Int. 2004;65(4):1408–1415. | ||
Kaysen GA, Yeun J, Depner T. Albumin synthesis, catabolism and distribution in dialysis patients. Miner Electrolyte Metab. 1997;23(3–6):218–224. | ||
Blaese RM, Strober W, Levy AL, Waldmann TA. Hypercatabolism of IgG, IgA, IgM, and albumin in the Wiskott-Aldrich syndrome. A unique disorder of serum protein metabolism. J Clin Invest. 1971;50(11): | ||
Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci. 2013;1285:26–43. | ||
Mundel P, Reiser J. Proteinuria: an enzymatic disease of the podocyte? Kidney Int. 2010;77(7):571–580. | ||
Peterson PA, Evrin PE, Berggard I. Differentiation of glomerular, tubular, and normal proteinuria: determinations of urinary excretion of beta-2-macroglobulin, albumin, and total protein. J Clin Invest. 1969;48(7):1189–1198. | ||
Maack T, Johnson V, Kau ST, Figueiredo J, Sigulem D. Renal filtration, transport, and metabolism of low-molecular-weight proteins: a review. Kidney Int. 1979;16(3):251–270. | ||
Park CH, Maack T. Albumin absorption and catabolism by isolated perfused proximal convoluted tubules of the rabbit. J Clin Invest. 1984;73(3):767–777. | ||
Gekle M. Renal tubule albumin transport. Annu Rev Physiol. 2005; | ||
Amsellem S, Gburek J, Hamard G, et al. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J Am Soc Nephrol. 2010;21(11):1859–1867. | ||
Castrop H. Reply to “Letter to the editor: ‘Quantifying albumin permeability with multiphoton microscopy: why the difference?’” Am J Physiol Renal Physiol. 2014;306(9):F1101–F1103. | ||
Schiessl IM, Castrop H. Angiotensin II AT2 receptor activation attenuates AT1 receptor-induced increases in the glomerular filtration of albumin: a multiphoton microscopy study. Am J Physiol Renal Physiol. 2013;305(8):F1189–F1200. | ||
Norden AG, Lapsley M, Lee PJ, et al. Glomerular protein sieving and implications for renal failure in Fanconi syndrome. Kidney Int. 2001;60(5):1885–1892. | ||
Orth SR, Ritz E. The nephrotic syndrome. N Engl J Med. 1998;338(17):1202–1211. | ||
Jensen H, Jarnum S, Hart Hansen JP. Gastrointestinal protein loss and intestinal function in the nephrotic syndrome. Nephron. 1966;3(4):209–220. | ||
Schultze G, Ahuja S, Faber U, Molzahn M. Gastrointestinal protein loss in the nephrotic syndrome studied with 51Cr-albumin. Nephron. 1980;25(5):227–230. | ||
Jensen H, Rossing N, Andersen SB, Jarnum S. Albumin metabolism in the nephrotic syndrome in adults. Clin Sci. 1967;33(3):445–457. | ||
Kaysen GA, Gambertoglio J, Felts J, Hutchison FN. Albumin synthesis, albuminuria and hyperlipemia in nephrotic patients. Kidney Int. 1987;31(6):1368–1376. | ||
Kaysen GA, Jones H Jr, Martin V, Hutchison FN. A low-protein diet restricts albumin synthesis in nephrotic rats. J Clin Invest. 1989;83(5):1623–1629. | ||
Kaysen GA, Gambertoglio J, Jimenez I, Jones H, Hutchison FN. Effect of dietary protein intake on albumin homeostasis in nephrotic patients. Kidney Int. 1986;29(2):572–577. | ||
Greenwald DA. Protein-losing gastroenteropathy. In: Feldman M, Friedman LS, Brandt LJ, editors. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. Vol 1. 10th ed. Philadelphia, PA: Saunders; 2016:464–470. | ||
Steinfeld JL, Davidson JD, Gordon RS Jr., Greene FE. The mechanism of hypoproteinemia in patients with regional enteritis and ulcerative colitis. Am J Med. 1960;29:405–415. | ||
Waldmann TA. Gastrointestinal protein loss demonstrated by Cr-51-labelled albumin. Lancet. 1961;2(7194):121–123. | ||
Waldmann TA, Wochner RD, Strober W. The role of the gastrointestinal tract in plasma protein metabolism. Studies with 51Cr-albumin. Am J Med. 1969;46(2):275–285. | ||
Wochner RD, Weissman SM, Waldmann TA, Houston D, Berlin NI. Direct measurement of the rates of synthesis of plasma proteins in control subjects and patients with gastrointestinal protein loss. J Clin Invest. 1968;47(5):971–982. | ||
Bernier JJ, Florent C, Desmazures C, Aymes C, L’Hirondel C. Diagnosis of protein-losing enteropathy by gastrointestinal clearance of alpha1-antitrypsin. Lancet. 1978;2(8093):763–764. | ||
Quigley EM, Ross IN, Haeney MR, Holbrook IB, Marsh MN. Reassessment of faecal alpha-1-antitrypsin excretion for use as screening test for intestinal protein loss. J Clin Pathol. 1987;40(1):61–66. | ||
Davidson JD, Waldmann TA, Goodman DS, Gordon RS Jr. Protein-losing gastroenteropathy in congestive heart-failure. Lancet. 1961;1(7183):899–902. | ||
Johnson JN, Driscoll DJ, O’Leary PW. Protein-losing enteropathy and the Fontan operation. Nutr Clin Pract. 2012;27(3):375–384. | ||
Becheur H, Pauwels A, Mostefa-Kara N, et al. [Gastric protein loss and alcoholic cirrhosis. Study by measurement of gastric clearance of alpha 1-antitrypsin]. Gastroenterol Clin Biol. 1996;20(8–9):669–673. French. | ||
Davcev P, Vanovski B, Sestakov D, Tadzer I. Protein-losing enteropathy in patients with liver cirrhosis. Digestion. 1969;2(1):17–22. | ||
Georgopoulos P, Mowat C, McMillan DC, Kingstone K, Ghosh S, Stanley AJ. Is portal hypertension associated with protein-losing enteropathy? J Gastroenterol Hepatol. 2005;20(1):103–107. | ||
Conn HO. Is protein-losing enteropathy a significant complication of portal hypertension? Am J Gastroenterol. 1998;93(1):127–128. | ||
Stanley AJ, Gilmour HM, Ghosh S, Ferguson A, McGilchrist AJ. Transjugular intrahepatic portosystemic shunt as a treatment for protein-losing enteropathy caused by portal hypertension. Gastroenterology. 1996;111(6):1679–1682. | ||
Wong WM, Hui CK, Yuen MF, et al. Reversal of protein-losing enteropathy by liver transplantation. J Clin Gastroenterol. 2003;36(1): | ||
Maxwell JL, Terracio L, Borg TK, Baynes JW, Thorpe SR. A fluorescent residualizing label for studies on protein uptake and catabolism in vivo and in vitro. Biochem J. 1990;267(1):155–162. | ||
Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I-related receptor FcRn. Annu Rev Immunol. 2000;18:739–766. | ||
Chaudhury C, Mehnaz S, Robinson JM, et al. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J Exp Med. 2003;197(3):315–322. | ||
Kim J, Bronson CL, Hayton WL, et al. Albumin turnover: FcRn-mediated recycling saves as much albumin from degradation as the liver produces. Am J Physiol Gastrointest Liver Physiol. 2006;290(2): | ||
Freeman T, Gordon AH. Albumin catabolism in hypoproteinaemic states studies with 131-I-albumin. Bibl Haematol. 1965;23:1108–1115. | ||
Roopenian DC, Low BE, Christianson GJ, Proetzel G, Sproule TJ, Wiles MV. Albumin-deficient mouse models for studying metabolism of human albumin and pharmacokinetics of albumin-based drugs. MAbs. 2015;7(2):344–351. | ||
Rondeau P, Bourdon E. The glycation of albumin: structural and functional impacts. Biochimie. 2011;93(4):645–658. | ||
Tamada D, Otsuki M, Kitamura T, et al. Effects of growth hormone excess on glycated albumin concentrations: analysis in acromegalic patients. Clin Chim Acta. 2015;440:93–96. | ||
Anguizola J, Matsuda R, Barnaby OS, et al. Review: glycation of human serum albumin. Clin Chim Acta. 2013;425:64–76. | ||
Nishikawa M, Yamashita F, Takakura Y, Hashida M, Sezaki H. Demonstration of the receptor-mediated hepatic uptake of dextran in mice. J Pharm Pharmacol. 1992;44(5):396–401. | ||
Koga M, Murai J, Saito H, Matsumoto S, Kasayama S. Effects of thyroid hormone on serum glycated albumin levels: study on non-diabetic subjects. Diabetes Res Clin Pract. 2009;84(2):163–167. | ||
Parving HH, Hansen JM, Nielsen SL, Rossing N, Munck O, Lassen NA. Mechanisms of edema formation in myxedema – increased protein extravasation and relatively slow lymphatic drainage. N Engl J Med. 1979;301(9):460–465. | ||
Starling EH. On the absorption of fluids from the connective tissue spaces. J Physiol. 1896;19(4):312–326. | ||
Tuma P, Hubbard AL. Transcytosis: crossing cellular barriers. Physiol Rev. 2003;83(3):871–932. | ||
Levick JR, Michel CC. Microvascular fluid exchange and the revised Starling principle. Cardiovasc Res. 2010;87(2):198–210. | ||
Woodcock TE, Woodcock TM. Revised Starling equation and the glycocalyx model of transvascular fluid exchange: an improved paradigm for prescribing intravenous fluid therapy. Br J Anaesth. 2012;108(3):384–394. | ||
Beeken WL, Volwiler W, Goldsworthy PD, et al. Studies of I-131-albumin catabolism and distribution in normal young male adults. J Clin Invest. 1962;41:1312–1333. | ||
Takeda Y, Reeve EB. Studies of the metabolism and distribution of albumin with autologous I131-albumin in healthy men. J Lab Clin Med. 1963;61:183–202. | ||
Fleck A, Raines G, Hawker F, et al. Increased vascular permeability: a major cause of hypoalbuminaemia in disease and injury. Lancet. 1985;1(8432):781–784. | ||
Ballmer PE, Ochsenbein AF, Schutz-Hofmann S. Transcapillary escape rate of albumin positively correlates with plasma albumin concentration in acute but not in chronic inflammatory disease. Metabolism. 1994;43(6):697–705. | ||
Berrington de Gonzalez A, Hartge P, Cerhan JR, et al. Body-mass index and mortality among 1.46 million white adults. N Engl J Med. 2010;363(23):2211–2219. | ||
Hedlund J. Community-acquired pneumonia requiring hospitalisation. Factors of importance for the short- and long term prognosis. Scand J Infect Dis Suppl. 1995;97:1–60. | ||
Gunen H, Hacievliyagil SS, Kosar F, et al. Factors affecting survival of hospitalised patients with COPD. Eur Respir J. 2005;26(2):234–241. | ||
Robert JH, Frossard JL, Mermillod B, et al. Early prediction of acute pancreatitis: prospective study comparing computed tomography scans, Ranson, Glascow, Acute Physiology and Chronic Health Evaluation II scores, and various serum markers. World J Surg. 2002;26(5):612–619. | ||
Chakravarty BJ. Predictors and the rate of medical treatment failure in ulcerative colitis. Am J Gastroenterol. 1993;88(6):852–855. | ||
Nelson JJ, Liao D, Sharrett AR, et al. Serum albumin level as a predictor of incident coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) study. Am J Epidemiol. 2000;151(5):468–477. | ||
Ishii H, Aoyama T, Takahashi H, et al. Serum albumin and C-reactive protein levels predict clinical outcome in hemodialysis patients undergoing endovascular therapy for peripheral artery disease. Atherosclerosis. 2013;227(1):130–134. | ||
Llewellyn DJ, Langa KM, Friedland RP, Lang IA. Serum albumin concentration and cognitive impairment. Curr Alzheimer Res. 2010;7(1):91–96. | ||
Gupta D, Lis CG. Pretreatment serum albumin as a predictor of cancer survival: a systematic review of the epidemiological literature. Nutr J. 2010;9:69. | ||
Lyons O, Whelan B, Bennett K, O’Riordan D, Silke B. Serum albumin as an outcome predictor in hospital emergency medical admissions. Eur J Intern Med. 2010;21(1):17–20. | ||
Devi PU, Murugan S. Biochemical derangements in patients with schizophrenia: a case-control study. J Clin Diagn Res. 2008;2(4): | ||
Huang TL. Lower serum albumin levels in patients with mood disorders. Chang Gung Med J. 2002;25(8):509–513. | ||
Berruti A, Dogliotti L, Bitossi R, et al. Incidence of skeletal complications in patients with bone metastatic prostate cancer and hormone refractory disease: predictive role of bone resorption and formation markers evaluated at baseline. J Urol. 2000;164(4):1248–1253. | ||
D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44(1):217–231. | ||
Nagase S, Shimamune K, Shumiya S. Albumin-deficient rat mutant. Science. 1979;205(4406):590–591. | ||
Alderson P, Bunn F, Lefebvre C, et al. Human albumin solution for resuscitation and volume expansion in critically ill patients. Cochrane Database Syst Rev. 2002;(1):CD001208. | ||
Alderson P, Schierhout G, Roberts I, Bunn F. Colloids versus crystalloids for fluid resuscitation in critically ill patients. Cochrane Database Syst Rev. 2000;(2):CD000567. | ||
Cochrane Injuries Group Albumin R. Human albumin administration in critically ill patients: systematic review of randomised controlled trials. BMJ. 1998;317(7153):235–240. | ||
Schierhout G, Roberts I. Fluid resuscitation with colloid or crystalloid solutions in critically ill patients: a systematic review of randomised trials. BMJ. 1998;316(7136):961–964. | ||
Boluyt N, Bollen CW, Bos AP, Kok JH, Offringa M. Fluid resuscitation in neonatal and pediatric hypovolemic shock: a Dutch Pediatric Society evidence-based clinical practice guideline. Intensive Care Med. 2006;32(7):995–1003. | ||
Oca MJ, Nelson M, Donn SM. Randomized trial of normal saline versus 5% albumin for the treatment of neonatal hypotension. J Perinatol. 2003;23(6):473–476. | ||
Ahlfors CE, Wennberg RP. Bilirubin-albumin binding and neonatal jaundice. Semin Perinatol. 2004;28(5):334–339. | ||
Takahashi M, Sugiyama K, Shumiya S, Nagase S. Penetration of bilirubin into the brain in albumin-deficient and jaundiced rats (AJR) and Nagase analbuminemic rats (NAR). J Biochem. 1984;96(6):1705–1712. | ||
Wilkinson P, Mendenhall CL. Serum albumin turnover in normal subjects and patients with cirrhosis measured by 131i-labelled human albumin. Clin Sci. 1963;25:281–292. | ||
Rossing N. The normal metabolism of 131I-labelled albumin in man. Clin Sci. 1967;33:593–602. | ||
Hasch E, Jarnum S, Tygstrup N. Albumin synthesis rate as a measure of liver function in patient with cirrhosis. Acta Med Scand. 1967;182(1):83–92. | ||
Kaysen GA, Schoenfeld PY. Albumin homeostasis in patients undergoing continuous ambulatory peritoneal dialysis. Kidney Int. 1984;25(1):107–114. | ||
Ballmer PE, McNurlan MA, Milne E, et al. Measurement of albumin synthesis in humans: a new approach employing stable isotopes. Am J Physiol. 1990;259(6 Pt 1):E797–E803. | ||
Ballmer PE, Walshe D, McNurlan MA, Watson H, Brunt PW, Garlick PJ. Albumin synthesis rates in cirrhosis: correlation with Child-Turcotte classification. Hepatology. 1993;18(2):292–297. | ||
Gersovitz M, Munro HN, Udall J, Young VR. Albumin synthesis in young and elderly subjects using a new stable isotope methodology: response to level of protein intake. Metabolism. 1980;29(11): | ||
Hoffenberg R, Black E, Brock JF. Albumin and gamma-globulin tracer studies in protein depletion states. J Clin Invest. 1966;45(1):143–152. | ||
Kelman L, Saunders SJ, Frith L, Wicht S, Corrigal A. Effects of dietary protein restriction on albumin synthesis, albumin catabolism, and the plasma aminogram. Am J Clin Nutr. 1972;25(11):1174–1178. | ||
Tessari P, Barazzoni R, Kiwanuka E, et al. Impairment of albumin and whole body postprandial protein synthesis in compensated liver cirrhosis. Am J Physiol Endocrinol Metab. 2002;282(2):E304–E311. | ||
Hunter KA, Ballmer PE, Anderson SE, Broom J, Garlick PJ, McNurlan MA. Acute stimulation of albumin synthesis rate with oral meal feeding in healthy subjects measured with [ring-2H5]phenylalanine. Clin Sci (Lond). 1995;88(2):235–242. | ||
Caso G, Feiner J, Mileva I, et al. Response of albumin synthesis to oral nutrients in young and elderly subjects. Am J Clin Nutr. 2007;85(2):446–451. | ||
De Feo P, Gaisano MG, Haymond MW. Differential effects of insulin deficiency on albumin and fibrinogen synthesis in humans. J Clin Invest. 1991;88(3):833–840. | ||
Sterling K. Serum albumin turnover in Laennec’s cirrhosis as measured by I131-tagged albumin. J Clin Invest. 1951;30(11):1238–1242. | ||
Ballmer PE, Reichen J, McNurlan MA, Sterchi AB, Anderson SE, Garlick PJ. Albumin but not fibrinogen synthesis correlates with galactose elimination capacity in patients with cirrhosis of the liver. Hepatology. 1996;24(1):53–59. | ||
Mayer G, Schomerus H. Synthesis rates of albumin and fibrinogen during and after acute hepatitis. Digestion. 1975;13(5):261–271. | ||
Cain GD, Mayer G, Jones EA. Augmentation of albumin but not fibrinogen synthesis by corticosteroids in patients with hepatocellular disease. J Clin Invest. 1970;49(12):2198–2204. | ||
Schomerus H, Mayer G. Synthesis rates of fibrinogen and albumin in patients with rheumatoid arthritis. Acta Hepatogastroenterol (Stuttg). 1977;24(1):11–14. | ||
Mayer G, Schomerus H. Synthesis rates of albumin and fibrinogen in patients with cardiac and pulmonary cachexia. Acta Hepatogastroenterol (Stuttg). 1977;24(2):82–85. | ||
Bianchi R, Mariani G, Pilo A. Albumin synthesis measurement by means of an improved two tracer method in patients with chronic renal failure. Effects of low protein diet. J Nucl Biol Med. 1970;14(4):136–144. | ||
Moshage HJ, Janssen JA, Franssen JH, Hafkenscheid JC, Yap SH. Study of the molecular mechanism of decreased liver synthesis of albumin in inflammation. J Clin Invest. 1987;79(6):1635–1641. | ||
Fearon KC, Falconer JS, Slater C, McMillan DC, Ross JA, Preston T. Albumin synthesis rates are not decreased in hypoalbuminemic cachectic cancer patients with an ongoing acute-phase protein response. Ann Surg. 1998;227(2):249–254. | ||
Mansoor O, Cayol M, Gachon P, et al. Albumin and fibrinogen syntheses increase while muscle protein synthesis decreases in head-injured patients. Am J Physiol. 1997;273(5 Pt 1):E898–E902. | ||
Barle H, Gamrin L, Essen P, McNurlan MA, Garlick PJ, Wernerman J. Growth hormone does not affect albumin synthesis in the critically ill. Intensive Care Med. 2001;27(5):836–843. | ||
Barle H, Hammarqvist F, Westman B, et al. Synthesis rates of total liver protein and albumin are both increased in patients with an acute inflammatory response. Clin Sci (Lond). 2006;110(1):93–99. | ||
Ballmer PE, Weber BK, Roy-Chaudhury P, et al. Elevation of albumin synthesis rates in nephrotic patients measured with [1-13C]leucine. Kidney Int. 1992;41(1):132–138. |
Supplementary materials
Measurements of albumin synthesis and catabolism
There are two fundamentally different approaches that have been used to measure human albumin synthesis and catabolism. The first approach is based on the analysis of the clearance of a bolus injection of a tracer of albumin. In the steady state, this clearance (ie, catabolism) must be equal to the albumin synthesis rate. Since albumin has a half-time of about 17 days, this approach requires measurements of tracer albumin over 2–3 weeks during which it is assumed that there is steady state, conditions that may not be valid in many disease states. The second approach uses labeled albumin precursors to measure the acute rate of liver albumin synthesis over a period of a few hours. This latter approach does not provide any direct measurement of albumin catabolic rates. Each of these methods is discussed here.
Notation:
D = tracer albumin dose
Cp(t) = tracer plasma albumin concentration as function of time
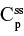 = steady state, nonlabeled, plasma albumin concentration
= steady state, nonlabeled, plasma albumin concentration
Q = total body metabolic rate
MT = total amount of nonlabeled albumin in body
M1(t) = amount of tracer in compartment 1 (plasma) as function of time
M2(t) = amount of tracer in compartment 2 (extravascular) as function of time
kout = Q/MT = fractional turnover rate for whole body
k1 = metabolic rate constant of compartment 1
k2 = metabolic rate constant of compartment 2
k12 = rate constant for exchange from compartment 1 to compartment 2
k21 = rate constant for exchange from compartment 2 to compartment 1
tT = turnover time = 1/kout
Clss = steady state clearance
Vss = steady-state volume of distribution
A1, A2 = coefficient of first (second) exponential describing tracer concentration
a1, a2 = rate constant of first (second) exponential describing tracer concentration
Steady-state measurement of albumin clearance and volume of distribution
The standard tracer pharmacokinetic experiment is to inject a tracer dose of albumin (eg, 131I-albumin) and determine the plasma tracer concentration (C(t)) over a time period that is long enough to be able to accurately extrapolate to infinity. The goal of these tracer measurements is to determine the two fundamental steady-state physiological parameters: Q, the total rate of metabolism of albumin, and MT, the total amount of albumin in the body. These two factors are determined experimentally from two related pharmacokinetic parameters determined from the tracer kinetic experiment, the steady-state clearance (Clss) and volume of distribution (Vss), defined by:
|
|

where 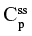 is the steady-state plasma albumin concentration. In addition, one can define the “fractional turnover rate” and the “turnover time”:
is the steady-state plasma albumin concentration. In addition, one can define the “fractional turnover rate” and the “turnover time”:
|
|
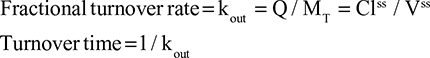
There are two different theoretical approaches that have been used to determine the albumin pharmacokinetics (compartmental and noncompartmental), and each of these is discussed in the Supplementary material. The compartmental approach used in the older literature describes the kinetics by an equivalent two- or three-compartmental system and determines Clss and Vss from the exponential decay at long durations (from 5 to 17 days). The major assumption required for this compartmental approach is that the exchange between the plasma and extravascular space is fast relative to the metabolic rate. This is a good assumption for albumin where the turnover rate is about 25 days while the intercompartmental exchange rate is about 1 day.1 The noncompartmental approach, first described by Meier and Zierler2 in 1955, has been used in the more recent publications. The only theoretical assumption required for this approach is that the albumin metabolism occurs in the plasma compartment, that is, the effective albumin concentration at the site of metabolism is equal to the plasma concentration. There is some experimental evidence that indirectly supports this assumption, and it is usually assumed that it is valid for albumin.
Noncompartmental pharmacokinetic analysis
This is a general approach that does not require any assumptions about the exchange kinetics between plasma and the different tissue interstitial spaces. It is assumed that the system is linear. The basic experiment is to input a tracer dose D and then measure the tracer concentration C(t) as a function of time. It can be shown that under very general conditions the steady-state clearance (Clss) is described by the following relation:
|
|
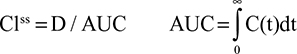
This is valid for an arbitrary input time course of the dose D and metabolism of albumin in any or all of the different extravascular spaces.
There is a similar, but less general, expression for Vss:
|
|
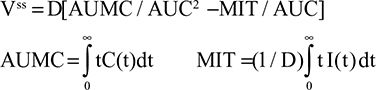
where I(t) is the time course of tracer input. Usually, it can be assumed that the input time course is a bolus input (I(t) = δ(t)) with very short time course compared to the albumin distribution:
|
|
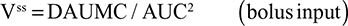
This relation is less general than the Clss relation (Equation S3) and is only valid if the metabolism occurs in the plasma compartment. If the metabolism occurs in an extravascular compartment, Vss cannot be determined without some additional assumptions about the pharmacokinetics in the extravascular space.3 As shown later for the two compartment models (Figure S1), this Vss relationship should be a good approximation for albumin even if the metabolism occurs in the extravascular space because the blood–tissue exchange rate is relatively fast (about 1 day) compared with the metabolic rate (about 20 days).
 | Figure S1 Schematic diagram of the two different compartmental models. Notes: I(t): input to system as function of time t; Mi(t): amount of solute in compartment i; ki: rate constant for excretion from compartment i; kij: rate constant for exchange from compartment i to compartment j; Q: amount of excretion. |
Compartment pharmacokinetic analysis
In this approach, it is assumed that the kinetics can be described by a compartmental model from which the clearance can then be determined. This is discussed in detail here for a two-compartment model, but the same analysis would apply to more complicated three-compartment models.
Two different limiting cases of the two-compartment model is shown earlier, differing in the site of albumin metabolism. M1 and M2 are the amounts of solute in the two compartments, V1 is the volume of compartment 1 and k12 and k21 are exchange rates from compartment 1 to compartment 2 and in the opposite direction, respectively. The concentration in compartment 1 is equal to:
|
|
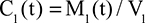
In Model 1, the albumin metabolism (Q) occurs from compartment 1:
|
|
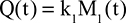
In Model 2, the metabolism occurs in compartment 2:
|
|
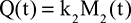
For either model, C1(t) for a bolus dose D is described by a sum of two exponentials:
|
|
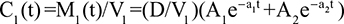
Substituting Equation S9 into the noncompartmental Equations S3 and S4:
|
|
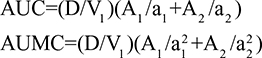
Model 1 is described by the following two differential equations:
|
|
where I1(t) is the time-dependent input to compartment 1. Solving Equation S11 for the case where I1(t) is a bolus input of dose D (I1=Dδ(t)), M1(t) is described by a two-exponential function:
|
|
This is a surprisingly complex expression, and it can be seen that the coefficients for the two exponentials do not, in general, have a simple relationship to the parameters describing the two individual compartments.
The steady-state clearance can be determined directly from the steady-state solution to Equation S11. Using Equation S7 for the definition of Q:
|
|
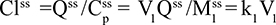
The steady-state volume of distribution is defined by Equation S1:
|
|
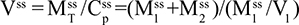
Since at steady state from Equation S11, M2ss=(k12/k21)M1ss:
|
|
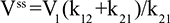
One obtains exactly the same values for Clss and Vss using the general noncompartmental relations (Equations S3–S5 for bolus input) and using Equation S10 with the coefficients for Model 1 (Equation S12). This is expected since in Model 1 the metabolism occurs in the plasma compartment, and for this condition, these noncompartmental relations are always satisfied.
The corresponding differential equations for Model 2 are:
|
|
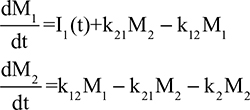
Solving, Equation S16 for the case where I1(t) is a bolus input of dose D (I1=D δ(t)), M1(t) is described by a two-exponential function:
|
|
The steady-state relation between M1 and M2 is obtained from Equation S16:
|
|
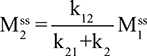
Solving Equation S16 for the steady-state clearance:
|
|
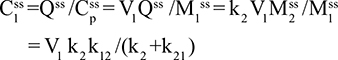
Using the general noncompartmental relations (Equations S3–S5 for bolus input) and using Equation S10 with the coefficients for Model 2 (Equation S17), one obtains exactly the same value for Clss. This is expected since the noncompartment Clss relation is valid even if the metabolism is occurring in an extravascular compartment, as it is for Model 2.
Substituting Equation S18 in Equation S14, the steady-state volume of distribution for Model 2 is:
|
|
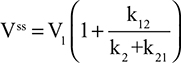
The noncompartment relationship for Vss should not be valid for Model 2 because the metabolism does not occur in the plasma compartment. As predicted, substituting the Model 2 coefficients (Equation S17 in Equations S10 and S5), the noncompartmental prediction  is incorrect (ie, differs from Equation S20):
is incorrect (ie, differs from Equation S20):
|
|
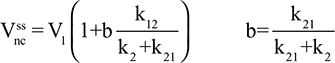
It can be seen that if k21>>k2 (the exchange between the two compartments is fast compared with the metabolic rate), then the noncompartmental expression becomes equal to the true Vss. This is a good approximation for albumin where the metabolic rate is about 20 days while the intercompartmental exchange is about 1 day.
At durations greater than ∼4 days, the second term in Equation S12 or S17 becomes negligible relative to the first and the concentration in compartment 1 (eg, plasma) can be approximated by:
|
|
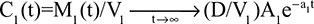
Although not always explicitly stated, the albumin compartmental kinetic analysis depends on the assumption that the exchange between the two compartments is fast compared with the metabolic rate, that is, k1<<k12 (Model 1) or k2<<k21 (Model 2). Using this assumption, the expression for B in Equation S12 can be approximated by:
|
|
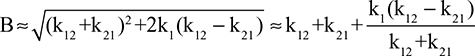
Substituting this approximation for B into the Model 1 expression for a1 (Equation S12):
|
|
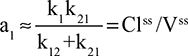
where the last equality uses the Model 1 steady-state clearance and volume of distribution relations (Equations S13 and S15). Substituting this approximation for B in the Model 1 expression for A1 (Equation S12) and assuming k1<<k12:
|
|
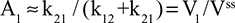
Finally, substituting these expressions for a1 and A1 into Equation S22:
|
|
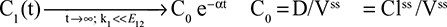
Although this was explicitly derived for Model 1, it can be shown that this same approximation is valid for Model 2 at durations greater than ∼4 days and assuming that k2<<k21. In summary, with this assumption the tracer plasma concentration for the two-compartmental model at durations greater than ∼4 days can be approximated by a single exponential with a rate constant α equal to the “fractional turnover” rate for the whole body (Equation S2) and the zero time intercept equal to the tracer-injected dose (D) diluted in the steady-state volume of distribution, allowing the experimental determination of both Clss and Vss. This result provides the theoretical justification for most of the older albumin tracer kinetic analysis.
Acute measurements of albumin synthesis
In this approach, labeled albumin precursors are given as a bolus injection and the rate of appearance of these labels in albumin is measured over a period of few hours. This provides a direct measurement of the short-term fraction of intravascular (ie, plasma) albumin synthesized per hour referred to as the “fractional synthesis rate.” Fractional synthesis is related to the plasma albumin clearance (ClP) and plasma volume (Vp) by the relation:
|
|
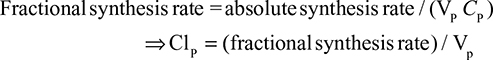
The absolute synthesis rate is then determined from Equation S27 with the plasma volume measured by some technique such as 125I albumin distribution.
Initially, studies used 14carbonate infusions to label the arginine guanido carbon via urea cycle metabolism with the assumption that the specific activity of the arginine precursor for albumin synthesis is identical to the urea-specific activity.4
|
|
The urea synthesis rate is directly measured using 14C urea. These were short-term experiments, with the specific activity usually measured at 5 hours (additional small corrections are made for albumin removal from plasma during this 5-hour period).
In recent years, the standard approach for measuring albumin synthesis has been based on the precursor “flooding technique” in which a large bolus of ([1-13C] leucine5 or [2Hring] phenyalanine6) is injected, uniformly labeling all the precursor pools, and albumin isotopic enrichment is measured versus time.5 There is a lag period (the “secretion time”) in the appearance of enriched albumin of about 30 minutes. The fractional synthesis rate is determined from:
|
|

The two time points are selected for a period when the enrichment is increasing roughly linearly (after the lag period) – usually between about 40 and 80 minutes after the bolus injection. A major advantage of using this relatively short time interval (ie, 40 minutes) is that no correction is required for loss of albumin from the intravascular space by metabolism or diffusion.5
References
Takeda Y, Reeve EB. Studies of the metabolism and distribution of albumin with autologous I131-albumin in healthy men. J Lab Clin Med. 1963;61:183–202. | ||
Meier P, Zierler KL. On the theory of the indicator-dilution method for measurement of blood flow and volume. J Appl Physiol. 1954;6(12):731–744. | ||
Nakashima E, Benet LZ. An integrated approach to pharmacokinetic analysis for linear mammillary systems in which input and exit may occur in/from any compartment. J Pharmacokinet Biopharm. 1989;17(6):673–686. | ||
McFarlane AS, Irons L, Koj A, Regoeczi E. The measurement of synthesis rates of albumin and fibrinogen in rabbits. Biochem J. 1965;95:536–540. | ||
Ballmer PE, McNurlan MA, Milne E, Heys SD, Buchan V, Calder AG, Garlick PJ. Measurement of albumin synthesis in humans: a new approach employing stable isotopes. Am J Physiol. 1990;259(6 Pt 1):E797–E803. | ||
Ballmer PE, Reichen J, McNurlan MA, Sterchi AB, Anderson SE, Garlick PJ. Albumin but not fibrinogen synthesis correlates with galactose elimination capacity in patients with cirrhosis of the liver. Hepatology. 1996;24(1):53–59. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.