")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Human factors evaluation of a novel digital medicine system in psychiatry
Authors Peters-Strickland T, Hatch A, Adenwala A , Atkinson K , Bartfeld B
Received 15 November 2017
Accepted for publication 17 January 2018
Published 16 February 2018 Volume 2018:14 Pages 553—565
DOI https://doi.org/10.2147/NDT.S157102
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Timothy Peters-Strickland,1 Ainslie Hatch,2 Anke Adenwala,3 Katie Atkinson,4 Benjamin Bartfeld5
1Global Clinical Development, Otsuka Pharmaceutical Development & Commercialization, Inc., Princeton, NJ, USA; 2Clinical Sciences, Digital Medicine, Otsuka America Pharmaceutical, Inc., Princeton, NJ, USA; 3Biostatistics, GfK Custom Research, LLC, Chicago, IL, USA; 4Human Factors Engineering, Proteus Digital Health, Redwood, CA, USA; 5Industrial Design Specialist, Otsuka Pharmaceutical Development & Commercialization, Inc., Princeton, NJ, USA
Background: The digital medicine system (DMS), a drug–device combination developed for patients with serious mental illness, integrates adherence measurement with pharmacologic treatment by embedding an ingestible sensor in a pill, allowing for information sharing among patients, health care providers (HCPs), and caregivers via a mobile interface. Studies conducted during the DMS development process aimed to minimize cognitive burden and use-related risks and demonstrated effective use of the technology.
Methods: Human factors (HF) studies assessed the system’s safe and effective use by the intended users for the intended uses. The patient interface was tested in six formative HF studies followed by a validation study. The HCP/caregiver interface was tested in one study before validation. All tasks critical to safety or necessary for effective use were included. Formative studies identified use-related risks and the causes of use problems to guide design modification. Validation of the patient and HCP/caregiver interfaces assessed risks of the final product.
Results: During the patient formative studies, design improvements were made to address problems and mitigate risks thought to be associated with a suboptimal system design or patient understanding of the system. In the validation study of the patient interface, 35 patients attempted 23 performance tasks, for a total of 805 attempts; 783/805 attempts were completed with success. One close call, 15 failures, and 6 difficulties occurred on these user tasks; only 3 of these were on a critical task. Residual risks resistant to mitigation were found to be of low severity based on the US Food and Drug Administration 2016 guidance.
Conclusion: The final design of the DMS reflects input by the intended user populations through a comprehensive development methodology. In alignment with the US Food and Drug Administration goals for HF studies, the system was found to be safe and effective for the intended users, uses, and use environments.
Keywords: digital medicine system, drug–device combination, schizophrenia, bipolar disorder, major depressive disorder, aripiprazole, serious mental illness, usability
Plain language summary
A new digital medicine system provides objective medication adherence information based on whether a patient takes a pill that is embedded with a sensor. The system records medication taking when the sensor is activated in the stomach and sends a signal to a patch worn on the skin, which in turn sends the information to the patient’s phone. Adherence information is then available to the patient and his or her health care provider. In order to assess and improve the ability of the system to be used properly while minimizing medication- or device-associated risks, iterative human factors studies were performed with progressively updated versions of the system until the remaining risks were considered of low severity. These studies showed that the system modifications reduced risks to the lowest level possible and improved the ability of the intended user population – patients with schizophrenia, bipolar disorder, or major depressive disorder – to safely use the system.
Introduction
Poor adherence to medication is a common problem in patients with serious mental illness (SMI) such as schizophrenia, bipolar I disorder, and major depressive disorder (MDD)1–3 and has been consistently associated with suboptimal treatment response, including relapse, rehospitalization, and poor health outcomes.4–6 Adherence is most often assessed by patient self-report or health care provider (HCP) assessment.7 These subjective methods often exaggerate the degree of medication taking and result in overestimation of patient adherence by HCPs.8 Objective assessment of adherence is very difficult in practice. Directly observed medication ingestion is the gold standard but can be used only in a limited number of settings such as hospitals or nursing homes. Currently available and more broadly applicable options include pill counts, pharmacy refill records, technology-assisted monitoring of pill containers such as Medication Event Monitoring System bottle caps, and biological assays from bodily fluids.9 These objective methods have limitations and do not provide an accurate measure of the actual medication ingestion event. Capsule photographs taken with cellular phones have been reported as a simple tool to assess adherence but, similar to the other modalities, cannot confirm medication ingestion.10 Use of long-acting injectable (LAI) formulations of antipsychotics eliminates the need for adherence monitoring because of directly observable injections at the clinic, as long as the patient does not discontinue treatment. However, the potential benefits of LAIs are limited only to patients who accept the LAI antipsychotic as a therapeutic option.
The digital medicine system (DMS) is an innovative drug–device combination developed for patients with SMI, which integrates adherence measurement capabilities as part of the drug formulation via an embedded ingestible sensor. It objectively measures medication ingestion and reports adherence to oral aripiprazole, an atypical antipsychotic indicated as monotherapy for the treatment of schizophrenia, acute treatment of manic and mixed episodes associated with bipolar I disorder, and for the treatment of MDD adjunctive to antidepressants.11 The system provides continuous feedback to patients on medication ingestion and also collects data on patient activity, patient rest, self-rated mood, and self-rated rest quality that may improve patient engagement and inform an HCP’s decision making.12 Early clinical experience with the system has been reported in healthy volunteers13; patients with tuberculosis, heart failure, or hypertension14; and in patients with schizophrenia, MDD, or bipolar disorder.15–17 The DMS consists of a digital sensor–enabled medication, a wearable sensor (patch), and software applications that enable secure collection and sharing of information using a patient mobile interface (ie, patient application) and corresponding web-based interface (dashboard) for HCPs and caregivers (Figure 1). Self-management systems based on mobile device applications developed for patients with schizophrenia have been reported,18,19 but no currently marketed product offers a combination of functions comparable with that of the DMS.
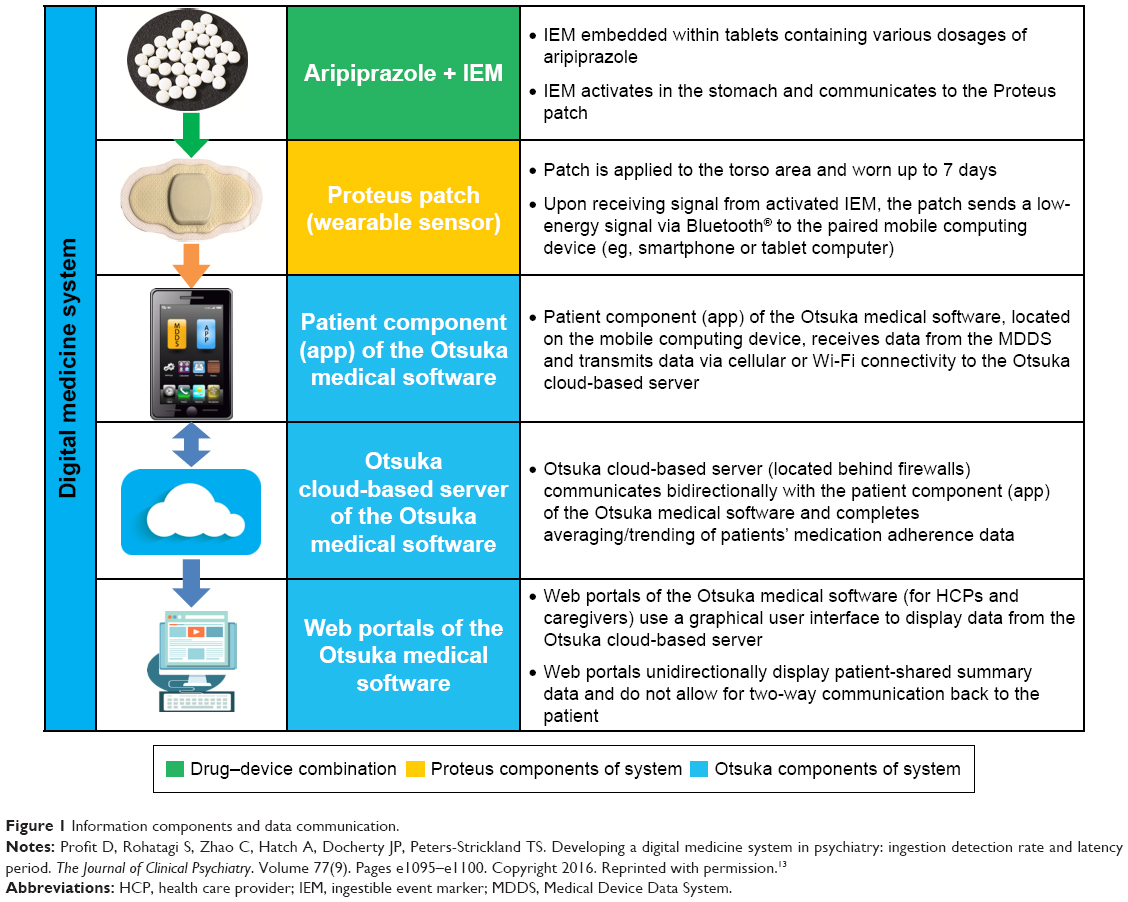 | Figure 1 Information components and data communication. |
The absence of directly applicable user experience from a comparable existing product highlights the importance of a comprehensive development program that includes an analysis of use-related risks and device optimization through human factors (HF) studies. HF is defined as the application of knowledge about human capabilities (physical, sensory, emotional, and intellectual) and limitations to the design and development of tools, devices, systems, environments, and organizations.20 The objective of HF studies is to assess the safe and effective use of a system by the intended users for the intended uses following the method recommended in the US Food and Drug Administration (FDA) guidance for developing safe and effective drug–device combinations.21,22 The methodology of HF studies differs from that of clinical trials because 1) testing is designed to reveal and highlight use errors to inform an improved design and 2) the collected data are qualitative and not statistically evaluated because the focus is on the severity of consequences associated with individual use errors.21 The HF studies conducted as part of the DMS development aimed to assess whether the three intended groups of users (patients, HCPs, and caregivers) can appropriately use the technology. Digital health applications created for users with SMI require specific product design characteristics that allow effective use of the product by the target population.23
This article describes two stages of HF studies: formative studies that help optimize design of the product and validation studies that evaluate the final product. Formative and validation studies are also called iterative and summative studies, respectively.20 The formative and validation studies were conducted with psychiatric patients, HCPs, and caregivers during the process of developing the DMS. To the best of our knowledge, the DMS represents the first integrative digital health product developed in psychiatry that has undergone a comprehensive HF assessment to support the FDA regulatory submission.
Methods
The HF studies were conducted in accordance with FDA recommendations21 and the FDA-recognized standards on HF and usability engineering of medical devices.20 The HF study process assesses and iteratively mitigates use-related hazards that may otherwise lead to unsafe or ineffective use of the product by the intended users (Figure 2A).21 The iterative process of testing modified designs continues until the risk related to the use of the system is eliminated or empirically assessed to be minimal. Validation studies conclude the HF study process and aim to demonstrate that the final product has been optimized for safe and effective use by the intended users in the expected use environments.21
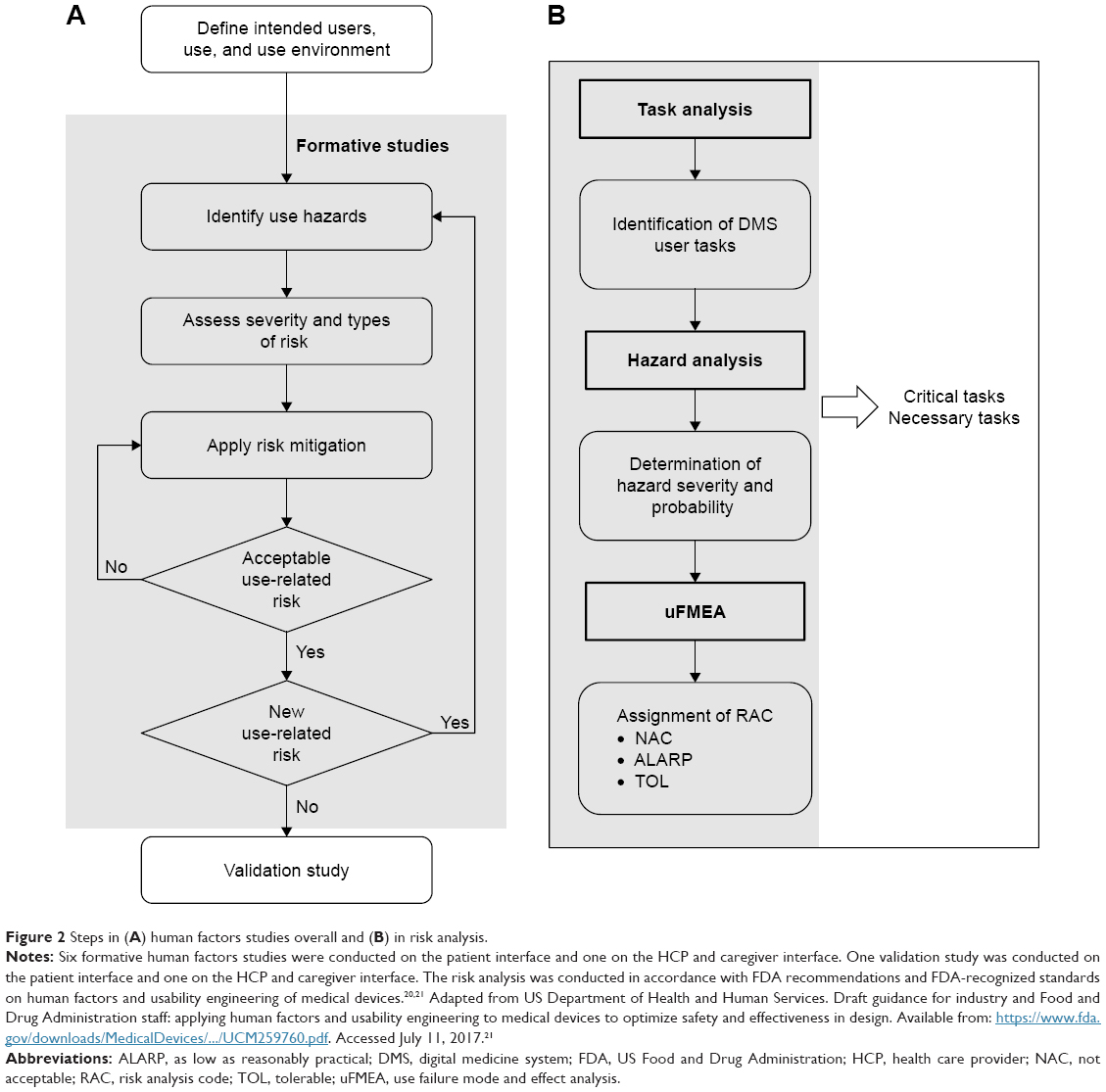 | Figure 2 Steps in (A) human factors studies overall and (B) in risk analysis. |
The patient interface of the DMS was tested in six formative user-centered HF studies followed by a validation study. The HCP/caregiver interface was tested in a single formative study before the validation study. The studies were approved by an institutional review board (Core Human Factors, Inc., Bala Cynwyd, PA, USA). All participants provided written informed consent.
Iterative studies on patient interface
The primary objectives of the formative studies on the patient interface were to assess the effectiveness of mitigations from previous DMS usability studies, identify steps in the use process that may result in new unforeseen risks, and understand the root cause of performance failures or difficulties to help optimize the design of the product (Figure 2A). A use-risk analysis was performed before and after each study to identify remaining potential use-related risks associated with the system and inform product design iterations. The risk analysis, schematically shown in Figure 2B,20,21 included task analysis, hazard analysis, and use failure mode and effect analysis (uFMEA). The task analysis identified all reasonably foreseeable user steps during use of the DMS. The hazard analysis determined potential use-related hazards resulting from unsuccessful completion of use-related tasks and assessed probability and severity of the hazards. A uFMEA provided a risk-analysis code (RAC) for each potential failure mode representing the acceptability of the harm (not acceptable, as low as reasonably practical, or tolerable); the RAC was used to prioritize efforts to mitigate risks during the redesign process.
The risk analysis identified critical and necessary tasks for safe and effective use of the DMS, which were tested in validation studies. Critical tasks are required for safe and effective use of the product, and when performed either incorrectly or not at all can cause serious harm to the user, whereas necessary tasks are required to achieve the medical benefit but do not constitute a serious safety risk if omitted or performed incorrectly.
The patient interface was tested in six formative studies (Table 1). Prior to validation, patients were chosen based on their conformity with the intended users of the system as per FDA guidance on sampling methodology. Patients with schizophrenia, bipolar I disorder, and MDD were eligible. Participants were selected based on the additional criteria that they were capable of participating in a simulated use study and being a user of the system, as determined by the independent recruiter with a psychiatrist’s professional assessment. Per FDA and American National Standards Institute (ANSI) guidance, the sample size for the formative stage was determined according to study needs consistent with the intent of the research. The first two studies consisted of single 60-minute individual sessions. The remaining four studies included a 60-minute individual onboarding session on day 1 that simulated a first day with the system and a 60-minute individual session on day 2 that simulated regular use. The day 1 activities comprised initial steps required for the system setup (installing the application on a mobile device, logging into the software, creating an account, inputting user settings, and sending an invitation to share information with an HCP), applying the first wearable sensor, and pairing it with the mobile application. The day 2 regular-use activities included weekly maintenance steps, such as replacing the wearable sensor and pairing the new sensor to the patient mobile application. The participants in the third and fourth studies of the patient interface were randomly assigned to one of two arms, either HCP-assisted or HCP-unassisted onboarding that balanced diagnoses (schizophrenia, bipolar I disorder, MDD) between the two groups. Some of the critical tasks assessed during formative studies are listed in Table 2. For formative study 5, the definitions of critical and necessary tasks were aligned with the updated regulatory guidelines on HF studies,21 and all tasks on which failures or difficulties were observed in the previous formative study were tested. This study was conducted in three waves or sprints, allowing design changes in the interim to mitigate any observed usability issues. At the conclusion of formative study 5, only three issues remained that could potentially be mitigated through design. A final formative study, study 6, assessed these remaining failures and difficulties related to the interface design and concluded that minor design changes could address these issues.
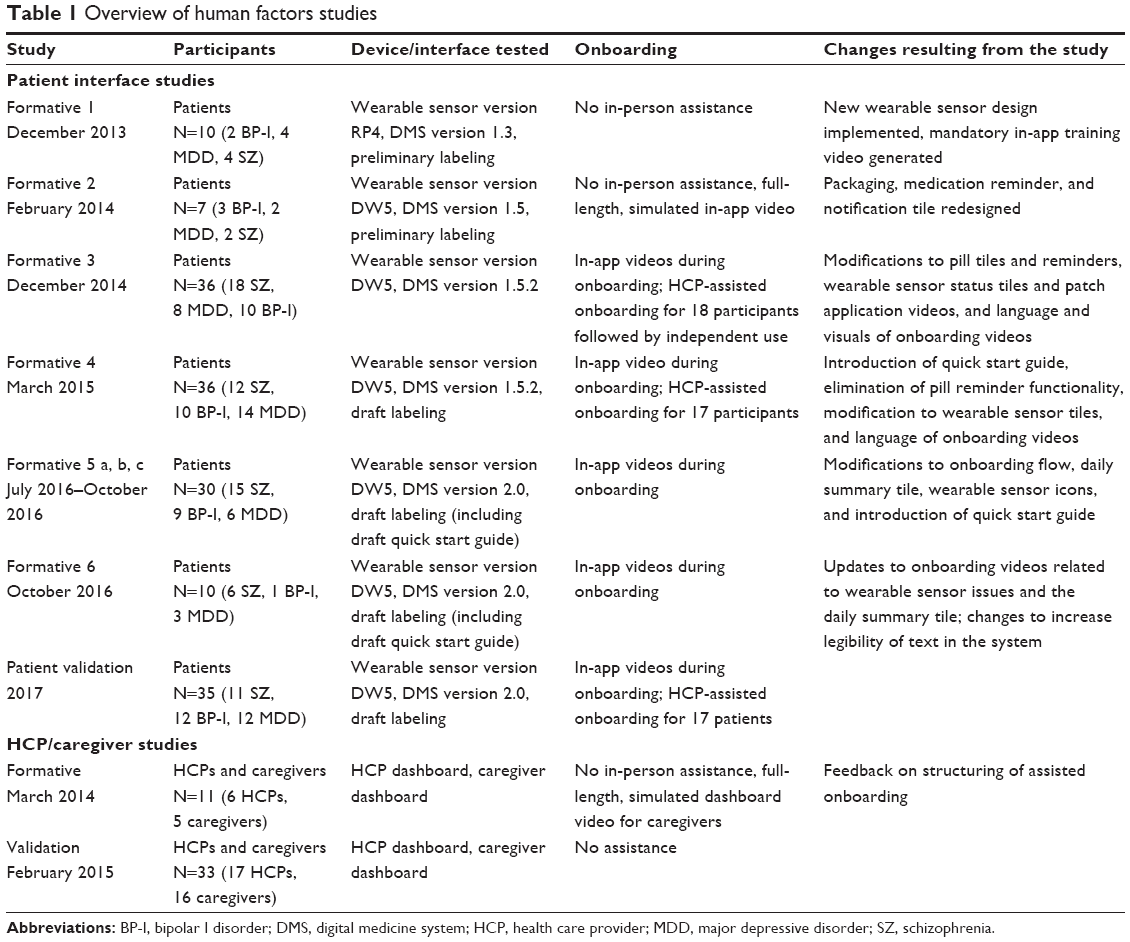 | Table 1 Overview of human factors studies |
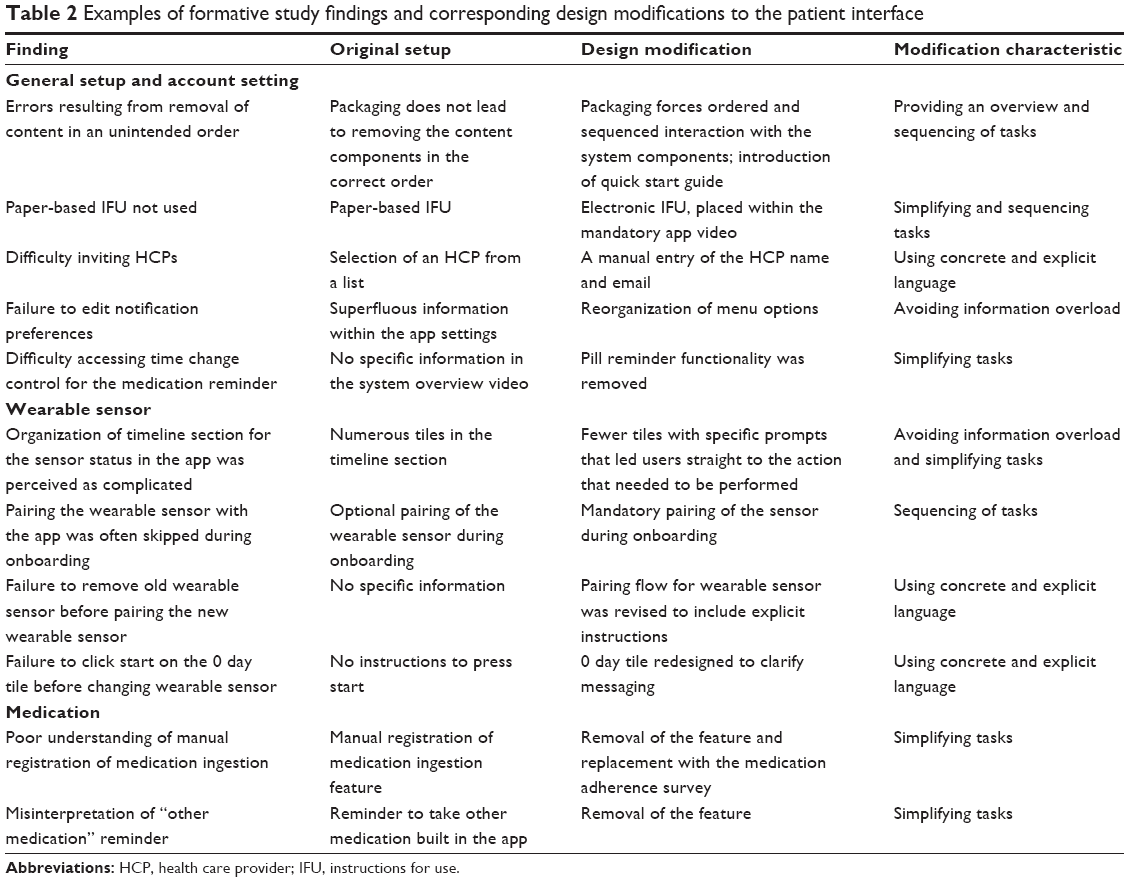 | Table 2 Examples of formative study findings and corresponding design modifications to the patient interface |
Validation study on patient interface
In the patient interface validation study, participants’ performance was assessed on tasks that represented critical, necessary, and desirable tasks within the DMS; desirable tasks are part of the overall DMS experience but do not impact its purpose of measuring medication adherence. Critical and necessary tasks are typically the focus of validation studies as these constitute the tasks that are required for the safe and effective use of a system. Tasks are designated “critical” or “necessary” based on the maximum potential harm associated in the uFMEA; in this case, only 3 of the 23 user tasks evaluated in the study were deemed “critical” because they were linked to a potential harm of “critical” severity in the system’s risk analysis. Each of these three tasks represented interactions with the app in which a user could misinterpret app content and adjust his/her pill-taking schedule as a result.
The sample size for the validation study was consistent with ANSI/Association for the Advancement of Medical Instrumentation (AAMI) HE guidance, which specifies a minimum of 15 participants per distinct user group. A representative sample of the intended user population was randomly assigned to each onboarding arm. Participants completed tasks in a simulated environment designed to mimic the real-world setting to the degree necessary for effective testing. The tasks were set up to simulate 7 days of system use. The smartphone application was controlled by an HF software tool designed specifically to allow the moderator to trigger notifications and application functions similar to those experienced in actual use, consistent with a simulated use methodology. For example, because no pill was ingested during the study, this was one of the key system feedbacks that had to be simulated via this software tool. Skin placement of the wearable sensor was simulated by the participant placing the sensor on a flexible wrap worn on the abdomen.
The participants were evenly divided into assisted and unassisted onboarding groups. Both participant groups viewed within-app video segments during onboarding, and participants in the assisted group received HCP-provided assistance as necessary. Both groups subsequently completed the same independent-use assessments.
Participants
Study participants were selected from the patient database of a clinic in California and were representative of the intended population of DMS users, ie, male and female patients aged 18–65 years with a confirmed diagnosis of schizophrenia, bipolar I disorder, or MDD. The participants only needed to be potential users of the system and did not have to be stable on oral aripiprazole in order to take part in the study. Individuals assessed by a referring clinician to be mildly to moderately ill (Clinical Global Impression-Severity of Illness score ≤4), stable in their condition, deemed capable of engaging in the two required sessions, and not previously involved in any study related to the DMS were eligible for enrollment. In addition, all participants were required to own and use a smartphone. Candidates were excluded if they had any personal or commercial interest in a pharmaceutical or medical device company or did not understand and speak English.
Procedures
Participants were transported to an independent research facility with individual testing rooms set up to simulate a simple home or office environment. Each participant completed two sessions lasting ≤75 minutes (day 1 onboarding and day 2 regular use) separated by 24 hours. As in the formative studies, day 1 onboarding included tasks performed during first-day use of the product, whereas day 2 regular use included tasks experienced with the use of the product over time. For the validation study, the sample size for each onboarding arm (assisted or unassisted) was determined based on the ANSI/AAMI HE75 2009 guidance recommending ≥15 participants per distinct user group.20,21 A minimum of five participants with each of the three intended diagnoses were assigned to each arm. On both study days, a moderator provided necessary background information, assessed errors, and probed for root causes but did not interfere with the participants’ activities.
Assessments
Each interview was conducted by a team of HF professionals, including a moderator and a notetaker. Performance difficulties, close calls, or failures on critical or necessary tasks were identified via observation of participants’ performance and follow-up questions. This approach was intended to capture unanticipated use errors observed by the moderator or articulated by the participant. Investigators rated task performance as either 1) success, when the user performed the assigned task correctly and independently regardless of time taken; 2) performed with difficulty, when the user displayed visible confusion or insufficient understanding of the interface beyond mere exploration or avoided failure through vigilance or self-corrected actions or responses in a situation in which the failure would not have led to serious harm; 3) close call, when the user, through vigilance, self-corrected actions or responses that otherwise would have resulted in a failure and the failure could have led to serious harm had it occurred; 4) failed performance, when the user did not complete a task successfully or stated that he/she was done with the task without successfully completing the assigned task, stated that he/she needed to give up and did not attempt any further assistance from support materials, required assistance from the moderator to proceed to the subsequent task, or stated an incorrect interpretation of necessary or critical tasks; and 5) not attempted, when the user did not attempt a task because of time constraints or a previous error that did not allow performance of a task. In addition to the performance tasks, the moderator asked specific questions to evaluate participants’ understanding of messages contained within the system tied to necessary or critical tasks and cautionary statements on labeling and packaging materials. Targeted discussions with participants investigated the causes of performance failures, close calls, and difficulties.
Study on HCP and caregiver interface before validation
The primary objectives of the study on the HCP and caregiver interface were to identify steps in the use process that resulted in risks or confusion and to understand the root cause of performance failures or difficulties to help redesign the product. HCP and caregiver use of the system comprised initial setup steps and periodic viewing of patient data. A use-risk analysis was performed before and after the study as previously described for formative studies on the patient interface.
Validation study on HCP and caregiver interface
In the validation study on the HCP and caregiver interface, the participants completed all critical and necessary tasks identified in the risk analysis. The participants completed the study tasks in an office-like environment simulating conditions found during treatment and routine care for patients with SMI; the simulated environment was controlled by the research team.
Participants
For the HCP and caregiver interface, the two groups of intended DMS users include HCPs active in health care of patients with SMI and nonprofessional caregivers of patients with SMI. Both HCPs and caregivers were identified and contacted by professional recruiters using databases of potential respondents and specified recruitment criteria. HCPs such as psychiatrists, nurse practitioners, nurses, and social workers with ≥5 years of professional experience and nonprofessional caregivers such as adult family members and friends with ≥3 years of experience caring for patients with SMI were eligible for enrollment. All participants were also required to have an email address and to use a computer, tablet, or smartphone. Candidates were excluded if they had any personal or commercial interest in a pharmaceutical or medical device company or previous experience related to the DMS.
Procedures
The performance of HCPs and caregivers was assessed separately. Each participant completed an individual 60-minute session. A moderator described the study objectives, explained the intended use of the system, and provided high-level context about the system components. The study assessed all previously identified critical and necessary tasks. The critical tasks tested in the HCP study were selection of patients and review of data; in the caregiver study, no tasks were rated as critical. For both HCPs and caregivers, performance on each task was designated as “success”, “with difficulty”, “failure”, or “did not complete”. Following each task, the moderator asked questions to investigate the root causes of any difficulties completing the task and conducted a post-assessment interview.
Results and discussion
Formative studies on the patient interface
A total of 129 patients with SMI were enrolled across the six formative studies conducted sequentially to evaluate the patient interface (Table 1). Observed performance was similar among patient populations regardless of the diagnoses of schizophrenia, bipolar I disorder, or MDD. To address identified risks in critical and necessary tasks, design improvements were made in areas that comprised general setup and use (including instructions for use and packaging, and notification setting), wearable sensor, medication, and interpretation of information (Table S1). Findings collected during the formative studies and the corresponding design modifications are shown in Table 2. For example, pairing of the wearable sensor with the app was made a mandatory step during onboarding because the step was often skipped when it was optional. Use errors were most frequently observed on tasks related to the wearable sensor. Modifications implemented to address these risks included simplification of content and tasks, forced sequencing of tasks, avoiding information overload, and use of concrete and explicit language.23 Risk analysis performed after the final formative study indicated that the mitigation efforts eliminated potential failure modes that might lead to unacceptable risks. Additional changes, deemed unlikely to introduce new risks, were made to improve readability in the app and packaging and were tested in the validation study.
Validation study on the patient interface
The validation study enrolled 35 stable, mildly to moderately ill patients diagnosed with SMI (schizophrenia, n=11; bipolar I disorder, n=12; MDD, n=12). The mean age was 37.1 years (range, 19–63); 33 of 35 participants were right handed, and 14 of 35 required glasses to correct their vision. On day 1, patient tasks and critical messaging tasks were tested during onboarding with 17 patients assisted by an HCP and 18 patients performing the tasks independently. On day 2, regular-use tasks were tested with all patients without HCP participation. Eleven study participants, 4 in the assisted onboarding and 7 in the unassisted onboarding group, experienced a failure, difficulty, or close call on one or more performance tasks. These 11 patients accounted for 15 failures, 1 close call, and 6 difficulties, of which only 3 were observed on a critical task. Importantly, the maximum potential harm to the patients from these failures was deemed minor per risk assessment guidelines. The highest number of failures or difficulties was observed on the task of pairing the replacement wearable sensor (n=6). However, these represented confusion on the participants’ part and were resolved by the participants’ action or proposed action, thus not necessitating design changes. Table 3 shows performance assessment for various task categories. Participant performance in the assisted versus unassisted onboarding was not significantly different, although failures/difficulties were numerically higher in the unassisted group.
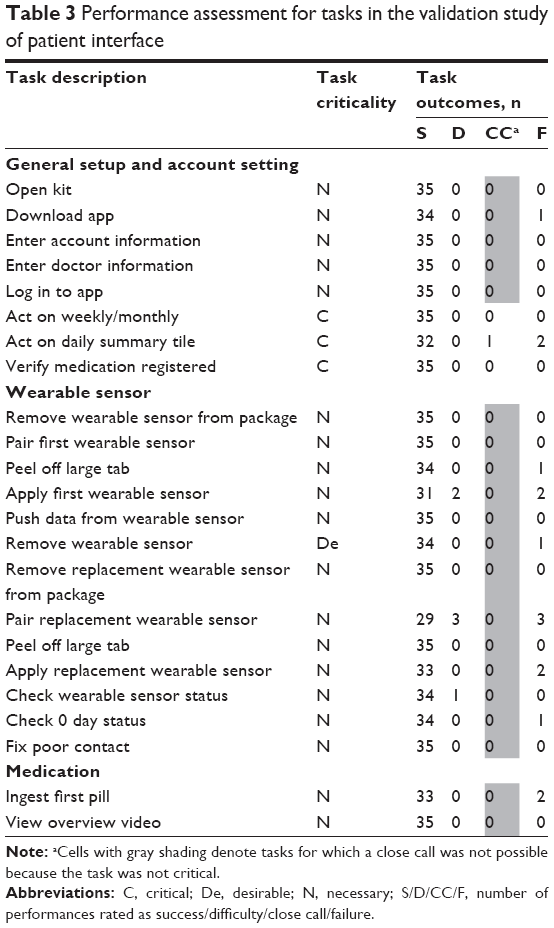 | Table 3 Performance assessment for tasks in the validation study of patient interface |
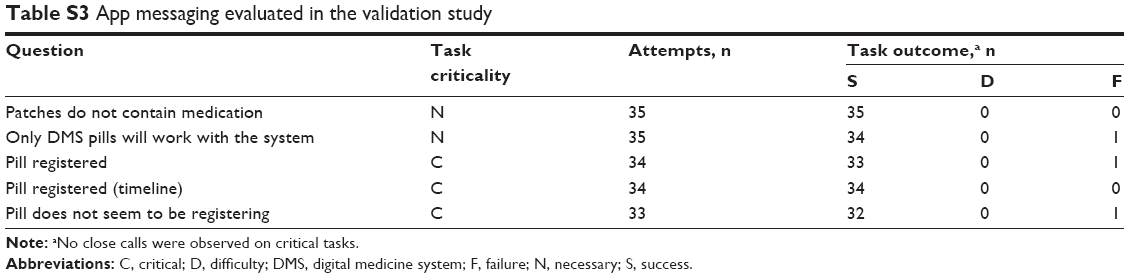
Knowledge assessment questions were asked to assess messaging within the app and comprehension of cautionary statements. Cautionary statements were classified into two categories, those linked with potential for harm of serious severity or higher, and those that were not. Performance on knowledge assessment tasks of the cautionary statements linked to serious or higher severity (Table S2) and app messages (Table S3) identified six instances of failure or difficulty. Four additional failures or difficulties were observed on cautionary statements associated with potential for harm lower than serious. Of the 10 total difficulties or failures observed, 6 were from the same two participants. Two failures were on knowledge assessments that were linked to critical tasks, but both of these would have led to an outcome associated with no more than minor harm. Language modification for clarity was recommended for one statement. No trends requiring design modifications were otherwise identified. Potential harm, if any, to participants from the remaining app messaging failures was likely to be minor. As stated by the FDA, the use of any medical device is always associated with some amount of residual risk resulting from use errors, and it is impossible to make any device error-proof or risk-free.21 Based on an analysis of the observed use errors and prior design changes resulting from the formative studies, the remaining risk was determined to be resistant to elimination or mitigation through further modifications of the patient interface or labeling. For example, although the patient interface could be simplified to some degree to reduce cognitive overload observed during iterative testing, this kind of a change could prove counterproductive because there is always a possibility of introducing new, unforeseen use errors when any change is made.
Based on the observations from the validation study, the residual risks that remain in the system may constitute at worst a minor patient safety hazard resulting from patients taking a one-pill extra dose. In the uFMEA, a one-pill extra dose was considered a minor severity hazard in the context of the known safety profile of aripiprazole. A possible scenario leading to a potential one-pill extra dose could include failure to respond to wearable sensor notifications of poor skin contact that could result in the system’s inability to register medication ingestion, therefore potentially causing a user to take another pill once notified on the next day that a pill had not registered. However, in addition to a notification of poor skin contact, the system would also trigger a daily adherence survey, provide contact details for customer support, and keep the issue of poor skin contact on the application display for the user to address at the next opportunity. An adherence survey gives the patient an option to self-report medication taking. In addition, it is likely that the patient’s HCP would receive a notification of missed doses via the HCP interface because of the unique design of the DMS, which requires patients to share medication ingestion data with their HCPs. This provides an additional level of protection and an opportunity for mitigation through communication with the HCP. Therefore, the identified residual risks will likely be minimized by the multilevel connectivity of the functions and outweighed by the anticipated product benefits. An assessment of the residual risk that considered the system’s functions, patient safety hazard, and the possibility of user performance failure suggested that patients with SMI can use the interface safely and effectively.
Study on the HCP and caregiver interface before validation
The study was conducted with six HCPs and five caregivers. Participant performance on all tasks within the HCP and caregiver interface, such as registration, navigation in the system, and review of patient data, was generally successful. No use errors leading to an increased risk were observed, and thus, no further design modifications to the HCP and caregiver interface were required.
Validation study on the HCP and caregiver interface
The study enrolled 17 HCPs (6 psychiatrists, 6 registered nurses, 1 nurse practitioner, and 4 social workers) and 16 caregivers representing a sample of intended and likely users. The same 11 tasks were assessed for both the HCP and caregiver interfaces. Two tasks were rated as critical for HCPs (patient selection and data review), and the remaining nine were rated as necessary; all caregiver tasks were rated as necessary.
HCPs and caregivers correctly completed a large majority of tasks (174/6/3 and 166/2/7, respectively, expressed as correct/with difficulty/failure), which suggests that the intended users can navigate the system and view and interpret patient data without use errors. Overall, the results showed that the product design was effective in supporting successful completion of the tested tasks by the intended users. Use errors observed in the task of patient selection (2 HCPs and 3 caregivers) and data review (1 HCP and 2 caregivers) indicate that small residual risk related to potential misinterpretation of patient data remains. However, this level of residual risk can be expected for any type of data display and does not require mitigation.
Limitations
As in all HF studies, simulated instead of real-world product use was tested. The simulated steps included ingestion of medication, placement of the wearable sensor and its communication with the application, setup of the user profile, and various scenarios related to the application notifications. Such an approach allows for observation of a participant’s performance and collection of subjective data but is inherently associated with well-known limitations. The study was conducted outside of the actual use environment (home or clinical setting), and the HCPs assisting with onboarding were not necessarily members of the participant’s treatment team. However, it should be noted that this is an inherent limitation of HF engineering studies, and the system has in fact been tested in patients with SMI.16,17 Another study limitation is that the enrolled patients were clinically stable, and all had some experience using mobile devices. Inclusion of clinically stable patients only was essential for collecting useful data on usability of the system without major interference from the illness, but this means that the results cannot be generalized to the most ill patient population.
Conclusion
To the best of our knowledge, the DMS represents the first integrative digital health product developed in psychiatry that has undergone a comprehensive HF assessment in support of the FDA regulatory submission. The patient interface was substantially improved in a systematic iterative manner, as demonstrated by a reduction in the percentage of failures on performance tasks and knowledge assessments from the formative studies to the validation study (Figure 3). The final design was informed by the intended user populations through the comprehensive development methodology. The modifications to the design of the patient interface focused on sequencing of tasks, simplifying content/tasks, avoiding information overload, and using concrete and explicit language. The use-related risks were assessed and mitigated in a series of iterative studies in accordance with FDA guidance and industry standards. It is important to note that use errors that might lead to a patient taking a one-pill extra dose were rare in the validation study. The aim of developing the DMS is to demonstrate substantial benefits of objectively measuring medication ingestion adherence over time while not affecting the risk.
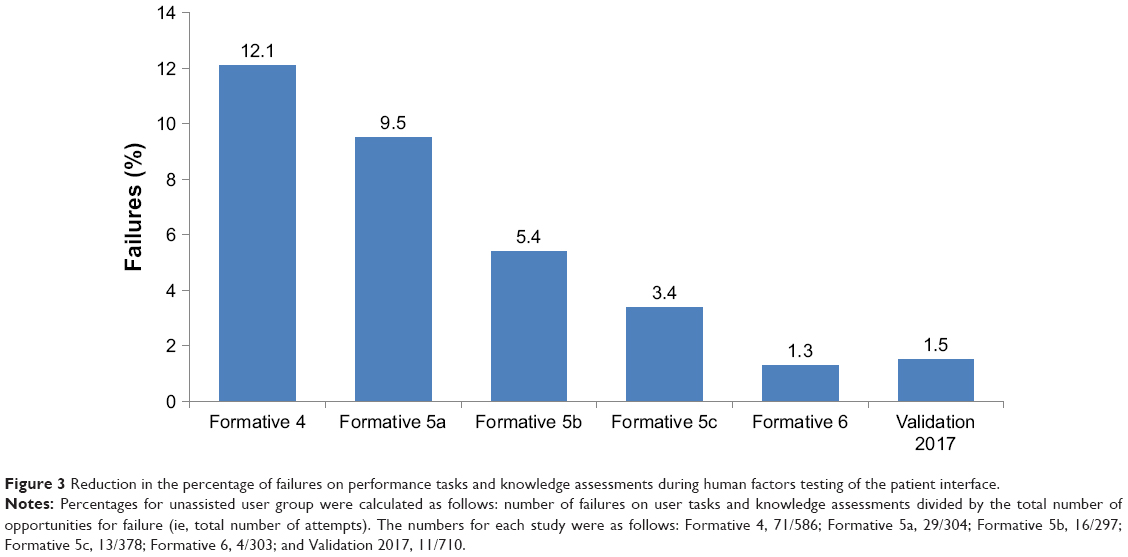 | Figure 3 Reduction in the percentage of failures on performance tasks and knowledge assessments during human factors testing of the patient interface. |
Overall, in alignment with the FDA HF guidance, the results of the validation studies and analysis of residual risk demonstrate acceptable safety and good usability of the DMS for the intended user population – patients with schizophrenia, bipolar I disorder, and MDD, and their HCPs and caregivers.
Acknowledgments
Editorial support for development of this manuscript was provided by Pavel Kramata, PhD, and Vandana Sharma, PhD, of C4 MedSolutions, LLC (Yardley, PA, USA), a CHC Group company, and was funded by Otsuka Pharmaceutical Development & Commercialization, Inc.
Disclosure
Timothy Peters-Strickland is an employee of Otsuka Pharmaceutical Development & Commercialization, Inc. Ainslie Hatch is an employee of Otsuka America Pharmaceutical, Inc., and was an employee of Otsuka Pharmaceutical Development & Commercialization, Inc., during this research project. Anke Adenwala is an employee of GfK Custom Research, LLC. Katie Atkinson is an employee of Proteus Digital Health. Benjamin Bartfeld is a contract worker for Otsuka Pharmaceutical Development & Commercialization, Inc. The authors report no other conflicts of interest in this work.
References
Lacro JP, Dunn LB, Dolder CR, Leckband SG, Jeste DV. Prevalence of and risk factors for medication nonadherence in patients with schizophrenia: a comprehensive review of recent literature. J Clin Psychiatry. 2002;63(10):892–909. | ||
Sajatovic M, Valenstein M, Blow FC, Ganoczy D, Ignacio RV. Treatment adherence with antipsychotic medications in bipolar disorder. Bipolar Disord. 2006;8(3):232–241. | ||
Hung CI. Factors predicting adherence to antidepressant treatment. Curr Opin Psychiatry. 2014;27(5):344–349. | ||
Ascher-Svanum H, Faries DE, Zhu B, Ernst FR, Swartz MS, Swanson JW. Medication adherence and long-term functional outcomes in the treatment of schizophrenia in usual care. J Clin Psychiatry. 2006;67(3):453–460. | ||
Hong J, Reed C, Novick D, Haro JM, Aguado J. Clinical and economic consequences of medication non-adherence in the treatment of patients with a manic/mixed episode of bipolar disorder: results from the European Mania in Bipolar Longitudinal Evaluation of Medication (EMBLEM) study. Psychiatry Res. 2011;190(1):110–114. | ||
Scott J, Pope M. Self-reported adherence to treatment with mood stabilizers, plasma levels, and psychiatric hospitalization. Am J Psychiatry. 2002;159(11):1927–1929. | ||
Velligan DI, Lam YW, Glahn DC, et al. Defining and assessing adherence to oral antipsychotics: a review of the literature. Schizophr Bull. 2006;32(4):724–742. | ||
Velligan D, Wang M, Diamond P, et al. Relationships among subjective and objective measures of adherence to oral antipsychotic medications. Psychiatr Serv. 2007;58(9):1187–1192. | ||
Sajatovic M, Velligan DI, Weiden PJ, Valenstein MA, Ogedegbe G. Measurement of psychiatric treatment adherence. J Psychosom Res. 2010;69(6):591–599. | ||
Galloway GP, Coyle JR, Guillen JE, Flower K, Mendelson JE. A simple, novel method for assessing medication adherence: capsule photographs taken with cellular telephones. J Addict Med. 2011;5(3):170–174. | ||
Abilify® (aripiprazole) [prescribing information]. Tokyo, Japan: Otsuka Pharmaceutical Co., Ltd.; 2016. | ||
Shafrin J, Schwartz TT, Lakdawalla DN, Forma FM. Estimating the value of new technologies that provide more accurate drug adherence information to providers for their patients with schizophrenia. J Manag Care Spec Pharm. 2016;22(11):1285–1291. | ||
Profit D, Rohatagi S, Zhao C, Hatch A, Docherty JP, Peters-Strickland TS. Developing a digital medicine system in psychiatry: ingestion detection rate and latency period. J Clin Psychiatry. 2016;77(9):e1095–e1100. | ||
Au-Yeung KY, Moon GD, Robertson TL, et al. Early clinical experience with networked system for promoting patient self-management. Am J Manag Care. 2011;17(7):e277–e287. | ||
Kane JM, Perlis RH, DiCarlo LA, Au-Yeung K, Duong J, Petrides G. First experience with a wireless system incorporating physiologic assessments and direct confirmation of digital tablet ingestions in ambulatory patients with schizophrenia or bipolar disorder. J Clin Psychiatry. 2013;74(6):e533–e540. | ||
Peters-Strickland T, Pestreich L, Hatch A, et al. Usability of a novel digital medicine system in adults with schizophrenia treated with sensor-embedded tablets of aripiprazole. Neuropsychiatr Dis Treat. 2016;12:2587–2594. | ||
Rohatagi S, Profit D, Hatch A, Zhao C, Docherty JP, Peters-Strickland TS. Optimization of a digital medicine system in psychiatry. J Clin Psychiatry. 2016;77(9):e1101–e1107. | ||
Ben-Zeev D, Kaiser SM, Brenner CJ, Begale M, Duffecy J, Mohr DC. Development and usability testing of FOCUS: a smartphone system for self-management of schizophrenia. Psychiatr Rehabil J. 2013;36(4):289–296. | ||
Ben-Zeev D, Brenner CJ, Begale M, Duffecy J, Mohr DC, Mueser KT. Feasibility, acceptability, and preliminary efficacy of a smartphone intervention for schizophrenia. Schizophr Bull. 2014;40(6):1244–1253. | ||
Advancing Safety in Medical Technology. ANSI/AAMI HE75: Human Factors Engineering–Design of Medical Devices. Arlington, VA: Association for the Advancement of Medical Instrumentation; 2009. | ||
US Department of Health and Human Services, US Food and Drug Administration. Applying Human Factors and Usability Engineering to Medical Devices: Draft Guidance for Industry and Food and Drug Administration staff; 2016. Available from: https://www.fda.gov/downloads/MedicalDevices/.../UCM259760.pdf. Accessed July 11, 2017. | ||
US Department of Health and Human Services, US Food and Drug Administration, Center for Drug Evaluation and Research. Safety Considerations for Product Design to Minimize Medication Errors; 2016. Available from: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm331810.pdf. Accessed July 11, 2017. | ||
Rotondi AJ, Eack SM, Hanusa BH, Spring MB, Haas GL. Critical design elements of e-health applications for users with severe mental illness: singular focus, simple architecture, prominent contents, explicit navigation, and inclusive hyperlinks. Schizophr Bull. 2015;41(2):440–448. |
Supplementary materials
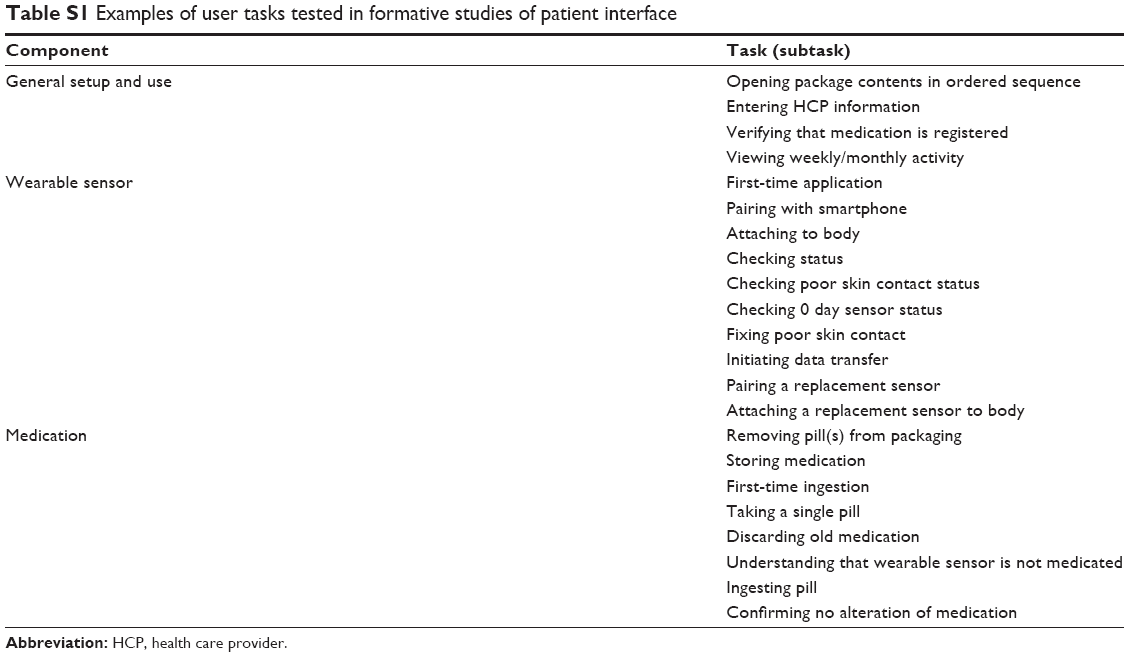 | Table S1 Examples of user tasks tested in formative studies of patient interface |
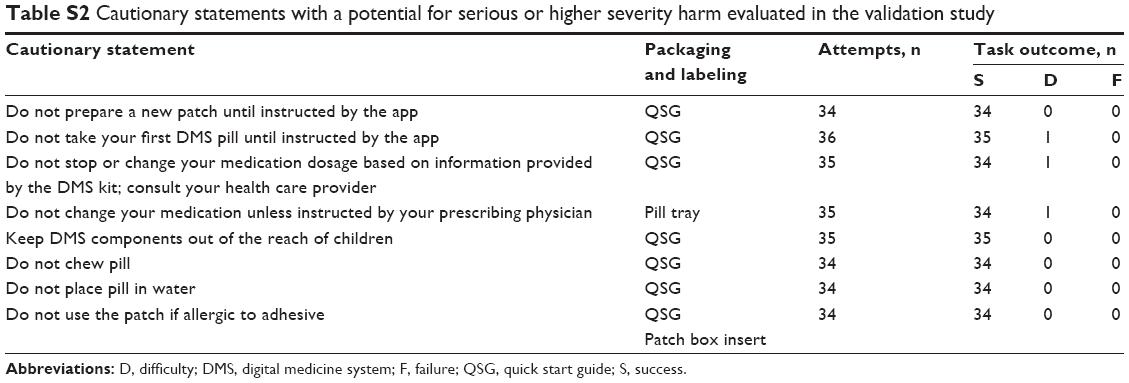 | Table S2 Cautionary statements with a potential for serious or higher severity harm evaluated in the validation study |
 | Table S3 App messaging evaluated in the validation study |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.