Back to Journals » Cancer Management and Research » Volume 10
Human epithelial growth factor receptor 2 in human salivary carcinoma ex pleomorphic adenoma: a potential therapeutic target
Authors Xia L, Wang Y, Hu Y, Zhang C, Gu T, Wang L, Li J ![]() , Yu W
, Yu W ![]() , Tian Z
, Tian Z
Received 4 August 2018
Accepted for publication 7 October 2018
Published 30 November 2018 Volume 2018:10 Pages 6571—6579
DOI https://doi.org/10.2147/CMAR.S182652
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Liang Xia,1,2,* Yang Wang,1,* Yuhua Hu,1,* Chunye Zhang,1 Ting Gu,1 Lizhen Wang,1 Jiang Li,1 Wenwen Yu,2 Zhen Tian1
1Department of Oral Pathology, Ninth People’s Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai Key Laboratory of Stomatology, Shanghai 200011, People’s Republic of China; 2Department of Oral and Cranio-maxillofacial Surgery, Ninth People’s Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai Key Laboratory of Stomatology, Shanghai 200011, People’s Republic of China
*These authors contributed equally to this work
Background: To inhibit human epithelial growth factor receptor 2 (HER2) in salivary carcinoma ex pleomorphic adenoma (CXPA) and investigate the effects on tumor cell proliferation, cell cycle, and apoptosis. To assess the possibility of blocking HER2 to improve the malignant biological behavior of CXPA.
Materials and methods: HER2 expression and amplification were examined using an immunofluorescence assay and fluorescence in situ hybridization in 2 CXPA cell lines (SM-AP1 and SM-AP4 cells). The effects on tumor cell proliferation, cell cycle, apoptosis, and HER2 downstream pathways were verified after the application of a HER2 inhibitor.
Results: HER2 was overexpressed and amplified in SM-AP1 and SM-AP4 cell lines. After blocking HER2, the tumor proliferation and cell cycle were significantly induced, and the apoptosis process was activated. Moreover, the downstream pathways PI3K/AKT and MAPK/ERK were significantly inhibited.
Conclusion: HER2 was overexpressed and amplified in CXPA cell lines and might thus play an important role in tumor development. Inhibiting HER2 may be a novel targeted therapy for poor biological behavior of CXPA.
Keywords: HER2, amplification, carcinoma ex pleomorphic adenoma, proliferation, apoptosis, target therapy
Background
PA is the most common benign neoplasm of the salivary gland. Long-lasting or recurrent PA can undergo malignant transformation into CXPA.1 According to the previous study, CXPA comprises ~4% of all salivary tumors and 12% of all salivary malignant tumors.2 On the basis of the statistics from our department, CXPA accounts for 8% of all salivary malignant tumors, representing the third most common salivary malignancy.3
HER2 is an epithelial growth factor receptor tyrosine kinase encoded by chromosome 17q12, which has been proven to be overexpressed in a variety of solid tumors.4,5 Previous studies have shown that HER2 is overexpressed in salivary malignant pleomorphic adenoma and is involved in multiple cellular pathways, including cell proliferation, adhesion, necrosis, and vascular growth and migration; it also plays an important role in changing the biological behavior of tumors.4 Previous studies have shown that HER2 overexpression or amplification is an indicator of poor prognosis in patients with salivary invasive CXPA, which is associated with high histological grade and metastasis of CXPA.6 HER2-targeted therapy has potential application value for HER2-positive patients with CXPA.7 In that case, the effect of blocking HER2 on cell proliferation, cell cycle, apoptosis, and other biological behaviors of CXPA tumor cells warrants further study.
Lapatinib, which is a small-molecule inhibitor of the tyrosine kinase activity of the HER2 protein, can combine with the ATP sites to prevent the phosphorylation and activation of tumor cells. Lapatinib has been approved by the US FDA for the treatment of HER2-positive breast cancer and is associated with improved survival outcomes in patients.8 Lapatinib combination therapy can significantly prolong disease progression time, improve drug response rate, and partially alleviate the symptoms of patients with trastuzumab resistance.9
In the present study, the HER2 inhibitor lapatinib was used to intervene with the tumor cells in vitro. Additionally, we evaluated whether the HER2 inhibitor could suppress the expression of HER2 in vitro and exert the inhibitory effect on the downstream signaling pathways of HER2 in CXPA cell lines, so as to improve the malignant biological behavior of the tumor.
Materials and methods
Cell lines and cell culture
The human salivary gland CXPA cell lines SM-AP1 and SM-AP4 were kindly provided by Niigata University.10 The human fibroblast HS683 cell line was purchased from the American Type Culture Collection. All cell lines were preserved by the Shanghai Key Laboratory of Stomatology, the Ninth People’s Hospital, Shanghai Jiao Tong University, School of Medicine. CXPA cells and fibroblasts were cultured with DMEM medium containing 10% FBS, 100 U/mL penicillin, 100 mg/mL streptomycin, and 2 mM glutamine in a 5% CO2 incubator at 37°C. The cells were digested by 0.25% trypsin with 0.02% EDTA.
Immunofluorescence microscopy
For immunofluorescence staining, cultured cells were rinsed three times with PBS and fixed with 3.7% formaldehyde and then permeabilized with 0.2% Triton X-100. After blocking in 1% BSA for 1 hour, the cells were incubated with the primary antibody in a moist, 4°C chamber overnight, washed, and then incubated for 30 minutes with fluorescein isothiocyanate (FITC)-labeled secondary antibody in the dark (dilution 1:1000; Thermo Fisher Scientific, Waltham, MA, USA), followed by staining with 5 µg/mL DAPI for 2 minutes at room temperature. The cells were washed and examined under a fluorescence microscope (E800; Nikon Instruments, Melville, NY, USA) at 400× magnification.
FISH
FISH was performed on the slides of cells with a HER2 DNA Probe Kit (PathVysion, Chicago, IL, USA) according to the manufacturer’s instructions.6 Two fluorescent-labeled DNA probes, HER2 spectrum orange and chromosome 17 enumeration control probe (CEP17) spectrum green, were included in the kit. The signal analysis was processed using an Olympus BX51 fluorescent microscope system. At least 20 tumor cell nuclei in a minimum of three different areas were counted to determine the HER2 numbers and CEP17 signals. The HER2/CEP17 ratios were consistently interpreted according to the American Society of Clinical Oncology/College of American Pathologists (ASCO/CAP) recommendations.11 HER2 gene amplification was classified as positive when HER2/CEP17 ratio is ≥2 or HER2 copy number is ≥6.0, and negative when HER2/CEP17 ratio is <2 and HER2 copy number is <4.0. If the HER2/CEP17 ratio is <2 and HER2 copy number is 4.0–6.0, another 20 tumor cells were counted.
Western blotting analysis
The levels of HER2 (#2165; Cell Signaling Technology, Danvers, MA, USA), phosphorylated HER2 (p-HER2, #6942; Cell Signaling Technology), AKT (#4628; Cell Signaling Technology), p-AKT (#4060; Cell Signaling Technology), ERK (#4695; Cell Signaling Technology), and p-ERK (#4370; Cell Signaling Technology) were determined by Western blotting assays. The SM-AP1 and SM-AP4 cell lines were washed with PBS after 1 hour of treatment with lapatinib (#S2111; Selleckchem, TX, USA) at 10−3, 10−2, 10−1, 1, and 10 µM concentration. The concentration of protein was determined by the routine process by using the protein lysate containing a phosphatase inhibitor. The available proteins were thermally denatured at 100°C for 10 minutes. The appropriate steps were followed as described in previous studies. The protein level was normalized to the internal control GAPDH (#SC-47724; Santa Cruz, CA, USA).
Proliferation assay
The CCK8 (#CK04; Dojindo, Japan) colorimetric assay was used to screen for cell proliferation. Cells were seeded in 96-well plates at a density of 2×103 cells per well. A total of 10 µL of CCK8 solution was added to each well, and cell culture was continued for 2 hours at 37°C in dark. The absorbance was measured using a microplate reader at a wavelength of 450 nm. These measurements were carried out for 5 consecutive days. Normal human fibroblast cells were used as negative control. The experiment was performed in triplicate.
Cell cycle analysis
Cells were collected and fixed in 75% ethanol, washed two times with ice-cold PBS, and resuspended in 500 µL of PBS. Cell suspensions were incubated with RNase A (50 µg/mL) for 30 minutes at 37°C and sequentially stained with PI (50 µg/mL) for 1 hour and then analyzed by flow cytometry. Normal human fibroblast cells were used as negative control. Three independent experiments were performed.
Apoptosis detection
The Annexin V-FITC/PI Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA) was used following the manufacturer’s instructions. Cells showing annexin V+/PI− staining were considered as early apoptotic cells, and those showing annexin V+/PI+ staining were considered as late apoptotic cells. After the staining, the cells were immediately analyzed using a BD FACS Calibur flow cytometer and the CellQuest software. Normal human fibroblast cells were used as negative control. The experiment was performed in triplicate.
Statistical analysis
All the statistical data were analyzed by the SPSS 16.0 (SPSS Inc., Chicago, IL, USA) and GraphPad Prism 6.0 software (San Diego, CA, USA). Quantitative data were presented as mean ± SD and were compared using Student’s t-test. P<0.05 was considered to be statistically significant. All the assays were performed at least three times independently.
Results
HER2 is expressed and amplified in CXPA cell lines
Immunofluorescence staining showed that the HER2 protein was present in both SM-AP1 and SM-AP4 cells, but not in fibroblast cells (Figure 1). FISH results showed that the HER2 gene was amplified in SM-AP1 cells with an average increasing gene copy number of 6.363 (Figure 2A), and in SM-AP4 cells with an average increasing gene copy number of 6.625 (Figure 2B). However, the HER2/CEP17 ratio was 1, indicating that no HER2 gene amplification occurred in the fibroblasts (Figure 2C).
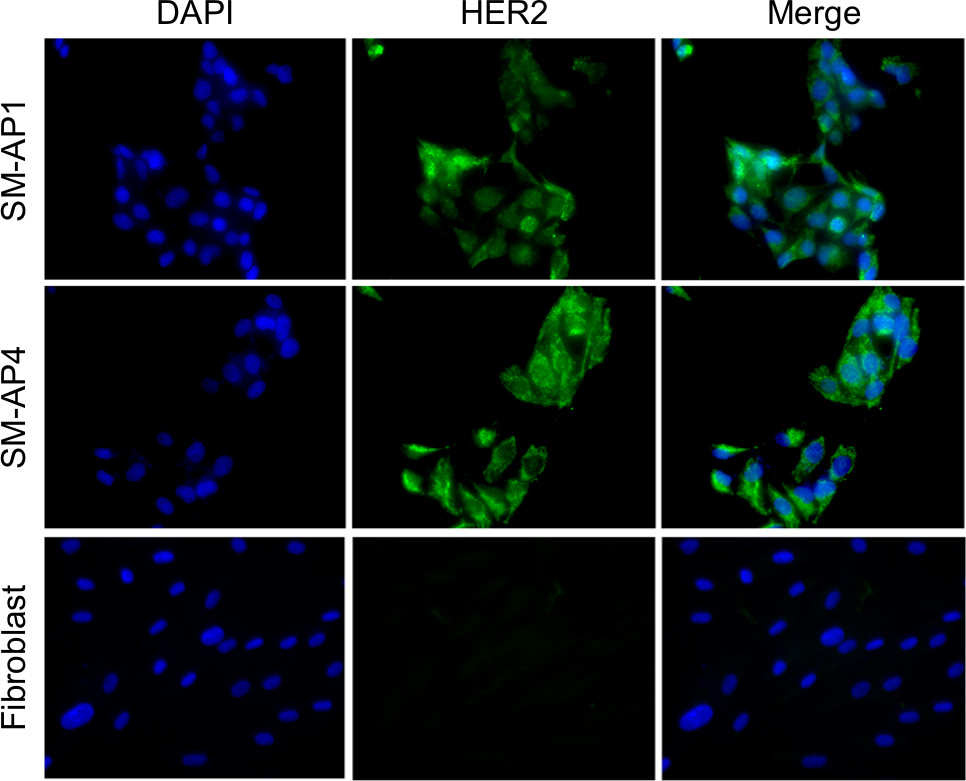 | Figure 1 The expression of the HER2 protein in SM-AP1, SM-AP4, and fibroblast cells. Notes: The HER2 protein was expressed in SM-AP1 and SM-AP4 cells, but not in the fibroblasts. Magnification 400×. Abbreviation: HER2, human epithelial growth factor receptor 2. |
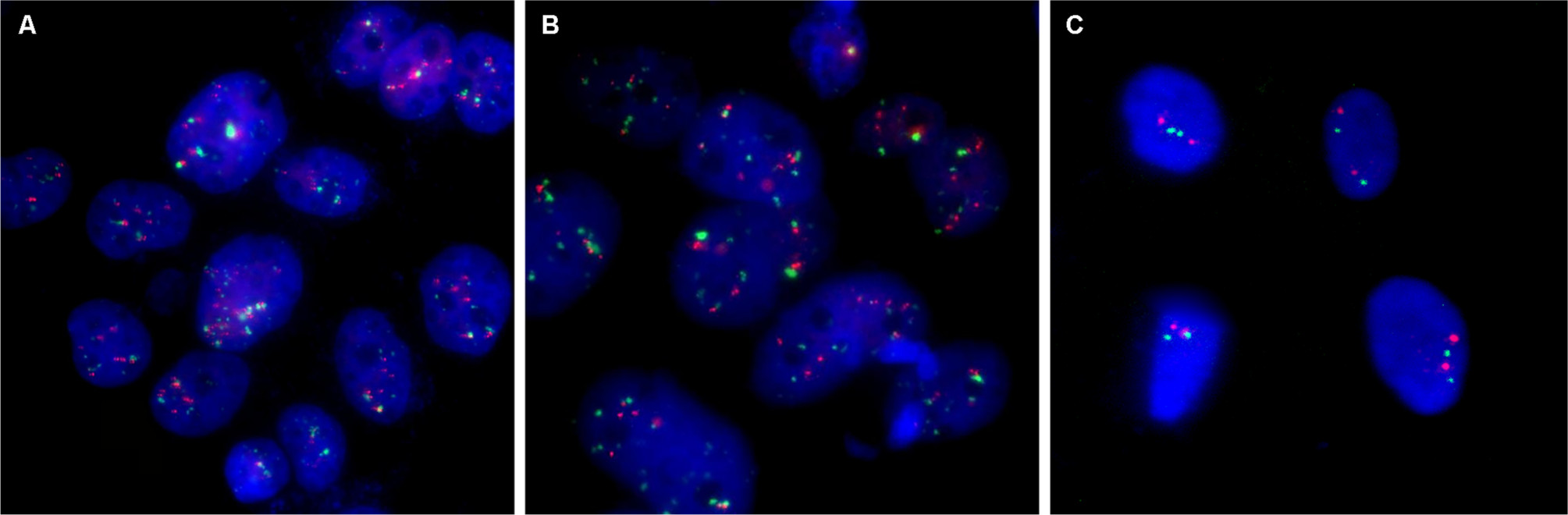 | Figure 2 HER2 amplification was detected in SM-AP1 (A) and SM-AP4 (B) cells, but not in the fibroblasts (C). Notes: Increased copy numbers (more than six) in cells are shown in A and B. HER2/CEP17 ratio =1 is shown in C. Magnification 1000×. Abbreviation: HER2, human epithelial growth factor receptor 2. |
Inhibitory effect of lapatinib on HER2 protein phosphorylation and its downstream pathway
After lapatinib treatment, HER2 protein expression in SM-AP1 and SM-AP4 cells was almost the same. However, the phosphorylation of HER2 was apparently suppressed. p-HER2 protein expression was inhibited by lapatinib in a dose-dependent manner. Higher lapatinib dose resulted in less expression of the p-HER2 protein (Figure 3A and B). We verified the inhibitory effect of lapatinib on the PI3K/AKT and MAPK/ERK pathways, which are downstream of HER2. In SM-AP1 cells, the expression of total AKT and p-AKT was inhibited by lapatinib in a dose-dependent manner, whereas lapatinib had little effect on the expression of total EKR, and p-ERK expression was inhibited in a dose-dependent manner. In SM-AP4 cells, although the inhibitory effect of lapatinib on total AKT, p-AKT, and total ERK expression was not obvious, p-ERK was apparently suppressed, especially when the concentration of lapatinib was higher than 1 µM (Figure 3A and B).
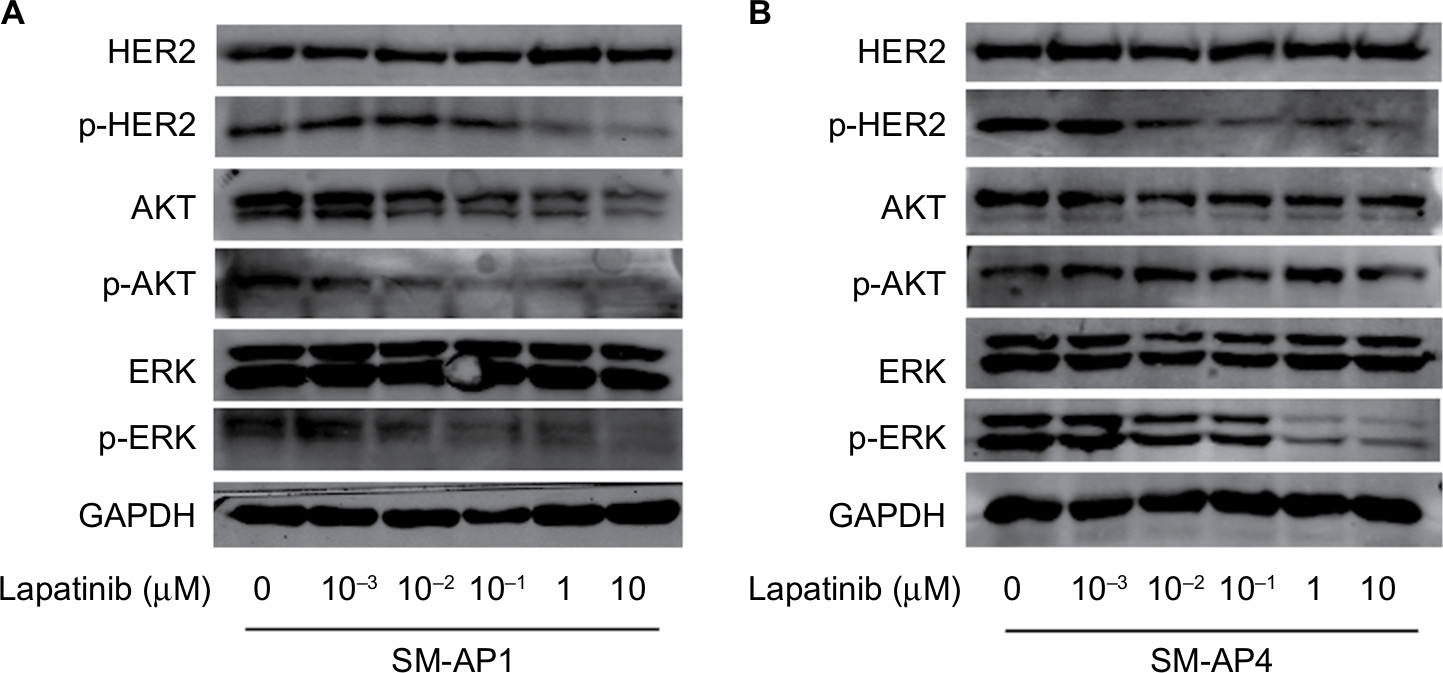 | Figure 3 The effect of lapatinib on HER2 expression and its downstream pathways in SM-AP1 (A) and SM-AP4 (B) cells. Notes: (A) p-HER2, AKT, p-AKT, and p-ERK were inhibited by lapatinib in a dose-dependent manner in SM-AP1 cells. (B) p-HER2 was inhibited by lapatinib in a dose-dependent manner, and p-ERK was suppressed significantly when the concentration of lapatinib reached at 1 µM in SM-AP4 cells. Abbreviations: HER2, human epithelial growth factor receptor 2; p-AKT, phosphorylated AKT; p-ERK, phosphorylated ERK; p-HER2, phosphorylated HER2. |
Effects of lapatinib on cell proliferation, cell cycle, and apoptosis
CCK8 results showed that the activity and proliferation of SM-AP1 and SM-AP4 cells were decreased significantly after lapatinib treatment (P<0.001) (Figure 4A and B). Fibroblast cells were used as a control group. Lapatinib did not significantly affect the proliferation ability of fibroblasts (Figure 4C), because there was no HER2 amplification in fibroblasts (Figure 2C). The results of flow cytometry showed that the cell cycle of SM-AP1 and SM-AP4 cells was significantly inhibited and was blocked in the G1 phase (Table 1; Figure 5A–D). However, the cell cycle of fibroblasts was not suppressed by lapatinib (Figure 5E and F). The apoptosis of tumor cells was increased significantly after lapatinib treatment (Figure 6A–D, G). Conversely, the apoptosis of fibroblasts was not obviously affected by lapatinib (Figure 6E–G).
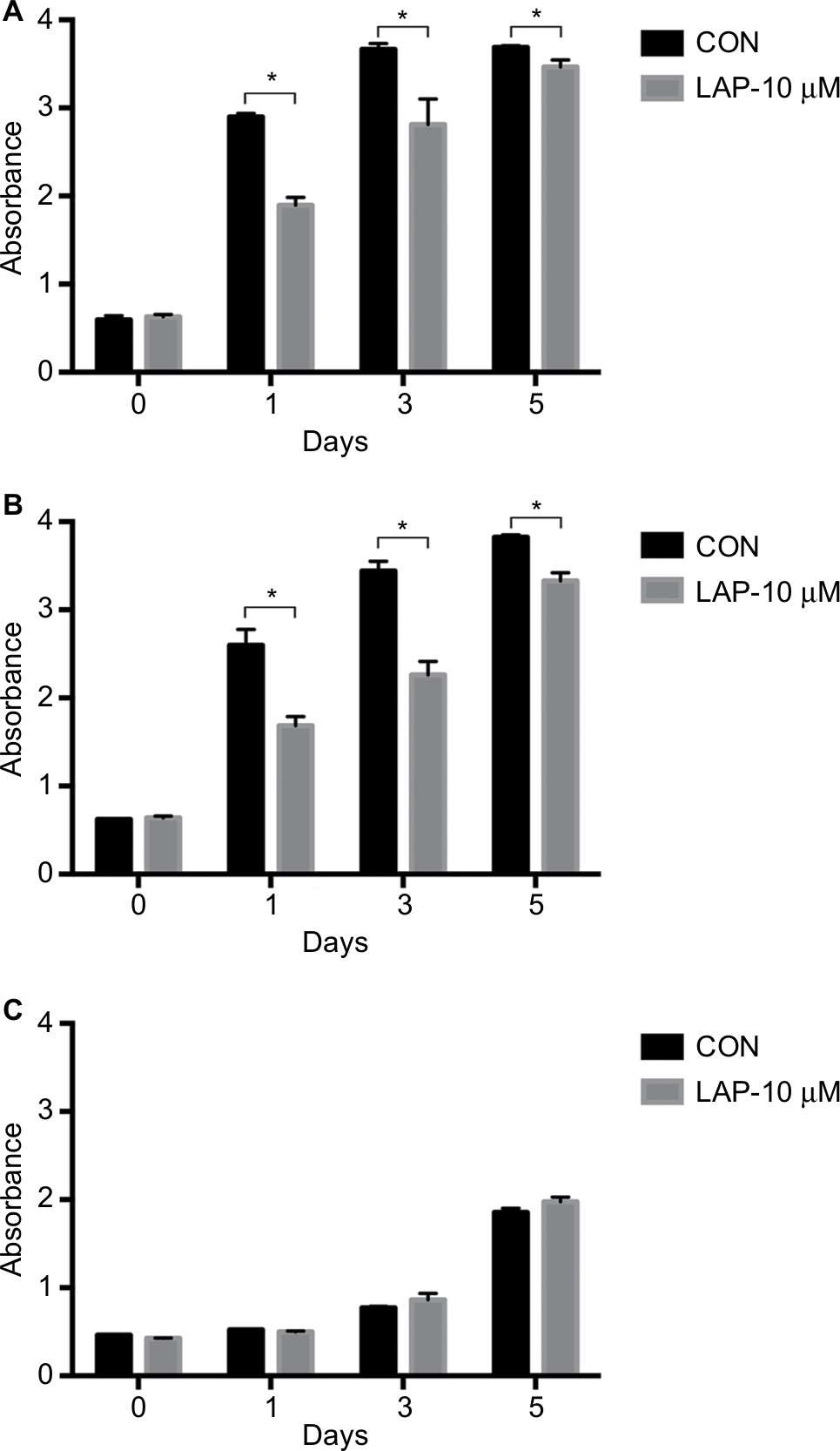 | Figure 4 Lapatinib inhibits the activity and proliferation of SM-AP1 (A) and SM-AP4 (B) cells, but not those of normal fibroblasts (C). Note: *Significant at P<0.001. Abbreviation: CON, control group. |
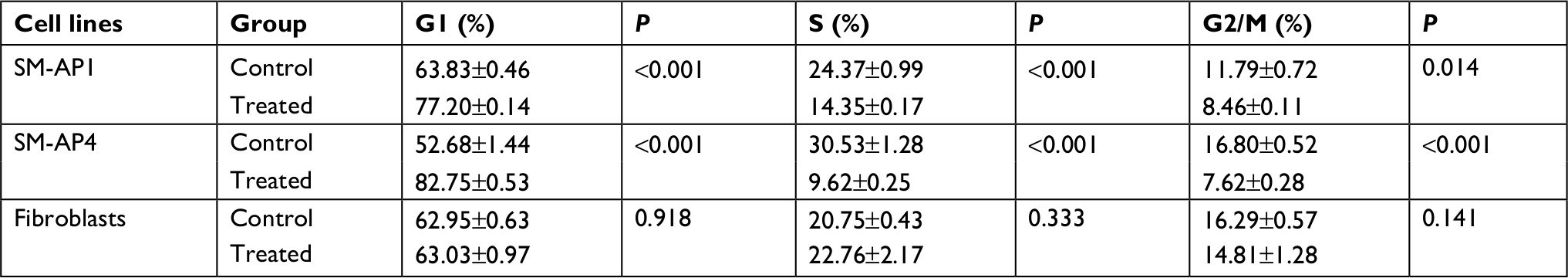 | Table 1 Cell cycle analysis Notes: Analysis of cell cycle in the control group and the lapatinib-treated group of SM-AP1, SM-AP4, and normal fibroblasts. Data represent mean ± SD of three independent experiments. |
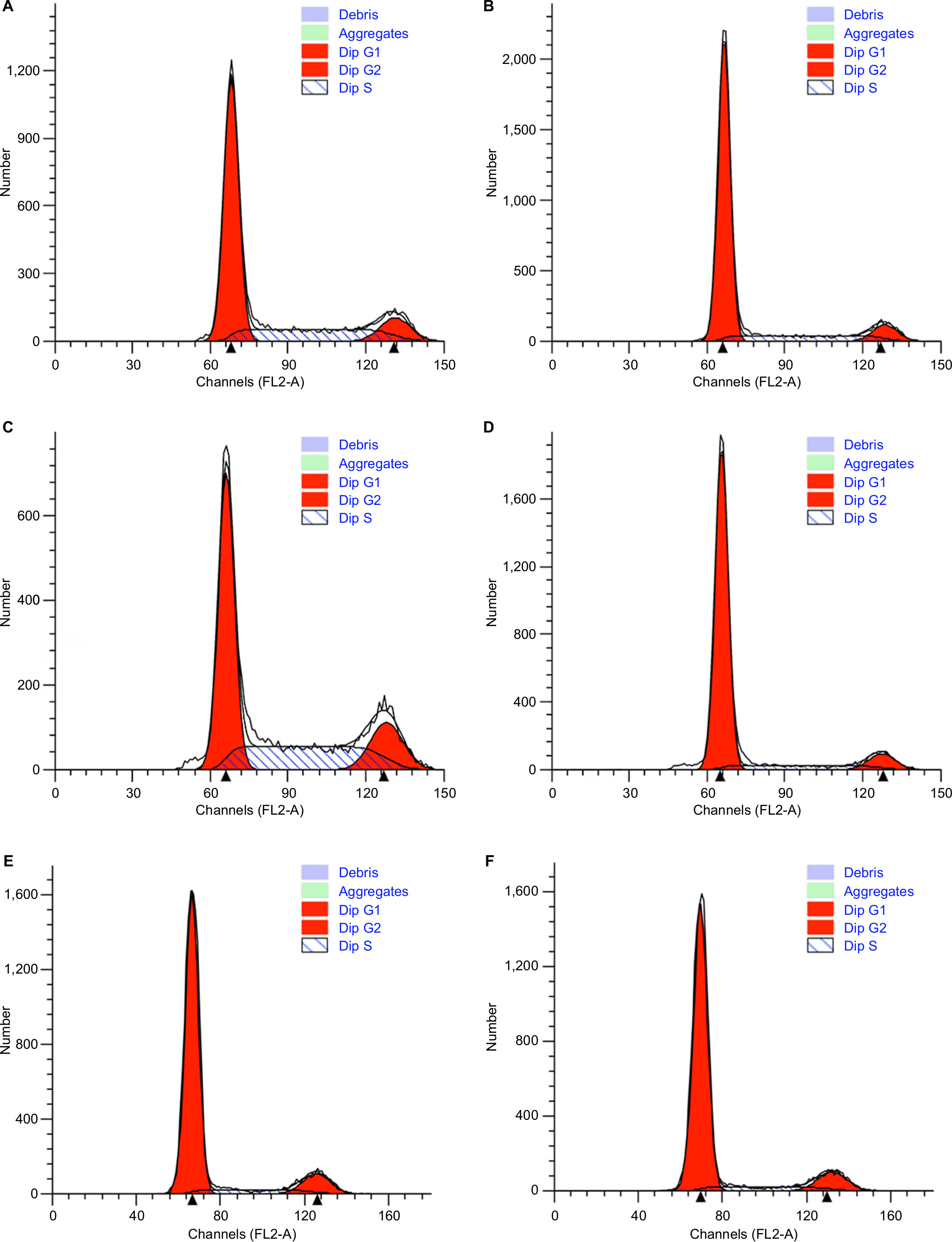 | Figure 5 The effect of lapatinib on cell cycle of SM-AP1 and SM-AP4 cells. Note: (A and B) SM-AP1 cell cycle arrested in the G1 phase after lapatinib treatment; (C and D) SM-AP4 cell cycle arrested in the G1 phase after lapatinib treatment; (E and F) normal fibroblast cell cycle was not affected by lapatinib treatment. |
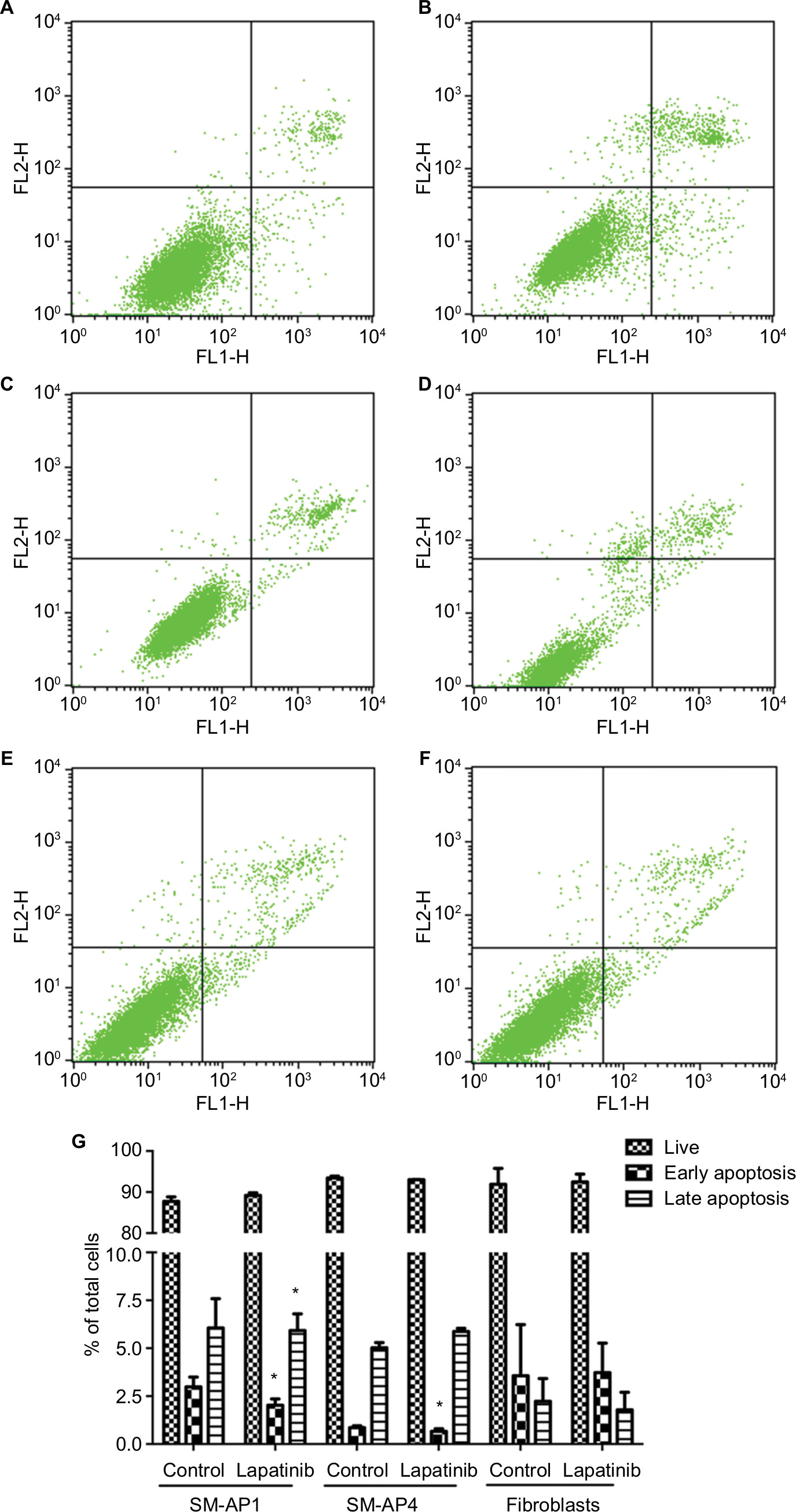 | Figure 6 Lapatinib promoted the apoptosis of SM-AP1 (A and B) and SM-AP4 cells (C and D), but not that of normal fibroblasts (E and F). The percentage of apoptotic cells was increased in SM-AP1 cells, whereas the percentage of early apoptotic cells was increased in SM-AP4 cells (G). The apoptosis of fibroblasts was not affected by lapatinib (G). Note: *Significant at P<0.001. |
Discussion
The HER2 protein, a receptor tyrosine kinase, belongs to the EGFR family. HER2 can phosphorylate tyrosine residues of itself or its substrate. Phosphorylated tyrosine residues can in turn activate the HER2 protein, which then lead to the activation of downstream signal molecules and subsequently cause a series of signaling cascade in cells. HER2 plays an important role in mediating various cell signal transduction pathways involved in cell proliferation, apoptosis, adhesion, angiogenesis, metastasis, and other physiological processes, which are crucial for the advancement of the malignant phenotype.5
In breast cancer, HER2 is an important prognostic factor. HER2 overexpression and amplification are an important factor to be considered in the selection of targeted therapy.12,13 It has been reported that the targeting therapeutic antibody herceptin could downregulate protein expression by targeting HER2 and thus reduce the activation of the PI3K/AKT and MAPK/ERK signaling pathways.14,15
The PI3K/AKT pathway exists widely in cells, and it is a signal transduction pathway involved in cell growth, proliferation, differentiation, cell survival, adhesion, migration, and apoptosis. Studies have demonstrated that HER2 is overexpressed and amplified in breast cancer, lung cancer, melanoma, ovarian cancer, and cervical cancer. Zhou et al demonstrated that the phosphorylation level of AKT increased from normal breast epithelium, atypical hyperplasia to malignant transformation and carcinomatous infiltration.16 Anti-HER2 antibodies can inhibit the proliferation of tumor cells by blocking the activation of the PI3K/AKT pathway.
The MAPK/ERK pathway is an important signal transduction system mediating extracellular signal to intracellular response, which can be activated by phosphorylation by a variety of growth factors and cytokines. ERK is a core signal molecule that regulates cell growth and development; it is located in the cytoplasm and can be transferred into the nucleus to regulate cell processes after activation. HER2 combines with ligands and leads to the dimerization reaction and the activation of protein tyrosine kinases, which bind to the SH2 and SH3 domains of Grb to recruit Sos, and then activate Ras and its downstream ERK to trigger the subsequent cellular biological responses.17
Lapatinib is an orally available, small-molecule, reversible inhibitor of both EGFR and HER2 tyrosine kinases, and it has been approved for use in combination with capecitabine for the treatment of advanced HER2-amplified breast cancer.18,19 Additionally, it is reported that lapatinib has been used in the targeted chemotherapy for salivary ductal carcinoma with HER2 gene amplification.20 It is necessary to elucidate the role of HER2 in tumor development and the mechanism by which lapatinib exerts effects on CXPA. We evaluated the HER2 protein expression and gene amplification in CXPA cells and found that HER2 might play a role in the progression of CXPA. In the in vitro experiments, we verified the blocking effect of lapatinib on HER2 phosphorylation and the activation of its downstream pathway; this result was similar to the findings of a previous study on breast cancer.21 In breast cancer, lapatinib could significantly downregulate p-HER2, p-EKR, and p-AKT in a dose- and time-dependent manner but could not downregulate total HER2, AKT, and ERK in SK-BR3 cells.
We also observed the subsequent inhibitory effect of lapatinib on cell viability and proliferation and induction of tumor apoptosis in CXPA cell lines. Lapatinib prohibited the activation of MAPK/ERK and PI3K/AKT in the downstream of HER2 to inhibit cell proliferation. This finding suggested that the activated HER2 promoted the development and malignant transformation of CXPA by activating the downstream MAPK/ERK and PI3K/AKT pathways. Phosphorylation of AKT can activate the NF-κB pathway, which plays an important role in tumorigenesis, tumor formation, proliferation, and apoptosis. We suggest that the indirect inhibition of NF-κB may also play an important role in inducing tumor cell apoptosis and inhibiting tumor proliferation.
A previous study has demonstrated gain in HER2 and EGFR gene copy number in CXPA.22 In that study, Nishijima et al concluded that HER2 overexpression and HER2 gene amplification were associated with CXPA extracapsular invasion, lymph node metastasis, and worse prognosis, but EGFR overexpression was not correlated with the aggressiveness of tumor. Interestingly, high polysomy of the EGFR gene tended to promote the invasive capacity of CXPA and was related to tumor metastasis and poor prognosis. There is a discrepancy between the prognostic impacts of EGFR overexpression and high polysomy of the EGFR gene, but the molecular basis underlying these phenomena is still unclear and needs to be revealed. It is known that EGFR activation could induce oncogenic effects. Compared with a single tyrosine kinase inhibitor, lapatinib can simultaneously inhibit HER2 and EGFR to control the tumor malignant phenotype; this indicated that lapatinib could be a potential therapeutic drug for the treatment of CXPA.
HER2 is expected to become a potential drug-targeting site and is worthy of follow-up experiments to investigate.
Conclusion
We found that the HER2 gene was amplified and the HER2 protein was overexpressed in CXPA cell lines. Thus, HER2 may play an important role in tumor development. HER2 plays an important role in mediating cell signal transduction pathways in cancer. We suggest that lapatinib is an effective inhibitor that is capable of suppressing cell proliferation and cell cycle and can induce cell apoptosis in CXPA cells with HER2 gene amplification. Inhibiting HER2 may be a novel targeted therapy for poor biological behavior of CXPA.
Abbreviations
CCK8, Cell Counting Kit 8
CXPA, carcinoma ex pleomorphic adenoma
FDA, US Food and Drug Administration
FISH, fluorescence in situ hybridization
HER2, human epithelial growth factor receptor 2
PA, pleomorphic adenoma
PI, propidium iodide
p-AKT, phosphorylated AKT
p-ERK, phosphorylated ERK
p-HER2, phosphorylated HER2
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Shanghai Ninth People’s Hospital affiliated to Shanghai Jiao Tong University, School of Medicine (Shanghai, People’s Republic of China). The ethics approval (number 2013-68) is attached.
Consent for publication
All authors have agreed to the contents of the manuscript in its submitted form.
Availability of data and material
All data generated or analyzed during this study are included in this published article.
Acknowledgments
The authors are grateful to Divisions of Oral Pathology, Department of Tissue Regeneration and Reconstruction, Niigata University, for establishing the CXPA cell lines, and to Professor Jun Cheng for kindly providing the CXPA cells lines SM-AP1 and SM-AP4. We also would like to thank Hanbing Fu for excellent technical assistance. This work was supported by grants from Shanghai Nature Science Foundation (grant nos. 18ZR1422200) and the Medicine and Engineering Interdisciplinary Research Fund of Shanghai Jiao Tong University (YG2015QN10).
Author contributions
All authors read and approved the final manuscript. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Hashimoto K, Yamamoto H, Shiratsuchi H, et al. HER-2/neu gene amplification in carcinoma ex pleomorphic adenoma in relation to progression and prognosis: a chromogenic in-situ hybridization study. Histopathology. 2012;60(6B):E131–E142. | ||
Thompson L. World Health Organization classification of tumours: pathology and genetics of head and neck tumours. Ear Nose Throat J. 2006;85(2):74. | ||
Tian Z, Li L, Wang L, Hu Y, Li J. Salivary gland neoplasms in oral and maxillofacial regions: a 23-year retrospective study of 6982 cases in an eastern Chinese population. Int J Oral Maxillofac Surg. 2010;39(3):235–242. | ||
Marx AH, Tharun L, Muth J, et al. HER-2 amplification is highly homogenous in gastric cancer. Hum Pathol. 2009;40(6):769–777. | ||
Glisson B, Colevas AD, Haddad R, et al. HER2 expression in salivary gland carcinomas: dependence on histological subtype. Clin Cancer Res. 2004;10(3):944–946. | ||
Xia L, Hu Y, Li J, et al. A low percentage of HER-2 amplification whereas indicates poor prognosis in salivary carcinoma ex pleomorphic adenoma: a study of 140 cases. J Oral Pathol Med. 2017;46(3):167–174. | ||
Yan M, Parker BA, Schwab R, Kurzrock R. HER2 aberrations in cancer: implications for therapy. Cancer Treat Rev. 2014;40(6):770–780. | ||
Molina MA, Sáez R, Ramsey EE, et al. NH(2)-terminal truncated HER-2 protein but not full-length receptor is associated with nodal metastasis in human breast cancer. Clin Cancer Res. 2002;8(2):347–353. | ||
Tevaarwerk AJ, Kolesar JM. Lapatinib: a small-molecule inhibitor of epidermal growth factor receptor and human epidermal growth factor receptor-2 tyrosine kinases used in the treatment of breast cancer. Clin Ther. 2009;31(Pt 2):2332–2348. | ||
Maruyama S, Cheng J, Shingaki S, et al. Establishment and characterization of pleomorphic adenoma cell systems: an in-vitro demonstration of carcinomas arising secondarily from adenomas in the salivary gland. BMC Cancer. 2009;9:247. | ||
Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. Arch Pathol Lab Med. 2014;138(2):241–256. | ||
Shih J, Bashir B, Gustafson KS, et al. Cancer Signature Investigation: ERBB2 (HER2)-Activating Mutation and Amplification-Positive Breast Carcinoma Mimicking Lung Primary. J Natl Compr Canc Netw. 2015;13(8):947–952. | ||
Ulas A, Kos T, Avci N, et al. Patients with HER2-positive early breast cancer receiving adjuvant trastuzumab: clinicopathological features, efficacy, and factors affecting survival. Asian Pac J Cancer Prev. 2015;16(4):1643–1649. | ||
Longva KE, Pedersen NM, Haslekås C, Stang E, Madshus IH. Herceptin-induced inhibition of ErbB2 signaling involves reduced phosphorylation of Akt but not endocytic down-regulation of ErbB2. Int J Cancer. 2005;116(3):359–367. | ||
Sakai K, Yokote H, Murakami-Murofushi K, Tamura T, Saijo N, Nishio K. Pertuzumab, a novel HER dimerization inhibitor, inhibits the growth of human lung cancer cells mediated by the HER3 signaling pathway. Cancer Sci. 2007;98(9):1498–1503. | ||
Zhou X, Tan M, Stone Hawthorne V, et al. Activation of the Akt/mammalian target of rapamycin/4E-BP1 pathway by ErbB2 overexpression predicts tumor progression in breast cancers. Clin Cancer Res. 2004;10(20):6779–6788. | ||
Ohtsuka T, Sakaguchi M, Yamamoto H, et al. Interaction of cytokeratin 19 head domain and HER2 in the cytoplasm leads to activation of HER2-Erk pathway. Sci Rep. 2016;6:39557. | ||
Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64(18):6652–6659. | ||
Nishimura R, Toh U, Tanaka M, et al. Role of HER2-Related Biomarkers (HER2, p95HER2, HER3, PTEN, and PIK3CA) in the Efficacy of Lapatinib plus Capecitabine in HER2-Positive Advanced Breast Cancer Refractory to Trastuzumab. Oncology. 2017;93(1):51–61. | ||
Falchook GS, Lippman SM, Bastida CC, Kurzrock R. Human epidermal receptor 2-amplified salivary duct carcinoma: regression with dual human epidermal receptor 2 inhibition and anti-vascular endothelial growth factor combination treatment. Head Neck. 2014;36(3):E25–E27. | ||
Burris HA. Dual kinase inhibition in the treatment of breast cancer: initial experience with the EGFR/ErbB-2 inhibitor lapatinib. Oncologist. 2004;9(Suppl 3):10–15. | ||
Nishijima T, Yamamoto H, Nakano T, et al. Dual gain of HER2 and EGFR gene copy numbers impacts the prognosis of carcinoma ex pleomorphic adenoma. Hum Pathol. 2015;46(11):1730–1743. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.