Back to Journals » Cancer Management and Research » Volume 11
HPV infection associated DNA damage correlated with cervical precancerous lesions and cancer in the highest area of cervical cancer mortality, Longnan, China
Authors Zhao J ![]() , Guo Z, Wang Q, Si T, Pei S, Qu H, Shang L, Yang Y, Wang L
, Guo Z, Wang Q, Si T, Pei S, Qu H, Shang L, Yang Y, Wang L ![]()
Received 14 January 2019
Accepted for publication 27 June 2019
Published 30 July 2019 Volume 2019:11 Pages 7197—7210
DOI https://doi.org/10.2147/CMAR.S201415
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Harikrishna Nakshatri
Jin Zhao,1,* Zhong Guo,1,* Qiang Wang,2 Tianbin Si,3 Shuyan Pei,1 Hongmei Qu,1 Lina Shang,1 Yuqing Yang,1 Lili Wang1
1Department of Medical Function, Medical College of Northwest Minzu University, Lanzhou 730030, People’s Republic of China; 2Department of Pathology, No. 1 Hospital of Longnan City, Longnan 746000, People’s Republic of China; 3Department of Gynecology and Oncology, Gansu Provincial Cancer Hospital, Lanzhou 730050, People’s Republic of China
*These authors contributed equally to this work
Objectives: This study was to assess whether human papillomavirus (HPV) resulting in genetic instability is one reason for the high incidence and mortality of cervical cancer in Longnan.
Methods: Between 2012 and 2016, a total of 346 samples from Longnan were collected and divided into four groups: cervicitis group (n=57), cervical intraepithelial neoplasia I group (CIN I, n=63), CIN II/III group (n=79) and invasive squamous cell carcinoma group (SCC, n=147). HPV E6/E7 mRNA was detected by Quantivirus® HPV E6/E7 RNA 3.0 assay (bDNA). The markers of DNA damage response (DDR) – ataxia telangiectasia mutated (ATM) pSer1981, H2AX pSer139 (γH2AX), Chk2 pThr68 and P53 – were analyzed by immunohistochemistry.
Results: The activation of ATM, γH2AX, Chk2 and P53 was increased with increasing severity of cervical lesion. A significant difference of ATM expression in simple infection was also shown accompanied by the cervical lesion. The expression of γH2AX between HPV16+ and HPV16- specimens, γH2AX and P53 between HPV58+ and HPV58- groups had statistical significance. The expression and copy number of HPV E6/7 mRNA increases with the cervical lesion severity. A significant difference was shown for P53 expression between HPV E6/7 mRNA+ and mRNA- specimens. A close correlation with CHK2 expression for HPV E6/7 mRNA+ and HPV16 E6/7 mRNA+ specimens and γH2AX and CHK2 expression for SCC specimens was shown between low and high viral load groups.
Conclusions: DDR, HPV genotypes and HPV E6/E7 oncogene expression correlated with the level of dysplasia of cervical lesions. HPV infection resulted in genetic instability may be one reason for the high incidence and mortality in Longnan.
Keywords: human papillomaviruses, E6/E7 oncogenes, DNA damage response, cervical cancer
Introduction
Cervical cancer (CC) is a common, often lethal malignancy of women, causing 311,365 deaths in 2018 in the world. Almost 90% of CC deaths occur in developing countries.1 The incidence of CC in People's Republic of China is high, with 132,300 new cases each year, yielding a rate of 27 per 100,000 women.2
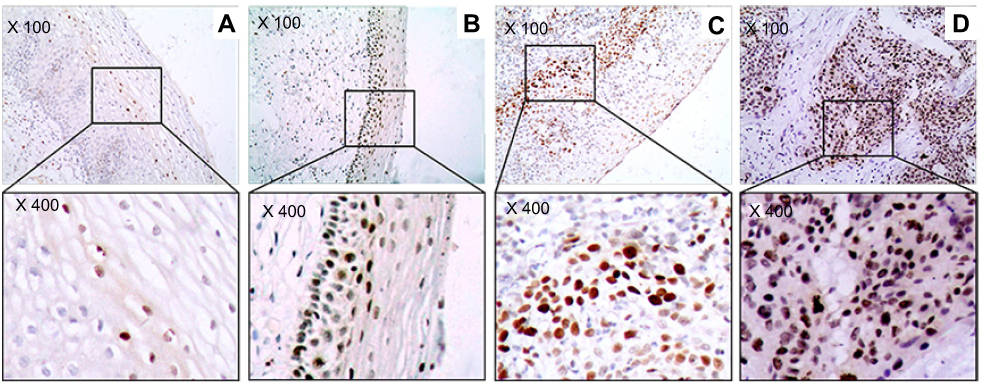 |
Figure 2 The marker (γH2AX) of DNA damage response in study sample. (A) Cervicitis; (B) CIN I; (C) CIN II/III; (D) SCC. Original magnification, ×100 and ×400. |
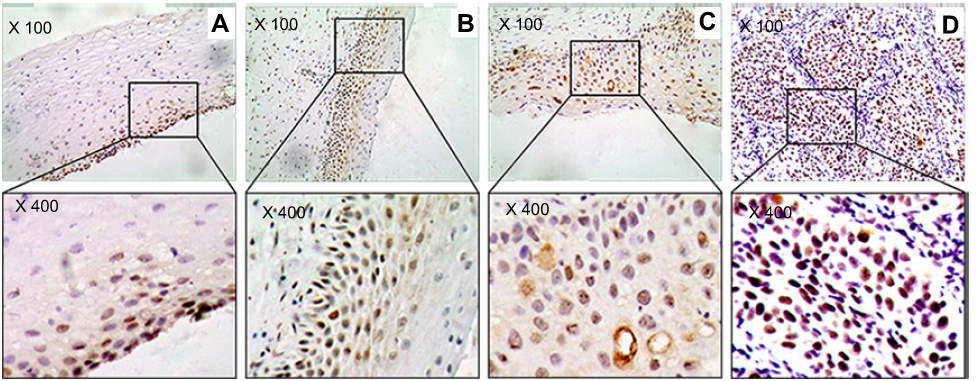 |
Figure 3 The marker (Chk2) of DNA damage response in study sample. (A) Cervicitis; (B) CIN I; (C) CIN II/III; (D) SCC. Original magnification, ×100 and ×400. |
The progression of CC is characterized by lesions that range from low to high grade and is accompanied by an increase in genetic instability.3 This instability is related to DNA damage response (DDR) in cervical cells.4 The ataxia telangiectasia mutated (ATM) kinase plays an important role in the coordination of DDR. ATM phosphorylation at serine 1981 (ATMpSer1981) by DNA damage activates ATM, which then subsequently phosphorylates H2AX at Ser139 (γH2AX), Chk2 at Thr68 (Chk2pThr68) and p53 at Ser15 (p53pSer15), maintaining genome integrity by coordinating cell cycle arrest, apoptosis and DNA damage repair.5
DDR which is relevant for cervical carcinogenesis involved with the persistent infection of human papillomavirus (HPV).6 Oncogenic transformation by HPV is mediated by the viral oncogenes HPV E6 and HPV E7.7 The E7 oncoprotein inactivates the retinoblastoma 1 tumor suppressor gene and E6 promotes ubiquitin-mediated degradation of the p53 tumor suppressor.8 Since p53 constitutes an integral component of the DDR, HPV positive CC cells show a significantly decreased ability to activate cell cycle checkpoints and to induce apoptosis upon DNA damage.9 Additionally, HPV oncogenes expression was reported to negatively impact various DNA repair pathways.10 Thus, HPV positive cancer cells show impaired control of the cell cycle in the context of DNA damage, and display damaged ability to repair DNA lesions.
Longnan of Gansu Province, located in the remote areas of Northwest of People's Republic of China, is a high incidence region of CC with CC mortality as high as 39 per 100,000, ranking first in People's Republic of China.11 Our previous study had shown that HPV16, 18 and 58 play a key role in the development of cervical intraepithelial neoplasia (CIN) and invasive squamous cell carcinoma (SCC) in Longnan women. However, the reason for the high incidence of CC and the high mortality is still not clear.12
The aim of this study was to analyze DDR, HPV genotypes and HPV E6/E7 oncogene expression and correlate these with the process of dysplastic cervical lesions. The results will help add new evidence that HPV-induced genetic instability of cervical cells relates to the high incidence of and mortality due to CC in Longnan patients.
Methods
Study subjects
A total of 346 Longnan patients, ages 17–79 years old, were initially included in the study between January 1, 2012 and January 30, 2016. All the samples were obtained from patients who underwent biopsies with colposcopy or surgical procedures. Of the samples, 305 were obtained from the No.1 Hospital of Longnan City and 24 samples from Gansu Provincial Cancer Hospital. All patients gave written informed consent for their participation. The inclusion criteria were patients 1) with cervicitis, CIN and SCC, pathology confirmed with cervical biopsy or hysterectomy or radical hysterectomy; 2) with HPV genotype testing results; 3) who were Han Chinese; 4) who had grown or lived in Longnan for more than 20 years. Cases were excluded when they met any of the following exclusion criteria: 1) re-examination after treatment of cervical lesions; 2) co-morbid endometrial lesion or ovary diseases or vaginal diseases; 3) pathological results of specimens from fractional curettage; 4) immunocompromised condition (eg, infection with human immunodeficiency virus). This study had been approved by the Ethics Committees of Northwest Minzu University prior to its start. All the specimens were formalin-fixed and paraffin-embedded (FFPE). All samples were evaluated by at least two experienced pathologists in their respective hospital pathology departments. Cervicitis, CIN I, CIN II/III and SCC were diagnosed according to the standard criteria.13
The HPV genotyping of all the samples was detected by Human Papillomavirus Genotyping kit and the results have been reported.12 All 346 specimens fell into one of four groups: cervicitis group (n=57. n=22 HPV+, n=35 HPV−), CIN I group (n=63. n=47 HPV+, n=16 HPV−), CIN II/III group (n=79. n=70 HPV+, n=9 HPV−) and SCC group (n=147. n=132 HPV+, n=15 HPV−). Of these, 218 specimens (n=28 cervicitis, n=30 CIN I, n=60 CINII/III and n=100 SCC) were selected for DDR study. In addition, 187 HPV positive specimens (n=20 cervicitis, n=30 CINI, n=43 CINII/III and n=94 SCC) were chosen for HPV E6/7 mRNA research.
HPV E6/E7 mRNA testing by Quantivirus® HPV E6/E7 RNA 3.0 assay (bDNA)
The Quantivirus® HPV E6/E7 RNA 3.0 assay (bDNA) (DiaCarta, CA, USA) is a sandwich nucleic acid hybridization procedure for the direct quantification of HPV RNA in FFPE samples without RNA purification and RT-PCR. Specimens containing HPV high-risk subtypes 16, 18, 31, 33, 35, 39, 45, 51, 52, 97 58, 59, 66, 68, and low risk subtypes 6, 11, 40, 42, 43, 44 have been validated for quantification in the assay.
The first step is the process of deparaffinization. Six pieces of 5-µm thick sections of FFPE cervical tissue from each paraffin block were used. After removing paraffin from FFPE cervical tissue samples, l mL of 100% xylene was added to the pieces of FFPE tissue sections, heated for 5 mins at 50°C to melt the paraffin, and then centrifuged for 2 mins at 20,000× g at room temperature to produce a tissue pellet. Then, 1 mL of 100% ethanol was added after the xylene was removed. After mixing, the sample was centrifuged for 2 mins at 20,000× g at room temperature and the ethanol was discarded without disturbing the pellet. After washing with ethanol twice, the pellet was air-dried for approximately 25 mins.
Five hundred microliter Tissue Lysis Mixture and 5 µL Proteinase K were added to the above sample and incubated at 65°C for 3 hrs. The sample tube was shaken for 2 mins every 20 mins during the incubation, and then each sample well was added 50 µL of Lysis Working Reagent. The RNA was captured onto a microwell by a set of specific, synthetic oligonucleotide capture probes. After each well of the Capture Plate was added 100 µL Pre-Amplifier Probes Working Reagent, the sealed Capture Plate was incubated at 55°C for 40 mins. Then, 100 µL Amplifier Probe Working Reagent was added to each well of the Capture Plate which was sealed and incubated at 55°C for 40 mins. Then, 100 µL Label Probe Working Reagent was added to each well of the Capture Plate which was sealed and incubated at 50°C for 40 mins. Finally, 100 µL Substrate Working Reagent was added to each well of the Capture Plate which was then sealed and incubated at 46°C for 20 mins. The plate was then read immediately (within 1 min) after being removed from the incubator by the System DiaCarta QuantiViurs Reader.
In the above steps, HPV genomic RNA was released from the virions. A set of target probes was hybridized to both the viral RNA and the pre-amplifier probes. Both the capture and the target probes were bonded to the 5-untranslated and E6 and E7 regions of the HPV genome. The amplifier probe subsequently hybridized to the pre-amplifier forming a branched DNA (bDNA) complex. Then, the multiple copies of an alkaline phosphatase (AP) labeled probe were hybridized to this immobilized complex. Detection was reached by incubating the AP-bound complex with a chemiluminescent substrate. Light emission was correlated directly to the amount of HPV RNA present in each sample, and results were recorded as relative light units (RLUs) by the System DiaCarta QuantiViurs Reader. The cutoff of 1.00 was used for determining positive in the assay. The positive control was defined by light emission from the control sample containing known concentrations of cells. HPV RNA concentrations of specimens were determined from this positive control. The result of positive or negative was generated based on RLUs from residual samples over the RLUs from the background.
Quantivirus® HPV E6/E7 RNA 3.0 assay (bDNA) was estimated using a two-part, eight-member panel with HPV target concentrations between 481 and 7,690,000 IU/mL (2500 and 40,000,000 HPV RNA copies/mL). This method was shown to be linear from 615 to 7,690,000 IU/mL (3200–40,000,000 HPV RNA copies/mL). The accuracy of this method, defined as the percent recovery (ratio of observed mean quantification to expected concentration), was from 92% to 108% across the quantification range.14,15
Immunohistochemical analysis of DDR
One representative tissue block for every patient, including the dysplastic region, invasive margin and tumor-associated non-neoplastic mucosa, was assayed by immunohistochemistry. In cases of large, late-stage tumors, various sections were assayed to include representative areas of the tumor center and lateral and deep invasive margins. The paraffin-embedded tissues that were used for the original hematoxylin and eosin stained sections were also chosen for immunohistochemistry.
Immunochemical staining was performed using an automatic immunostainer (DAKO Autostainer link-48) according to the manufacturer’s instructions. The primary antibodies used were as follows: anti-Ser1981 phosphorylated ATM (#05-740, Mouse Monoclonal Antibody; 1:200; Temecula, CA, USA); anti-Ser139 phosphorylated H2AX (#05–636, Mouse Monoclonal Antibody, 1:200, Milipore, Billerica, MA, USA); anti-Thr68 phosphorylated Chk2 (sc-16297-R, Rabbit polyclonal antibody; 1:200, Santa Cruz Biotechnology, Dallas, TX , USA), anti-p53 (DO-1, sc-126, Mouse Monoclonal Antibody; 1:200, Santa Cruz Biotechnology Inc. Dallas, TX, USA).
Cancer cells showing nuclear staining, regardless of the presence of cytoplasmic staining, were determined as positively immunostained for ATM, γH2AX, Chk2 and P53. Reduction or lack of staining was restricted to tumor cells. The stromal tissue surrounding tumor nests preserved the normal staining pattern, thereby providing an internal control on the same section.16
Evaluation of immunohistochemistry staining
Sections were scored semi-quantitatively as follows: (0+): 0% immunoreactive cells; (1+): <5% immunoreactive cells; (2+): 5–50% immunoreactive cells; (3+): greater 50% immunoreactive cells. At last, for statistical purposes and to define a cutoff level, slides with scores 0 and 1 were defined negative and those with scores 2 and 3 were considered positive. The slides were screened at lower power for any staining; higher magnification (100×) was used to determine the immunochemical scores.17
Statistical methods
Statistical analysis was performed using SPSS version 21.0 (SPSS, Chicago, IL, USA). Statistical analyses were performed with χ2 test, Fisher’s exact tests and multiple regression analyses. χ2 test and Fisher’s exact tests were used for the comparison of DDR activation, the positivity of HPV E6/7 mRNA and HPV E6/7 mRNA copy number between groups. A multiple regression analysis was performed to identify the correlation between HPV genotypes, the positivity of HPV E6/7 mRNA, copy number of HPV E6/7 mRNA and DDR activation. The OR and relative 95% CI were calculated. The significance level α was set at 0.05.
Results
Specific activation of DDR in CC carcinogenesis
To determine whether DDR relates to CC carcinogenesis, we compared the expression of ATM, γH2AX, Chk2 and P53 in cervicitis, CIN I, CIN II/III and SCC specimens (Figures 1–4). The activation of DDR was increased with increasing severity of the cervical lesion. The activation of ATM, γH2AX, Chk2 and P53 in cervicitis, CINI, CINII/II and SCC are shown in Figure 5. The difference between the four groups is significant (p=0.004, p=0.046, p=0.042, p=0.011).
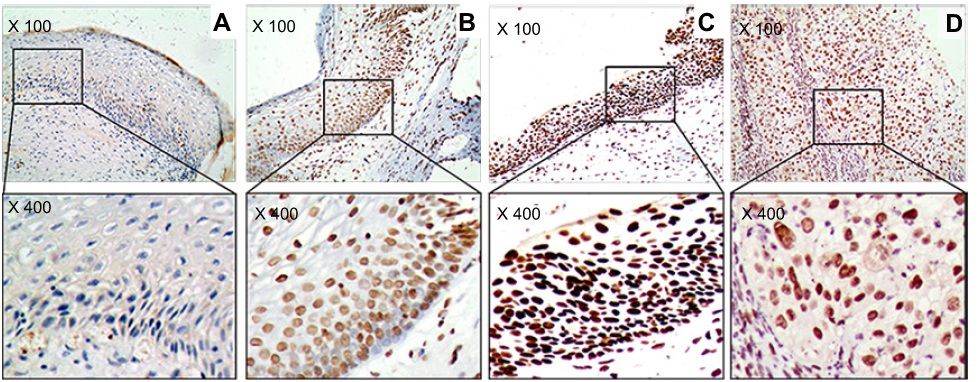 |
Figure 1 The marker (ATM) of DNA damage response in study sample. (A) Cervicitis; (B) CIN I; (C) CIN II/III; (D) SCC. Original magnification, ×100 and ×400. |
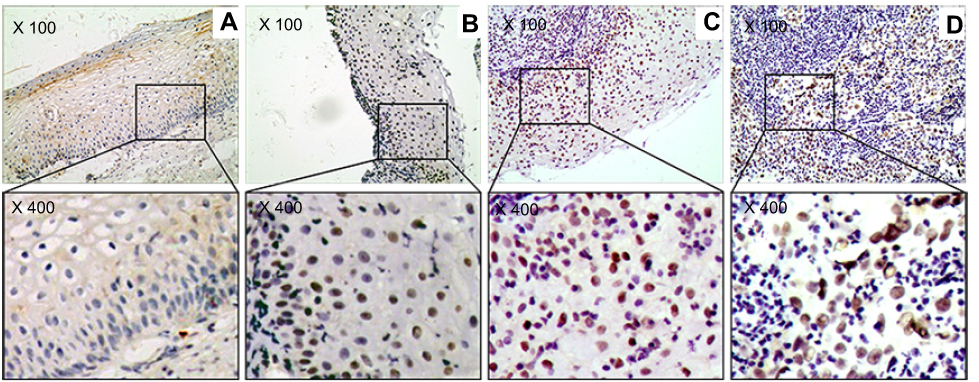 |
Figure 4 The marker (P53) of DNA damage response in study sample. (A) Cervicitis; (B) CIN I; (C) CIN II/III; (D) SCC. Original magnification, ×100 and ×400. |
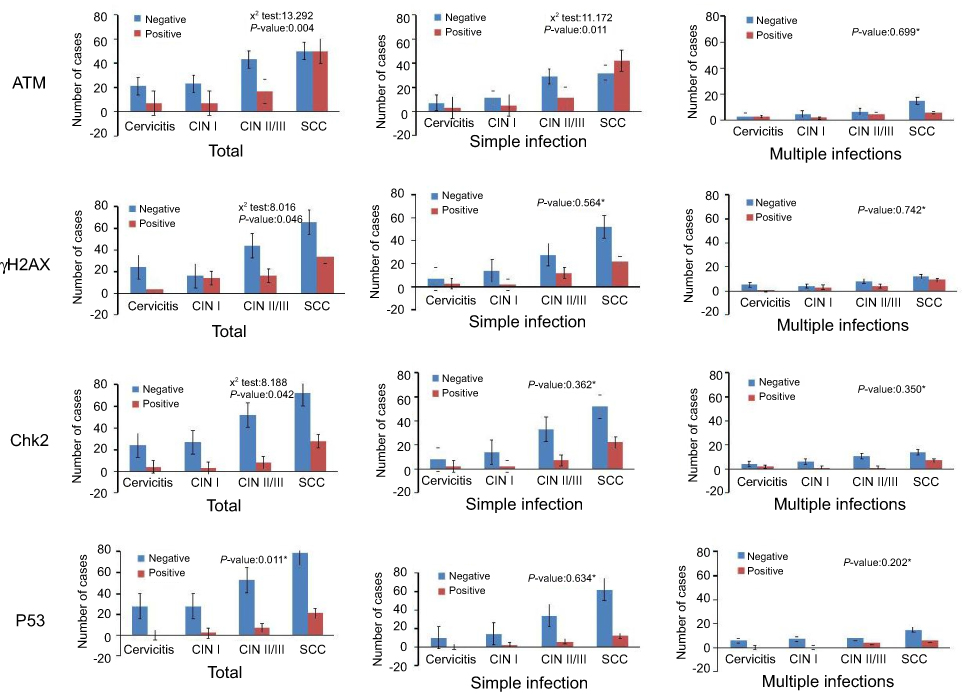 |
Figure 5 The activation of DNA damage response in study groups. *P-values are for Fisher’s exact tests. |
To further investigate DDR activation in different types of infection, we examined the expression of above DDR markers in simple and multiple infections. The expressions of ATM, γH2AX, Chk2 and P53 in simple and multiple infections are given in Figure 5. For simple infection, only ATM expression has significant difference (p=0.011, p=0.564, p=0.362, p=0.684); however, for multiple infections, no statistical significance was shown among the four groups.
The correlation between HPV genotypes and DDR activation
All the samples were divided into groups: HPV+ and HPV-, HPV16+ and HPV16-, HPV18+ and HPV18-, and HPV58+ and HPV58-. Some significant correlations between the activation of DDR and HPV genotypes were shown. When comparing the activation of DDR between HPV+ (n=186) and HPV- (n=32) specimens, the expression of ATM, γH2AX, Chk2 and P53 in HPV+ specimens was higher than in HPV- specimens (Table 1) but was not significant. The expression of DDR markers was higher in HPV16+ (n=116), HPV18+ (n=15) and HPV58+ (n=36) specimens compared to HPV16- (n=102), HPV18- (n=203) and HPV58- (n=182) specimens. However, the significant difference was only shown for the expression of γH2AX between HPV16+ and HPV16- groups (p=0.007), and γH2AX and P53 between HPV58+ and HPV58- groups (Table 1) (p=0.008, p=0.045).
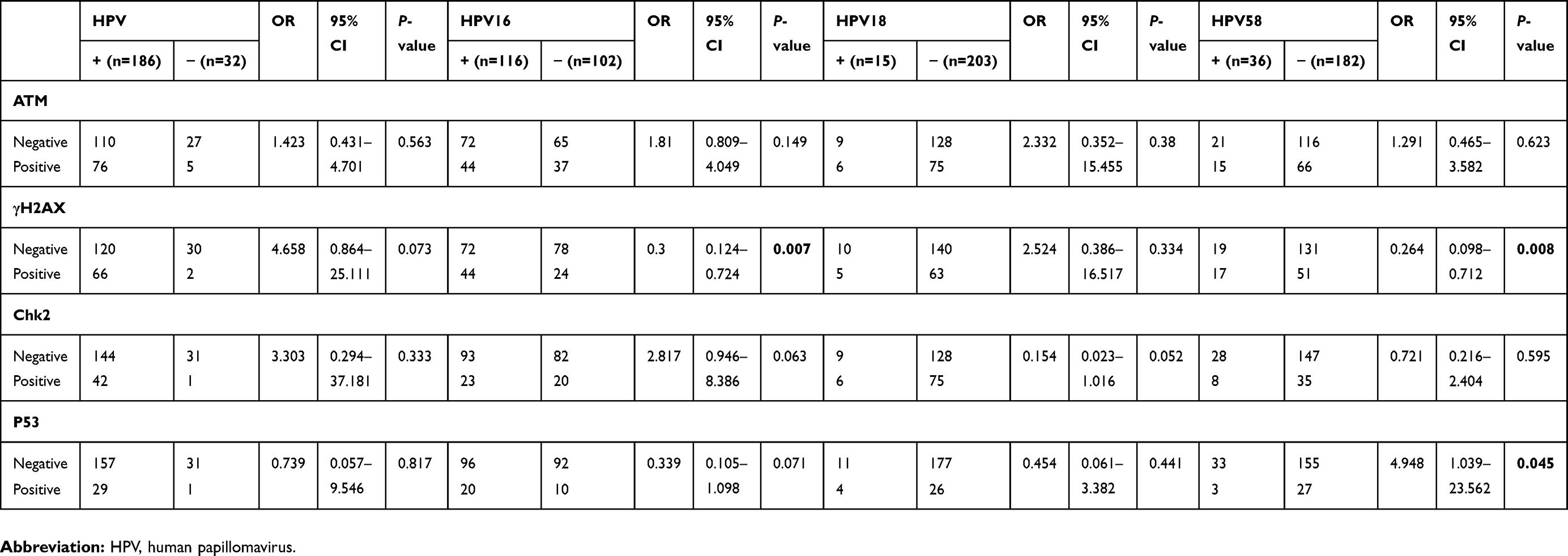 |
Table 1 Multivariate logistic regression analysis for HPV±, HPV16±, HPV18± and HPV58± groups |
The expression of HPV E6/7 mRNA test by histologic diagnosis
With the use of the Quantivirus® HPV E6/E7 RNA 3.0 assay (bDNA), HPV E6/7 mRNA was determined through the quantification of RLUs and concentrations of HPV RNA. Samples with a ratio of less <1 copy/mL were considered as negative. Results equal to or >1 copy/mL were considered positive. These were further divided into three groups: <1000 copy/mL (low viral load), 1000–10,000 copy/mL (medium viral load) and >10,000 copy/mL (high viral load). These criteria were modified from the method described by Dalstein and Adam.3,18
The HPV E6/7 mRNA expression in cervicitis, CINI, CINII/III and SCC is shown in Figure 6. The expression of HPV E6/7 mRNA increases with the severity of the histologic diagnosis. Among 187 specimens with histologic evaluation, 135 samples (72.19%, 135/187) were mRNA positive. Statistical significance was shown between four groups (p=0.000). Furthermore, increasing severity of cervical lesions correlated with a higher copy number of HPV E6/7 mRNA (Figure 6) (p=0.003).
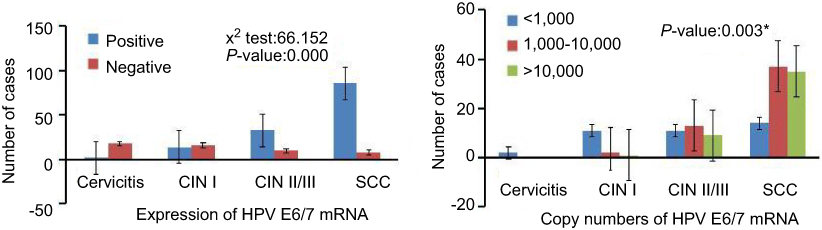 |
Figure 6 The expression and copy numbers of HPV E6/7 mRNA. *P-values are for Fisher’s exact tests. |
The correlation between the expression of HPV E6/7 mRNA and DDR activation
Comparing the activation of DDR between HPV E6/7 mRNA+ (n=135) and HPV E6/7 mRNA- (n=52) specimens, a significant difference was shown for P53 expression (p=0.015). However, the statistical analysis showed no significant differences for DDR activation when comparing HPV16 E6/7 mRNA+ (n=87), HPV18 E6/7 mRNA+ (n=7), HPV58 E6/7 mRNA+ (n=26) to HPV E6/7 mRNA- (n=52) specimens (Table 2).
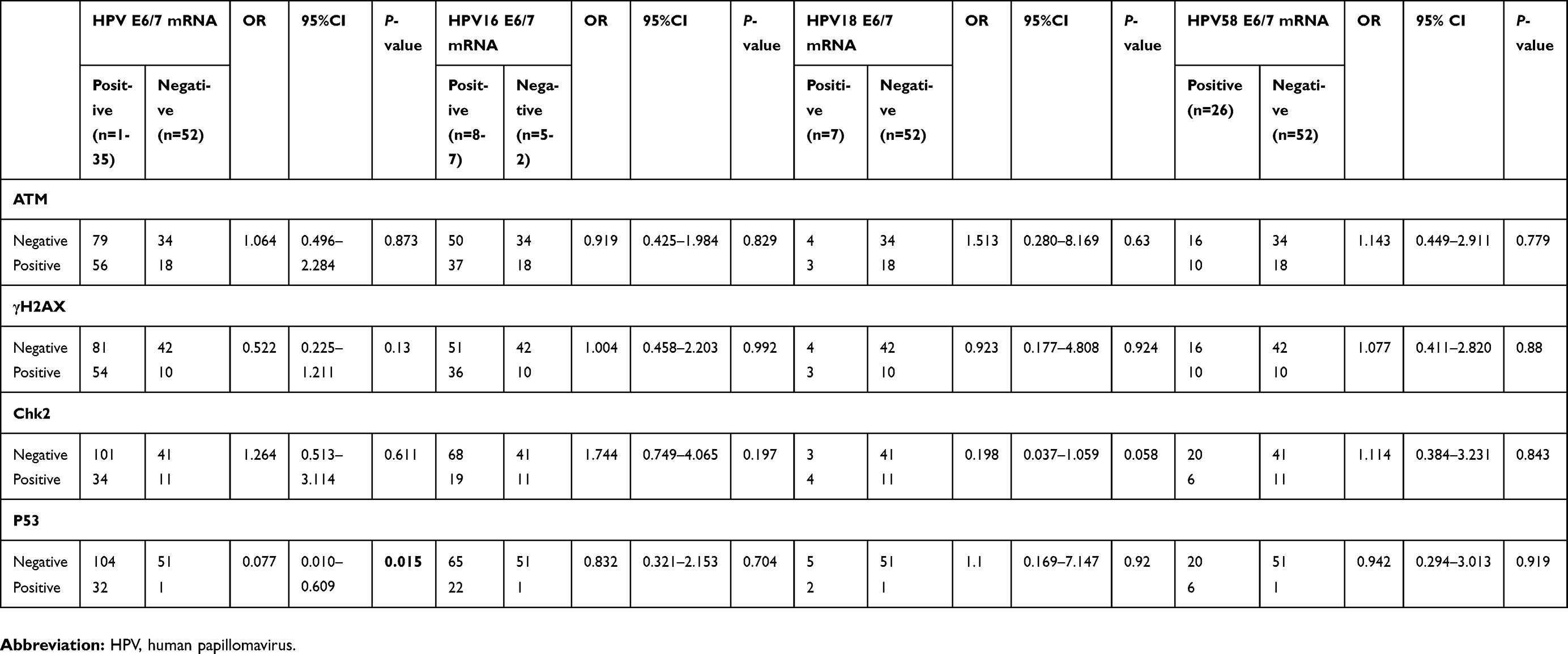 |
Table 2 Multivariate logistic regression analysis for the E6/7 mRNA expression of HPV, HPV16, HPV18 and HPV58 groups |
The correlation between the copy numbers of HPV E6/7 mRNA and DDR activation
When comparing the correlations between DDR activation and HPV E6/7 mRNA copy numbers in HPV E6/7 mRNA+, HPV16 E6/7 mRNA+, HPV58 E6/7 mRNA+ groups, no differences were shown for DDR activation between low viral load and medium viral load groups. However, a close correlation with CHK2 expression was shown between low viral load and high viral load groups in HPV E6/7 mRNA+ and HPV16 E6/7 mRNA+ groups (Table 3) (p=0.002, p=0.026). Furthermore, when the correlation between DDR activation and HPV E6/7 mRNA copy numbers was analyzed in CINⅡ/Ⅲ and SCC groups, no difference was shown in CINⅡ/Ⅲ groups (Table 3). However, high viral load groups in SCC groups had a significantly higher expression of γH2AX and CHK2 than low viral load groups (Table 3) (p=0.036, p=0.032). Because of low numbers of HPV18 E6/7 mRNA+ specimens and HPV E6/7 mRNA+ specimens in cervicitis and CIN, the correlation between DDR and mRNA E6/7 mRNA copy numbers was not included in this study.
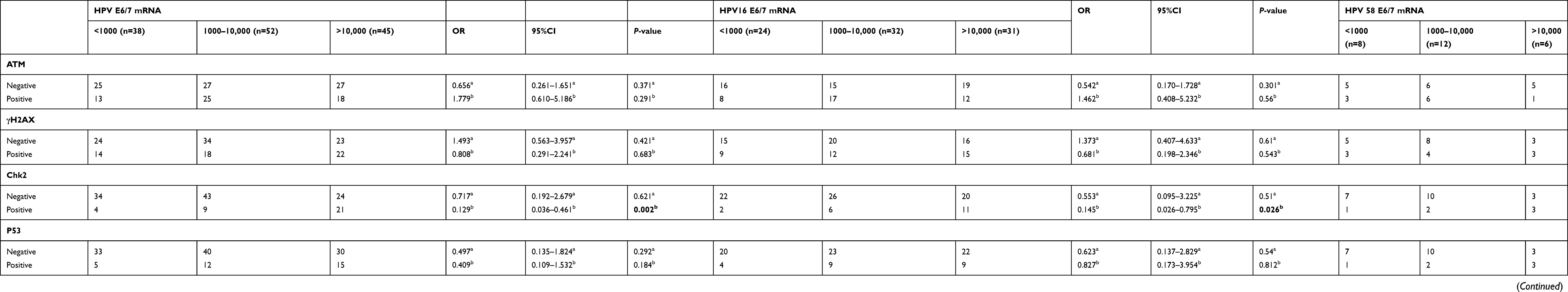 |
Table 3 Multivariate logistic regression analysis for the HPV E6/7 mRNA copy numbers in study groups (copy/mL) |
Discussion
Tumorigenesis is an evolutionary process that selects for genetic and epigenetic changes, allowing evasion of anti-proliferative and cell death inducing mechanisms which normally limit clone expansion of somatic cells.19 Most tumors acquire genetic instability and this takes place in the early stages of tumorigenesis, which lead to cell cycle blockade or apoptosis and thereby constraining tumor progression.20 Recently, our observations show that advanced carcinomas of stomach and cervix uteri reveal constitutive activation of γH2AX, an effector kinase within the DNA damage network that is activated by ATM in response to DNA double-strand breaks.21,22 Other research also shows the evolution of CC is accompanied by the genetic instability and higher levels of oxidative DNA damage correlated to the grade of cervical cell dysplasia.4 These results led us to hypothesize that DNA damage checkpoints might become activated in the process of cervical tumorigenesis in the Longnan patients.
We supply evidence that ATM is activated in earlier stages of cervical tumorigenesis, with activation levels of ATM increasing as cervicitis and CIN progress into carcinoma. This pattern of ATM activation reveals that earlier oncogenic events related to ATM activation and suggest an intriguing possibility – that ATM plays a role in tumor surveillance. The concept of ATM related to tumor surveillance is supported by other publications which demonstrated increase in ATM activation in lung and bladder precancerous lesions,23 and decrease in ATM function in prostate tumorigenesis.16 Consistent with these reports, we also found enhanced Chk2, γH2AX and P53 in cervical precancerous lesions. Even though γH2AX reached the highest levels in CINI and Chk2 and P53 were highest in SCC, the level of γH2AX was still elevated in SCC. This activation pattern of Chk2, γH2AX and P53 is consistent with their involvement in the surveillance of cervical tumorigenesis via ATM (Figure 5).
Evidence suggests that persistent infection of high-risk HPV genotypes causes genetic instability and DNA damage repair machinery of the host cell is destroyed by HPV infection.24 Fifteen carcinogenic HPV subtypes have been recognized, which together account for almost all CC cases.25 Of these subtypes, HPV16 and 18 are the most carcinogenic and are involved in 70% of the CCs.26 However, a comprehensive overview is lacking which compares the HPV oncogenes from the various HPV subtypes. The differences between the various HPV subtypes in the process of cervical carcinogenesis are not fully understood at the molecular level.
In this study, we evaluated the simple and multiple infections of HPV and DDR induced by various HPV subtypes in the process of cervical carcinogenesis (Figure 5, Table 1). No statistical significance was shown for the activation of DDR in multiple infections. However, ATM expression is statistically higher in simple infection with the process of cervical carcinogenesis (Figure 5). This pattern of DDR activation further reveals that earlier oncogenic events result in ATM activation and suggest ATM plays a role in tumor surveillance. Due to our relatively small specimen size of multiple infections, we are not able to exclude the possibility that ATM was also activated and further resulted in the activation of Chk2, γH2AX and P53. Our results also showed that ATM activation is not statistically higher in HPV+, HPV16+, HPV18+ and HPV58+ specimens. The fact that γH2AX and P53 remain elevated in the above positive specimens may be attributed to the possible longer half-life of γH2AX and P53 compared to the half-life of ATM activation or extra signals other than those of tumor surveillance in cervical tissue result in activation of Chk2, γH2AX and P53 (Table 1).
HPV is now conclusively identified as a major cause of CC. Despite its causal role in CC, the fact is that most of the HPV infections are transient, especially in younger age women.27 The type-specific persistence of oncogenic HPV is considered as the true precursor of the neoplastic process. HPV E6 and HPV E7 oncogene expression is necessary for the malignant transformation and maintenance of the neoplastic state.28 Therefore, the recognition of HPV E6 and HPV E7 mRNA of the respective HPV genotypes may serve as a better prognostic method for the development of cervical lesions.29 In our study, regarding the correlation between the expression and copy number of HPV mRNA and degree of cervical lesion, a statistically significant increase in copy number was found in cervical specimens of higher lesions (Figure 5), as published by others.28 According to Avanzi et al,30 repeated cycles of viral infection may add the number of genetically damaged cells and result in the gather of chromosome abnormalities. The viral genome integrated into the epithelial cell genome is a clastogenic event that could increase the number of micronucleated cells and introduce a degree of chromosome instability.31 It is, therefore, plausible that greater viral copy numbers lead to a greater possibility of genomic instability, as suggested by the present findings.
HPV infection has been linked to altered DNA damage and repair processes through various mechanisms. On one hand, HPV activated DNA damage response results in genomic instability. High-risk E6 and E7 proteins can independently produce DNA damage and result in numerical and chromosomal structural instability.32 Furthermore, high-risk E7 drives proliferation in the presence of DNA damage by inhibiting the DNA damage checkpoint response through proteolytic degradation of claspin.33 As reported by Bester et al, the expression of high-risk E6 and E7 reduces replication stress and DNA damage that result in genomic instability.34 In HPV-related cancers, it is frequently found that the viral genome is integrated into the host cell genome, resulting in deregulated expression of E6 and E7 that can further promote genomic instability.35 On the other hand, HPV manipulates the DNA damage repair pathway for viral replication. The Laimins lab reported that cells positive for high-risk HPV31 show constitutive activation of an ATM dependent, DDR was the first indication that HPV may manipulate DNA repair pathways for viral replication.36 ATM activation by HPV mainly through E7 and its activity seems to be primarily dependent upon differentiation to drive productive replication in a recombination-dependent manner. E7-dependent ATM activation may lead to increased protein stability of its downstream targets γH2AX, pChk2, 53BP1, RPA, Rad51 and BRCA1.
To our knowledge, no study has documented a relationship between HPV E6 and E7 oncogenes and DDR activation in the process of cervical carcinogenesis. While statistically significant activation of DDR is not seen in HPV16 E6/7 mRNA+, HPV18 E6/7mRNA+, HPV58 E6/7 mRNA + groups, the activation of P53 observed in the HPV E6/7 mRNA+ group, may suggest that the activation of DNA damage and repair is signaled in HPV infected cells (Table 2). Some studies show that the activation of DNA damage and repair results in p53 activation with a concomitant growth arrest that could be detrimental for viral replication in dividing cells. However, some studies also have shown that the ability of E6 to degrade and/or inactivate p53 is important for long-term genome maintenance.37 Furthermore, p53 inactivation or expression of a dominant negative p53 protein can complement genomes defective for E6 expression.38 The fact that we could detect the activation of Chk2 in the high viral load group of HPV E6/E7 mRNA+ specimens and HPV16 E6/E7 mRNA+ specimens and also the activation of γH2AX and Chk2 in the high viral load group of HPV E6/E7 mRNA+ SCC specimens suggests greater viral copy numbers lead to more DNA damage, as observed by our research; viral copy numbers are closely related to cervical carcinogenesis; the cells with higher copy number of E6 and E7 show a significantly impaired ability to activate cell cycle checkpoints and to induce apoptosis upon DNA damage. (Table 3)
There are some limitations in our study. First, the sample size of this study is relatively small. We only enrolled a total of 346 samples in the present research. Because of low numbers of HPV18 E6/7 mRNA+ specimens and HPV E6/7 mRNA+ specimens in cervicitis and CIN, the correlation between DDR and mRNA E6/7 mRNA copy numbers was not included in this study. In addition, some factors may be related to the progression of CIN to CC, such as age at first intercourse; high parity; smoking; endogenous and exogenous hormonal factors, including of parity, oral contraceptive use, obesity and so on; and infection with other sexually transmitted infectious agents; however, these were not included or discussed in the present study.
Overall, the results of the present study add to the evidence that HPV results in alterations on the genetic level and triggers the cervical carcinogenic process. HPV-related DDR may be one reason of the high incidence and mortality of CC in Longnan patients.
Ethics approval and consent to participate
This trial was approved by the ethics committee of Northwest Minzu University. The trial was registered in the Chinese Clinical Trial Registry (http://www.chictr.org.cn), registration no. ChiCTR-TRC-1800016405, principal investigator Zhong Guo, date of registration 31 May 2018. Written informed consent has been obtained from each patient to participate in this study. A parent or legal guardian provided written informed consent for any patient under the age of 18 years. This study was conducted in accordance with the Declaration of Helsinki.
Data availability
Individual patient data are confidential and thus they cannot be made publicly available.
Acknowledgments
This work was supported in part by Grants from the National Natural Science Foundation of China (Nos. 81260442, 81560508 and 31560254), the Program for Leading Talent of SEAC ([2016]57) and the Fundamental Research Funds for the Central Universities (Nos. 31920130043 and 31920170164).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Kim K, Zang R, Choi SC, et al. Current status of gynecological cancer in China. J Gynecol Oncol. 2009;20(2):72–76. doi:10.3802/jgo.2009.20.2.72
3. Adam ML, Pini C, Túlio S, et al. Assessment of the association between micronuclei and the degree of uterine lesions and viral load in women with human papillomavirus. Cancer Genomics Proteomics. 2015;12(2):67–71.
4. Visalli G, Riso R, Facciolà A, et al. Higher levels of oxidative DNA damage in cervical cells are correlated with the grade of dysplasia and HPV infection. J Med Virol. 2016;88(2):336–344.
5. Yu JS, Leng PF, Li YF, et al. Aryl hydrocarbon receptor suppresses the prostate cancer LNCaP cell growth and invasion by promoting DNA damage response under oxidative stress. DNA Cell Biol. 2017;36(11):1010–1017. doi:10.1089/dna.2017.3783
6. Walboomers JMM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189(1):12–19. doi:10.1002/(ISSN)1096-9896
7. von Knebel Doeberitz M, Rittmüller C, Zur Hausen H, et al. Inhibition of tumorigenicity of cervical cancer cells in nude mice by HPV E6–E7 anti-sense RNA. Int J Cancer. 1992;51(5):831–834.
8. Dyson N, Howley PM, Münger K, et al. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243(4893):934–937. doi:10.1126/science.2563173
9. Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10(8):550–560. doi:10.1038/nrc2886
10. Hufbauer M, Cooke J, van der Horst GTJ, et al. Human papillomavirus mediated inhibition of DNA damage sensing and repair drives skin carcinogenesis. Mol Cancer. 2015;14:183. doi:10.1186/s12943-014-0278-9
11. Yang L, Huangpu XM, Zhang SW, et al. Changes of mortality rate for cervical cancer during 1970’s and 1990’s periods in china. Acta Academiae Medicinae Sinicae. 2003;25(4):386–390.
12. Zhao J, Guo Z, Wang Q, et al. Human papillomavirus genotypes associated with cervical precancerous lesions and cancer in the highest area of cervical cancer mortality, Longnan, China. Infect Agent Cancer. 2017;12:8. doi:10.1186/s13027-017-0116-y
13. Kurman RJ, Ellenson LH, Ronnett BM. Blaustein’s Pathology of the Female Genital Tract.
14. Shen Y, Gong J, He Y, et al. Quantivirus® HPV E6/E7 RNA 3.0 assay (bDNA) is as sensitive, but less specific than Hybrid Capture 2 test. J Virol Methods. 2013;187(2):288–293. doi:10.1016/j.jviromet.2012.11.024
15. Liu TY, Xie R, Luo L, et al. Diagnostic validity of human papillomavirus E6/E7 mRNA test in cervical cytological samples. J Virol Methods. 2014;196:120–125. doi:10.1016/j.jviromet.2013.10.032
16. Bartkova J, Horejsí Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434(7035):864–870. doi:10.1038/nature03482
17. Fan C, Quan R, Feng X, et al. ATM activation is accompanied with earlier stages of prostate tumorigenesis. Biochim Biophys Acta. 2006;1763(10):1090–1097. doi:10.1016/j.bbamcr.2006.08.026
18. Dalstein V, Riethmuller D, Prétet JL, et al. Persistence and load of high-risk HPV are predictors for development of high-grade cervical lesions: a longitudinal French cohort study. Int J Cancer. 2003;106(3):396–403. doi:10.1002/ijc.11222
19. Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432(7015):307–315. doi:10.1038/nature03098
20. Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi:10.1038/25292
21. Guo Z, Pei S, Si T, et al. Expression of the γ-phosphorylated histone H2AX in gastric carcinoma and gastric precancerous lesions. Oncol Lett. 2015;9(4):1790–1794. doi:10.3892/ol.2015.2896
22. Zhao J, Wang Q, Li J, et al. Comparative study of phosphorylated histone H2AX expressions in the cervical cancer patients of pre- and postneoadjuvant chemotherapy. Eur J Gynaecol Oncol. 2015;36(3):318–322.
23. Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434(7035):907–913. doi:10.1038/nature03485
24. Hong SY. DNA damage response is hijacked by human papillomaviruses to complete their life cycle. J Zhejiang Univ Sci B. 2017;18(3):215–232. doi:10.1631/jzus.B1600306
25. Schiffman M, Castle PE, Jeronimo J, et al. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907. doi:10.1016/S0140-6736(07)61416-0
26. Smith JS, Lindsay L, Hoots B, et al. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: a meta-analysis update. Int J Cancer. 2007;121(3):621–632. doi:10.1002/ijc.22861
27. Lytwyn A, Sellors JW, Mahony JB, et al. Adjunctive human papillomavirus testing in the 2-year follow-up of women with low-grade cervical cytologic abnormalities: a randomized trial and economic evaluation. Arch Pathol Lab Med. 2003;127(9):1169–1175. doi:10.1043/1543-2165(2003)127<1169:AHPTIT>2.0.CO;2
28. Varnai AD, Bollmann M, Bankfalvi A, et al. Predictive testing of early cervical pre-cancer by detecting human papillomavirus E6/E7 mRNA in cervical cytologies up to high-grade squamous intraepithelial lesions: diagnostic and prognostic implications. Oncol Rep. 2008;19(2):457–465.
29. Molden T, Kraus I, Karlsen F, et al. Human papillomavirus E6/E7 mRNA expression in women younger than 30 years of age. Gynecol Oncol. 2006;100(1):95–100. doi:10.1016/j.ygyno.2005.07.108
30. Avanzi S, Alvisi G, Ripalti A. How virus persistence can initiate the tumorigenesis process. World J Virol. 2013;2(2):102–109. doi:10.5501/wjv.v2.i2.102
31. Muñoz N, Bosch FX, de Sanjosé S, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348(6):518–527. doi:10.1056/NEJMoa021641
32. Duensing S, Munger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002;62(23):7075–7082.
33. Spardy N, Covella K, Cha E, et al. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009;69(17):7022–7029. doi:10.1158/0008-5472.CAN-09-0925
34. Bester AC, Roniger M, Oren YS, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145(3):435–446. doi:10.1016/j.cell.2011.03.044
35. Anacker DC, Moody CA. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res. 2017;231:41–49. doi:10.1016/j.virusres.2016.11.006
36. Moody CA, Laimins LA. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoSPathog. 2009;5(10):e1000605.
37. Hollingworth R, Grand RJ. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses. 2015;7(6):2542–2591. doi:10.3390/v7052542
38. Williams JS, Kunkel TA. Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst). 2014;19:27–37. doi:10.1016/j.dnarep.2014.03.029
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.