")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
How Western Diet And Lifestyle Drive The Pandemic Of Obesity And Civilization Diseases
Authors Kopp W
Received 23 May 2019
Accepted for publication 13 September 2019
Published 24 October 2019 Volume 2019:12 Pages 2221—2236
DOI https://doi.org/10.2147/DMSO.S216791
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Wolfgang Kopp
Retired Head, Diagnostikzentrum Graz, Graz 8043, Austria
Correspondence: Wolfgang Kopp
Mariatrosterstraße 41, Graz 8043, Austria
Email [email protected]
Abstract: Westernized populations are plagued by a plethora of chronic non-infectious degenerative diseases, termed as “civilization diseases”, like obesity, diabetes, cardiovascular diseases, cancer, autoimmune diseases, Alzheimer’s disease and many more, diseases which are rare or virtually absent in hunter-gatherers and other non-westernized populations. There is a growing awareness that the cause of this amazing discrepancy lies in the profound changes in diet and lifestyle during recent human history. This paper shows that the transition from Paleolithic nutrition to Western diets, along with lack of corresponding genetic adaptations, cause significant distortions of the fine-tuned metabolism that has evolved over millions of years of human evolution in adaptation to Paleolithic diets. With the increasing spread of Western diet and lifestyle worldwide, overweight and civilization diseases are also rapidly increasing in developing countries. It is suggested that the diet-related key changes in the developmental process include an increased production of reactive oxygen species and oxidative stress, development of hyperinsulinemia and insulin resistance, low-grade inflammation and an abnormal activation of the sympathetic nervous system and the renin-angiotensin system, all of which play pivotal roles in the development of diseases of civilization. In addition, diet-related epigenetic changes and fetal programming play an important role. The suggested pathomechanism is also able to explain the well-known but not completely understood close relationship between obesity and the wide range of comorbidities, like type 2 diabetes mellitus, cardiovascular disease, etc., as diseases of the same etiopathology. Changing our lifestyle in accordance with our genetic makeup, including diet and physical activity, may help prevent or limit the development of these diseases.
Keywords: diabetes, obesity, metabolic syndrome, insulin hypersecretion, oxidative stress, paleolithic diet, pathogenesis
Introduction
Over the course of 6–8 generations, but especially in the last 2–3 generations, there has been an epidemic of obesity and non-infectious degenerative diseases known as “civilization diseases”. While infectious agents were the major causes of disease at the beginning of the 20th century, infectious diseases were replaced by type 2 diabetes mellitus (T2DM) and diabetic complications, cardiovascular diseases (CVD) and cancer as major causes of death by the 21st century. At present, westernized populations are plagued by a plethora of chronic degenerative diseases, including obesity, T2DM, atherosclerosis, coronary heart disease, stroke, autoimmune diseases, essential hypertension, cancer, osteoporosis and other more, and the number of these diseases is also rapidly increasing in developing countries.1–3
In striking contrast, obesity and civilization diseases are rare or virtually absent in hunter-gatherer (HG) and other non-westernized populations. Also, low serum insulin levels and persistently excellent insulin sensitivity are characteristic of HG, but only as long as these people adhere to their traditional “paleolithic” diets.2,4–7 Otherwise, transition to a “Western diet” (WD, as defined below) invariably leads to a dramatic increase in insulin resistance (IR) (defined here as an impaired ability of the hormone to suppress hepatic glucose output and to promote peripheral glucose disposal) and hyperinsulinemia as well as obesity, T2DM, hypertension, cancer and other more.4,6,7 On the other hand, a return to a traditional paleolithic diet is associated with marked improvement in IR and fasting insulin levels5,8,9 and glucose control and lipid profiles of T2DM.8,9 The common counter-argument that Stone Agers usually do not live long enough to develop degenerative diseases is not accurate. A conspectus of data on HG societies suggests that modal age of adult death is about seven decades (adaptive life span of 68–78 years). In contrast to most Westerners, these people tend to be healthy up to old age. Causes of death are predominantly infectious diseases, while chronic degenerative disorders are rare.10
Numerous authors have argued that a mismatch between our ancient physiology and the WD and lifestyle play a key role in the development of many of these degenerative diseases.2–4,7,11,12 Though, the question still remains: how do diet and lifestyle cause or contribute to the developmental process?
This paper shows that the significant changes in diet and lifestyle during recent human history cause severe metabolic distortions, which play a pivotal role in the development of degenerative diseases.
Important Differences Between Paleolithic And WDs
“Living organisms thrive best in the milieu and on the diet to which they were evolutionarily adapted; this is a fundamental axiom of biology”.11 This statement is based on the fact that the metabolism of each species has genetically adapted to a particular type of food over long periods of evolution. The specific diet guaranties health and survival.11,13 Man is no exception in this regard. Like all species, today’s humans are genetically adapted to the environment in which their ancestors survived and in which their genetic makeup was selected.4,13 The evolutionary adaptation process has produced a specific, well-adjusted metabolism in which numerous metabolic factors are intertwined and in balance with each other.
A comparison of 229 HG diets from around the world found that these diets are higher in protein (19–35% of energy) and low in carbohydrate (22–40% of energy) by normal western standards, whereas the fat intake would be comparable or higher (28–58% of energy) than values currently consumed in modern, industrialized societies.14 HG diets consisted mainly of game, fish and uncultivated plant-based foods such as roots, tubers, wild herbs, berries, nuts, vegetables, fruits and some honey, while cereals, refined sugars and dairy products were not part of it.4,7,12,14,15 More importantly, all carbohydrate foods (other than wild honey) that were consumed during the Paleolithic were low-glycemic and low-insulinemic in effect.4,12,15–17 Since the effect of protein and fat on insulin production is small too,18 postprandial glucose and insulin levels were low during most part of human evolution.
With the agricultural revolution about 10,000 years ago and especially since the industrial revolution 250 years ago, human nutrition has changed significantly. At present, carbohydrates play an enormously important role in the human diet. WDs consist of large amounts of high-glycemic/high-insulinemic carbohydrate foodstuff like refined cereals (currently, 85% of the cereals consumed in the US diet are highly processed refined grains4), corn, potatoes and sugars (in particular sucrose and fructose), dairy products (which also produce high postprandial insulin levels, despite a low GI, not significantly different from the Insulinemic Index of the reference bread18,19), as well as high amounts of fat and substantial amounts of protein. Glucose, which is abundantly derived from carbohydrate sources, especially from starchy foods and sugars, is currently the most important source of energy for the human body and accounts for about 40–75% of the energy intake.
Furthermore, due to agribusiness and modern agriculture, WDs contain excessive amounts of omega-6 polyunsaturated fatty acids (PUFAs) and only small amounts of omega-3 PUFAs, resulting in an unhealthy omega-6/omega-3 ratio of 20: 1 compared to a balanced ratio during the Paleolithic period. The consumption of the omega-6 PUFAs has dramatically increased in the western world primarily in the form of vegetable oils. A diet rich in omega-6 fatty acids is proinflammatory and prothrombotic, and has been implicated in the development of various degenerative diseases, including T2DM, CVD, cancer, obesity inflammatory bowel disease, major depression, Alzheimer’s disease and other more.20–22
However, while a higher intake of omega-3 fatty acids has been associated with reduced risks of cardiovascular disease and cancer in several observational studies, a recently published, randomized placebo-controlled study found that supplementation with n-3 fatty acids did not result in a lower incidence of major cardiovascular events or cancer than placebo.23
Lack Of Genetic Adaptation
While human nutrition has changed significantly over the last 10,000 years, and especially over the last 250 years, the human genome has remained largely unchanged. Changes in diet and lifestyle occurred too fast for the human genome to adapt to, therefore, humans are still biologically adapted to the environment of their preagricultural ancestors.13 It further has to be considered that an evolutionary adaptation process needs selection pressure (external agents which affect an organism’s ability to survive in a given environment) as a driving force. However, degenerative diseases such as atherosclerosis, hypertension, cancer, T2DM, etc. normally affect the postreproductive years. Even a high mortality rate due to these diseases after the reproduction phase will produce little or no selective pressure.
The significant changes in diet and lifestyle during recent human history cause severe metabolic distortions, including (but not limited to) a diet-related increased production of reactive oxygen species (ROS) and oxidative stress (OS), hyperinsulinemia and IR, low-grade inflammation and an abnormal activation of metabolic systems such as the sympathetic nervous system (SNS) and the renin-angiotensin-system (RAS), all of which play pivotal roles in the development of civilization diseases (Figure 1).
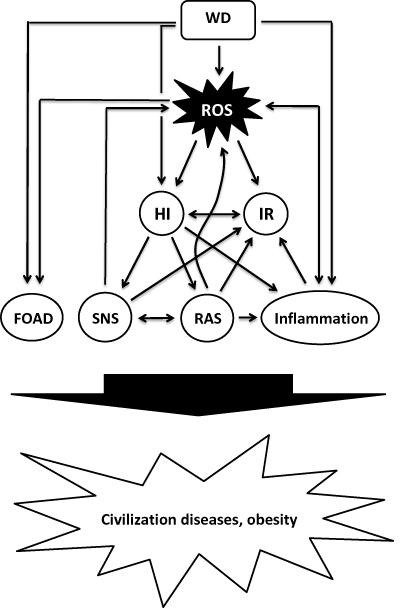 |
Figure 1 Proposed model of diet-induced development of obesity and civilization diseases. Abbreviations: FOAD, fetal origin of adult disease; HI, hyperinsulinemia; IR, insulin resistance; RAS, renin-angiotensin system; ROS, reactive oxygen species; SNS, sympathetic nervous system; WD, Western diet. |
Reactive Oxygen Species (ROS) – Friend And Foe
A growing body of evidence suggests that ROS play a dual role both as signaling molecules and as a damaging agent. On the one hand, ROS are an inevitable byproduct of the mitochondrial respiratory chain activity. Some of the electrons transferred along the electron transport chain (ETC) escape, and the sequential reduction of oxygen through the addition of electrons leads to the formation of a number of ROS, including superoxide, hydrogen peroxide (H2O2), hydroxyl radical, hydroxyl ion, and nitric oxide. On the other hand, ROS are deliberately produced in low concentrations in the form of the chemically less reactive H2O2 and serve as important signal molecules.24 Under physiological conditions, there is a balance between ROS generation and clearance, since eukaryotic cells have several antioxidative defense mechanisms, but these defenses are not perfect. Excessive formation of reactive species that is not counterbalanced by antioxidant defense mechanisms leads to OS which causes damage to lipids, proteins and DNA. In addition, increased ROS production and OS can interfere with intracellular signal transduction processes, which has been implicated in the development of IR, inflammation25,26 and various civilization diseases like cancer, CVD including atherosclerosis, cardiomyopathy, hypertension and heart failure, and other more.26–28
Oxidative Phosphorylation (OXPHOS) - A Demand-Driven Process
The primary role of mitochondria is the generation of energy and the regulation of cell metabolism. Mitochondria are responsible for the bulk of cellular adenosine triphosphate (ATP) production through OXPHOS. The macronutrients carbohydrates, fat and proteins, the primary catabolic substrates that provide the most energy to humans, are broken down into glucose, fatty acids and amino acids. Glucose and free fatty acids (FFA) serve as major fuels that can be oxidized for ATP production in mitochondria, while amino acids serve to form new proteins or as an energy source. Glucose and FFA are further degraded to acetyl-coenzyme A (acetyl-CoA). From acetyl-CoA, the reducing agents NADH and FADH2 are formed in the tricarboxylic acid cycle, which release electrons to the mitochondrial ETC. The sequential transport of electrons pumps protons through the inner mitochondrial membrane and creates an electrochemical gradient. The final acceptor of electrons in the ETC is molecular oxygen, yielding water and ATP.29 Basically, OXPHOS is a demand-driven process where energy supply is matched to ATP demand, regulated by adenosine diphosphate availability; therefore, a diet-related increase in electron delivery does not necessitate a proportional increase in ATP production. When electron supply exceeds demand for ATP, mitochondrial membrane potential rises, resulting in the production of increasing amounts of ROS.30–32 Therefore, nutrient excess can lead to increased production of ROS and OS.30,33
WDs, Western Lifestyle And Oxidative Stress
At present, human physiology is characterized by overeating, with frequent snacking and consumption of sucrose-containing soft drinks. “X-large” is the motto for meals and beverages. Specifically sugar is easily consumed in large quantities, not only in beverages, but also as sweets and as an additive in bakeries and ready meals. As a result, a significant part of the day is spent in the postprandial state, and the postprandial state is characterized by a persistent abundance of substrate in the circulation.34 Insulin levels are elevated for much of the day. Since foods with a high insulin response produce appetite and are less satiating,35–37 high-insulinemic WDs directly promote increased food intake and overeating. In addition, the energy density of food has a major impact on the regulation of food intake and body weight. Foods with high energy density content, like WDs, are more palatable, but less satiating and lead to a high energy intake.38 Also, calories from fluids are less satiating than those from solid foods and often lead to overconsumption.39
The glycemic index and the glycemic load (GL, the multiplicative product of carbohydrate amount and GI)16 of a meal are major determinants of post-prandial glucose excursion. WDs therefore often are associated with exaggerated supraphysiological postprandial peaks in blood sugar and lipids.32 Because OXPHOS is a demand-driven process, the bolus of energetic substrate in a high-calorie, low-energy situation can overwhelm the metabolic capabilities of mitochondria, resulting in an excess of the reduced form of nicotinamide adenine dinucleotide production, depolarization of mitochondrial membrane potential and OS.30–33 The level of OS is directly related to the increase in glucose and triglyceride levels after a meal.32
In addition, WDs and nutrient excess can cause mitochondrial dysfunction and increased ROS production through adverse effects on the mitochondrial life cycle40 and through mitochondrial overload and metabolic inflexibility.30
Consistent with the above, diets with a high GL,41 high-sucrose diets42 and WDs33,43,44 produce high levels of ROS. Postprandial glucose excursions correlate directly with the ensuing increase in free radicals.32,45 In contrast, Paleolithic and Mediterranean diets are associated with low OS.46
Diet-Related Development Of Hyperinsulinemia And IR
IR is of paramount importance for the pathogenesis of civilization diseases, but the mechanism is still not clearly identified. Visceral adiposity is traditionally considered to be the cause of IR and compensatory hyperinsulinemia, based on the assumption that increased adipose tissue mass causes increased release of FFA into the portal vein system, which in turn promotes IR in target tissues, but the picture is not as clear as it seems, and this assumption has been challenged for several reasons.30,47–50 Further, not all obese individuals are insulin resistant, while IR has been shown to exist in a significant proportion of the normal weight population.51 Alternatively, an excessive ß-cell secretory response has been suggested as a major cause of both, obesity and IR.48–50,52–54 Indeed, hyperinsulinemia has been found to precede the development of IR and obesity.49,54–58 Also, early hyperinsulinemia was the strongest predictor of T2DM in a 24 year study.59 Persistently elevated levels of insulin, regardless of their source, were shown to produce IR60–63 and to impair insulin-stimulated glucose uptake and its cellular signaling in a dose-dependent manner.63 An insulin-related overactivation of the SNS64 and RAS65,66 may also play an important role (see below).
A growing body of evidence suggests that increased ROS levels and OS play a major role in the development of hyperinsulinemia,48,67,68 IR28,40,63,69–73 and obesity.74
ROS are essential for the regulation and coordination of insulin production: nutrient-mediated ROS production (in the form of H2O2) is used in β-cells to couple nutrient (glucose, FFAs, amino acids) oxidation to insulin secretion.48,67 Pi et al67 showed that ROS (added as H2O2 or generated internally through addition of diethyl maleate) stimulate insulin secretion in a dose-dependent manner. While increased ROS production stimulates basal insulin secretion,67,68 scavenging ROS prevents both basal and stimulated insulin secretion.48,67,68
Elevated ROS levels from various sources, including nutrient/energy surplus,25 FFA,69 hyperinsulinemia,62 RAS,25,70 SNS,71,72,75 inflammation,72 metabolic inflexibility30 and mitochondrial fission40 were shown to be important triggers for IR. Otherwise, induction of insulin resistance in cultured cells is blocked when ROS are scavenged.73
WDs can play a causative role in the development of hyperinsulinemia and IR (prior to weight gain), as they produce high levels of both insulin and OS. On the one hand, WDs,33,43,44,76 high-sucrose diets,42,77 diets with a high GL41 and high-fructose diets78 produce high levels of ROS and OS which may cause IR and insulin hypersecretion. Increased ROS production and OS were shown to precede the onset of IR and obesity in mice fed a high-fat diet.79 Also, chronic exposure of β-cells to excess nutrients (in the form of glucose and oleate) has been shown to promote insulin hypersecretion, probably due to increased ROS production.68,69
On the other hand, WDs induce a high postprandial insulin response. Along with a western dietary pattern of frequent snacking and frequent consumption of sucrose-containing soft drinks, the associated high insulin requirement places strain on β-cells. Because beta cells are, from an evolutionary point of view, genetically poorly adapted to high insulin requirements, high-insulinemic diets may cause hypertrophy and dysfunction, finally resulting in hyper-responsiveness and hypersecretion in response to normal meals.49,50,52,57,80–82 Indeed, hyperinsulinemia was shown to be associated with pancreatic islet cell hyperplasia and enhanced secretory capacity.49,57,80–84 In line with this, an increased postprandial insulin response, probably due to ß-cell dysfunction and dysregulation, was the earliest metabolic change in the development of adiposity in adolescents in a study by Le Stunff,57 followed by the parallel, time-dependent development of IR and fasting hyperinsulinemia. A study of insulin dynamics among obese schoolchildren suggested that hypersecretion precedes development of insulin resistance by several years.85 Further, rat puppies fed a high carbohydrate-rich formula developed hyperinsulinemia, hyperphagia and concomitant body weight gain. Moreover, the metabolic alterations persisted in adult rats and were even passed on to the next generation.86 Prolonged feeding of Wistar rats with a diet high in glycemic index starch and fat, similar to WDs, resulted in insulin hypersecretion and weight gain.87 Wistar rats developed hyperinsulinemia on a high-sucrose diet.88 Also, Fisher rats fed ad libitum with a high-fat, refined sugar diet developed hyperinsulinemia and IR before gaining weight.89,90 Intriguingly, IR developed, regardless of whether dietary fat was high (39.5%) or low (9%), pointing to sucrose as the determining factor.90 Fructose, for example in the form of fructose corn syrup or as a constituent of sucrose (consisting equally of glucose and fructose), seems to play a particularly important role in the pathogenic process, especially given the enormous amounts consumed (as sweetener for soft drinks and additives for ready meals). The consumption of fructose corn syrup increased >1000% between 1970 and 1990.91 Feeding studies have shown that diets rich in fructose cause hyperinsulinemia,92 IR,78,93 and OS.78 Further, in a study on 8-year old schoolboys, high intake of (high-insulinemic) milk caused hyperinsulinemia, IR and weight gain.18
Finally, it is well established that IR also has a genetic component. Young healthy offspring of hypertensive parents and parents with T2DM are insulin resistant and have higher plasma insulin levels compared with matched healthy individuals with negative family history.94,95 WDs could be involved in this genetic inheritance: as mentioned above, rat puppies fed a high carbohydrate-rich formula developed IR, which was passed on to the next generation.86
Overactivation Of The SNS
The SNS is involved in the regulation of the microenvironment of virtually every major organ system of the body by releasing two catecholamine neuroeffector molecules, norepinephrine (NE) and epinephrine which act as both neurotransmitters and circulating hormones. NE is released primarily from the sympathetic nerves, while epinephrine is mainly secreted from the adrenal medulla. Adrenergic receptors are expressed on virtually every cell type in the body. A body of evidence supports the view that increased activation of the SNS is a hallmark of obesity and its associated metabolic disorders and may play an important role in a variety of other degenerative diseases.96
There is a close relationship between insulin and the SNS: insulin activates the SNS in a dose-dependent manner in normal individuals, as indicated by elevated NE plasma levels (due to sympathetic nerve endings spillover) and micro-neurographic studies.64 SNS activity is also influenced by food intake: among dietary substrates, ingestion of high-insulinemic carbohydrates, like starch and sugars, substantially enhances SNS activity, characterized by a significant increase in plasma NE levels,97 while protein or fat ingestion exert minimal effects on NE levels only.98 Diet composition therefore plays a major role in determining the level of SNS activity. In feeding studies, high carbohydrate/protein diets were associated with a significant increase in NE levels, while high fat/protein diets did not alter SNS activity.99,100
Furthermore, increased activation of the SNS, whether caused by lower body negative pressure or NE infusions promotes IR,71,101 probably due to OS,72,75 while ganglionic blockade in turn improved insulin-sensitivity.102 While sympathetic overactivation contributes to IR, the compensatory increase in insulin levels may contribute to greater sympathetic activation, consistent with the existence of a negative feedback loop.102 As with hyperinsulinemia56,57 and OS,79 elevated NE levels precede the development of IR.103
Overactivation Of The RAS
The RAS plays an important role in normal physiology as well as in pathologic conditions. The classical view of the RAS is presented as a hormonal circulating system, with angiotensin II (ANG II) as the major effector peptide. In addition to the classic hormonal circulating system, a local RAS exists in various organs and tissues, leading to production of ANG II, with autocrine and paracrine effects.104,105 Several metabolic factors can activate the RAS, including insulin65,66 and the SNS.63,106,107 Acute hyperinsulinemia increases plasma renin activity and plasma level of ANG II.65,66 The SNS can interact with the RAS in the form of a positive feedback loop: NE activates ANG II production through stimulation of renin secretion, whereas circulating ANG II interacts with the SNS at various sites and amplifies the response to sympathetic stimulation by presynaptic facilitatory modulation of NE release.106,107 Clinical and pharmacological studies have shown that ANG II is a critical promoter of IR,70,108 subclinical inflammation and OS.109,110
Overactivation of the SNS and the RAS plays an important role in a variety of degenerative diseases, including CVD, T2DM, cancer, and many more.103,109–113
Widespread Activation Of The Immune System And Low-Grade Inflammation
A fundamental feature of the immune system is to protect the host from pathogens. Inflammation is a central component of innate (non-specific) immunity. The inflammatory response serves to initiate the elimination of toxic agents and the repair of damaged tissues. After activation, innate immune system cells secrete proinflammatory cytokines that induce production of free radicals (ROS and reactive nitrogen species). The inflammatory response continues until the pathogens are eliminated and the tissue repair process is complete.114
An increasing body of evidence shows that chronic inflammation causes and advances many common diseases, including obesity, T2DM, CVD, inflammatory bowel disease, osteoarthritis, autoimmune diseases, to name a few.115
On the one hand, chronic inflammation causes OS, on the other hand, OS plays a crucial role in the development and perpetuation of inflammation, indicating that inflammation and OS are pathophysiological events that are closely related.116 In addition to OS, hyperinsulinemia,49,117,118 ANG II109 and activation of the SNS119 are also causally related to the development of chronic subclinical inflammation, linking insulin-resistant states like obesity and T2DM to inflammation. WDs were shown to reprogram the innate immune system and induce low-grade inflammation.21,120 In contrast, a low-GL diet reduces inflammation and tends to increase a beneficial adipokine (adiponectin) in overweight and obese but otherwise healthy adult men and women.121
Obesity And Civilization Diseases As A Manifestation Of (Diet-Related) Metabolic Changes (Figure 1)
The next section provides representative examples to explain the important role played by the metabolic alterations described above in the development of obesity and civilization diseases.
WDs And Obesity
Since 1980, the prevalence of obesity has doubled in more than 70 countries and continues to rise at an alarming rate in both developed and developing countries. While the prevalence of overweight and obesity among children and adolescents changed very little between the 1960s and early 1980s, dramatic increases were observed during the 1980s and 1990s. The obesity epidemic was first noted in the U.S, but during the last few decades, this phenomenon has spread to all parts of the world. Obesity is now considered as a pandemic condition, with serious health implications. Despite intensive research, the causes of the obesity epidemic remain incompletely understood.122–125 It is widely held that obesity results from a chronic surplus of energy intake compared to energy expenditure, which leads to storage of excessive amounts of triglycerides in adipose tissue. Multiple factors are thought to interact to produce a state of positive energy balance, including genetic and epigenetic factors, diet, lifestyle and lack of physical activity.122–125 Although genes may contribute to a person’s susceptibility to weight gain,126 genetic changes are unlikely to explain the rapid spread of obesity around the globe during the last decades.127
While high calorie intake combined with low physical activity certainly plays an important role in the development of obesity, diet-related metabolic alterations may be even more important. Mixed diets such as WDs (which provide high levels of glucose and FFA that compete for mitochondrial oxidation) are characterized by daily oscillations between glucose and FFA oxidation, which are coordinated by insulin and glucagon. During the fed state, a high insulin/glucagon ratio promotes lipid storage and suppression of gluconeogenesis, while in the fasted state a high glucagon/insulin ratio stimulates lipolysis and hepatic glucose production to provide glucose supply to glucose-dependent tissues. Hence, during the fed state, glucose serves as main fuel and fat is stored, while during the fasted state, fatty acids serve as main fuel and glucose is preserved for glucose-dependent tissues.30,35,128 Mobilization and oxidization of FFA in the fasted state (as well as during enhanced physical activity) are only possible as long as insulin levels are not elevated. As mentioned earlier, due to the westernization of diet and lifestyle, insulin levels are increased much of the day.34 High postprandial insulin levels drive fat storage and prevent lipolysis while maintaining FFA re-esterification.49,53,54,129 As a result, part of the (daily) stored FA can remain stored and contribute to a gradual increase in fat mass.49,53,54,125,129 However, the most important metabolic alterations may be the development of insulin hypersecretion and hyperinsulinemia, which significantly accelerates weight gain.49,50,54–57,125,130 Several studies in humans18,57,130,131 and animals56,58,89,90 support a causal role of hyperinsulinemia in the development of obesity. High rates of weight gain occurred in individuals who presented with a high acute insulin response to glucose and this effect was particularly manifested in insulin-sensitive individuals.130 The Da Qing Children Cohort Study showed that fasting insulin at the age of 5 years (even after adjustment for age, sex, birth weight, TV-viewing time and weight at baseline) predicted weight gain from age 5 to 10 years.131 Odelye et al55 demonstrated that fasting plasma insulin concentration correlates with the rate of weight gain. Also, a study of insulin dynamics among obese schoolchildren suggested that insulin hypersecretion precedes development of insulin resistance by several years.85 A study performed with knockout mice confirmed that hyperinsulinemia alters lipid metabolism and promotes obesity.49 Otherwise, inhibition of hyperinsulinemia with diazoxide or octreotide causes weight loss and decreases insulin levels without impairing glucose tolerance in obese humans.132 Development of IR prevents unlimited insulin-induced weight gain.133 As previously described, high-insulinemic diets, including WD, may play an important role in the development of insulin hypersecretion and (fasting) hyperinsulinemia.18,85–90 Also, several studies support a causal role of high-insulinemic diets in the development of obesity.89,134,135 In addition, high insulin levels lead to increased appetite, hyperphagia and carbohydrate craving.36,37,139 Several studies have also found that foods with high insulin responses are less satiating.35 Finally, high dietary intake of omega-6 PUFAs also has been implicated in the development of obesity.21
Genetic factors may account for differences in sensitivity and reactivity of β-cells to high-insulinemic foods, and thus for exaggerated insulin secretion (hypersecretion). This is supported by a study which found that variants in the insulin promoter gene were associated with insulin hypersecretion and strongly predicted weight gain.136
Furthermore, adipose tissue is not only the primary storage site for excess energy, but also a metabolically active organ that excretes a variety of biologically active molecules, which are believed to play a role in the development of a variety of metabolic alterations, such as OS, insulin resistance, low-grade inflammations, mitochondrial dysfunction, β-cell dysfunction, and other more, which may contribute to the development of civilization diseases.137,138
In summary, diet composition and lifestyle play an important role in the development of obesity. WDs cause a high postprandial insulin production and can produce insulin hypersecretion and hyperinsulinemia, which promote fat storage, prevent lipolysis and cause increased appetite, hyperphagia and weight gain.35–37,139 On the other hand, feeding studies that included only non-energy-restricted diets confirmed that caloric intake decreases spontaneously and weight loss is induced when carbohydrate intake is restricted.35,139,140 In addition, several human and animal intervention studies showed that Mediterranean or Paleolithic diets have highly beneficial effects on risk factors for diseases of civilization.2,141–143
Diabetes Mellitus
Global rates of diabetes mellitus have reached epidemic proportions and are associated with an ever-increasing health and socioeconomic burden. T2DM is a progressive disease due to IR and advancing β-cell dysfunction. OS is critically involved in the impairment of beta cell function due to the low antioxidant capacity of beta cells and has also been implicated in the progression of long-term T2DM complications, including microvascular and macrovascular dysfunction.144 ANG II, independently of its vasoconstrictor action, causes OS, β-cell inflammation and dysfunction through enhanced activation of cellular NADPH oxidase via ANG II receptors.112 The RAS is also considered to be involved in most of the pathological processes that result in diabetic nephropathy.145 SNS overactivity almost doubled the risk of T2DM over an eight-year follow-up period in the Atherosclerosis Risk in Communities Study with more than 8,000 middle-aged non-diabetic adults at baseline.146
Cardiovascular Diseases
Endothelial dysfunction is a key factor in the development of CVD. A dysfunctional endothelium promotes vascular inflammation, infiltration and activation of inflammatory cells, chemokine and cytokine secretion, vasoconstriction, lipoprotein oxidation, platelet aggregation, white blood cell adhesion and proliferation, and apoptosis of endothelial and vascular smooth muscle cells, key factors in atherogenesis and hypertension.147 OS,148 overactivity of the RAS,149 and IR and hyperinsulinemia150,151 are causally related to endothelial dysfunction. Even modest hyperinsulinemia, comparable to fasting hyperinsulinemia of insulin-resistant conditions, can cause severe endothelial dysfunction in large conduit arteries.150
Further, subclinical inflammation is involved in all stages of the atherosclerotic process, from the initiation of fatty streaks to the development of plaque rupture and thrombus formation.111,152 As mentioned before, hyperinsulinemia,117,118 the SNS119 and the RAS109 are causally related to the development of subclinical inflammation.
The RAS,109 the SNS,153 insulin-related endothelin-1 production154 and OS28 promote proliferation, migration, senescence, apoptosis, autophagy of vascular smooth muscle cell and vascular remodeling of resistance vessels of the systemic circulation and of renal vessels, as well as peripheral and renal vasoconstriction and peripheral vascular resistance, increase heart rate, stroke volume, renin secretion and tubular sodium reabsorption, and thereby contribute to the development of hypertension and atherosclerosis.111,149,155,156
Activation of the RAS stimulates accumulation of low-density lipoproteins, particularly the oxidatively modified form, in blood vessels which plays an important role in atherosclerotic plaque formation, progression and destabilization.157 According to the “oxidized linoleic acid theory of coronary heart disease”, dietary linoleic acid, especially when consumed from refined omega-6 vegetable oils, gets incorporated into all blood lipoproteins (such as LDL, VLDL and HDL), increasing the susceptibility of all lipoproteins to oxidize and hence increases cardiovascular risk.22 Further, plasma insulin concentrations are inversely related to HDL-cholesterol levels and positively associated with triglyceride levels, metabolic alterations strongly associated with atherosclerosis.158
Cancer
While the somatic mutation theory has been the prevalent theory in cancer research for the last 50 years, a body of evidence has accumulated showing that cancer is not only a genetic disease of uncontrolled cell proliferation, but also a metabolic disease.128,159 OS,27,44,116,160 hyperinsulinemia and IR,161 the SNS,113 the RAS162 and inflammation163 have been implicated as important causal factors for cancer development.
Epidemiological studies suggest that obesity and T2DM are positively correlated with both the risk of cancer. Among several factors that play a role, the most important link between obesity, T2DM, and cancer appears to be related to insulin resistance, hyperinsulinemia and increased levels of insulin-like growth factor (IGF). Hyperinsulinemia may affect cancer risk not only through direct mitogenic effects of insulin but also indirectly via increased production of IGF1. Hyperinsulinemia and augmented insulin and IGF1 signaling can enhance tumor development and growth.164
More……
Hyperinsulinemia, overactivation of the RAS and the SNS, inflammation and OS have been implicated in the development of Alzheimer disease72,165–167 and benign prostatic hyperplasia and hypertension.155 Hyperinsulinemia, overactivation of the SNS, inflammation and OS have been implicated in the development and polycystic ovary syndrome.160,168,169 Inflammation and OS contribute to the pathophysiology of a number of debilitating diseases, including nonalcoholic fatty liver, osteoarthritis, and autoimmune diseases, such as lupus and rheumatoid arthritis, Hashimoto thyroiditis, allergies, neurodegenerative disorders such as Parkinson’s disease, multiple sclerosis and Major Depressive Disorder, Graves’ disease, type 1 DM, psoriasis, asthma, inflammatory bowel disease, Crohn’s disease.115,118,170 Elevated NE tone has been suggested to be an etiological factor in open-angle glaucoma, osteoarthritis and rheumatoid arthritis, asthma, epilepsy, Parkinson disease.167 The RAS has been implicated in the development of neurodegenerative diseases, such as multiple sclerosis, Parkinson, Alzheimer and Huntington disease.171 In addition to its role in atherogenesis and hypertension, the RAS plays a critical role in the pathogenesis of many other types of CVDs including cardiomyopathy, valvular heart disease, aneurysms, infarction, stroke and renal disease.109,172 Hyperinsulinemia and low-grade inflammation have been implicated in the development of osteoarthritis.117
This listing could continue, but that would go beyond the scope of this publication.
There are certainly more metabolic alterations involved in the development of civilization diseases that are not mentioned in this article. On the one hand, however, it is assumed that the changes described are most important because they affect powerful metabolic systems such as the SNS, the RAS and the immune system, on the other hand, it is likely that (at least many) alterations that have not been addressed are likewise part of the disrupted metabolic balance described above.
Important Life-Style Factors
In addition to the dietary patterns described above, a variety of life-style factors have been implicated in disease development, including smoking, overuse of alcohol, drug abuse, lack of physical activity and inadequate relief of chronic stress.2,23,173–180
Cigarette smoking can cause or contribute to the development of a range of potentially fatal and disabling diseases and conditions including several types of cancer, CVD, chronic obstructive pulmonary disease. Intriguingly, smoking causes OS,173 IR,174 activation of SNS175 and the RAS,176 and low-grade inflammation,177 all of which have been implicated in the development of civilization diseases, as pointed out above.
Alcohol consumption, especially heavy drinking, is an important risk factor for many health problems and therefore contributes significantly to the global burden of disease. These include not only well-known outcomes of drinking such as liver cirrhosis or traffic accidents, but also categories of illnesses like infectious diseases, cancer, diabetes, neuropsychiatric disorders (including alcohol use disorders) and CVD. Most of the alcohol-related harm is caused by the regular consumption of alcohol. According to a study by the Centre for Addiction and Mental Health, about 40% of the world’s adult population consumes alcohol, and the average consumption per drinker is 17.1 litres per year.178,179
Finally, a sedentary lifestyle and lack of physical activity of today’s people has also been implicated in the development of civilization diseases.180
Microbiome
The human gut microbiota plays an important role in the health of the human host. An increasing body of evidence suggests that gut microbiota is adversely affected by a WD, which promotes inflammation resulting from both structural and behavioral changes in the resident microbiome, and that these changes are associated with obesity and metabolic diseases, including cancer, T2DM, asthma, psychiatric diseases and other more.181 Otherwise, a Mediterranean diet rich in plant foods favorably influenced the microbiome-related metabolome profiles in persons who previously consumed a WD.182
Further, increasing evidence supports an important role of genetic and epigenetic mechanisms in disease development. Epigenetics deals with changes in DNA and histone proteins that alter tissue and cell type-specific gene expression patterns, thereby causing certain diseases, particularly some cancers. Prenatal exposure to various environmental factors (maternal stress, smoking, viral infections, drugs and toxins, etc.) may affect the fetal epigenome. In particular, maternal nutrition has a profound effect on the epigenome and can determine gene expression patterns and health throughout the life course183,184
The Fetal Origins Of Adult Disease (FOAD) Hypothesis
It is meanwhile well established that prenatal development is a critical period in the etiology of human diseases: the fetal origins of adult disease hypothesis implies that adverse influences during early development, in especially maternal nutritional imbalance and metabolic disturbances during pregnancy, lead to developmental adaptations in the fetus that permanently alter the structure, physiology, and metabolism of the fetus (“fetal programming”) and make individuals susceptible to the development of obesity and related diseases, such as T2DM, hypertension, CVD and other, from childhood to adulthood.184–187
Proper nutrition is a key factor for the correct somatic growth and development of the fetus. The availability of nutrients to the fetus depends on placental supply and maternal nutrition. Maternal nutrition, food supply and metabolism during pregnancy and lactation have therefore marked implications on child development and long-term health.187,188
Both in humans and animals models, maternal malnutrition (protein and/or calorie restriction) as well as overnutrition (especially with diets rich in fat, refined grains and sucrose, typical of WDs) can lead to an adverse metabolic profile and a tendency for IR, metabolic syndrome, T2DM, obesity, and CVD during adulthood.186–190 Increasing evidence suggests that maternal hyperglycemia is also a risk factor for fetal programming and can down-regulate both fetal glucose tolerance and insulin sensitivity.185,189,191
Maternal obesity, excessive weight gain during pregnancy and gestational diabetes can also have significant adverse effects on fetal development, characterized by a tendency to obesity, insulin resistance, diabetes, hypertension and other more in later life.186,189,192,193 In an animal model of maternal obesity, adverse metabolic effects in the offspring were compounded by a high-fat diet during pregnancy and lactation.192
Increasing evidence suggests that OS and epigenetic mechanisms are the link between adverse intrauterine environment and disruption of the normal pattern of fetal development. During pregnancy, ROS serve as signaling molecules to allow for the normal progression of embryonic and fetal development. An excess production in ROS during the intrauterine period leads to altered placental function and plays a central role in altered fetal programming.186,194–197
Obesity itself is characterized by chronic low grade inflammation with permanently increased OS. Maternal obesity is therefore associated with metabolic alterations and dysregulation of redox balance in the mother-placenta-fetus unit.198 In addition, both maternal malnutrition and overeating are associated with OS and DNA methylation of selected gene promoter regions.188,193,195–198 Diet composition plays an important role in this regard: WDs,33,43,44,76 high-sucrose diets,42,77 diets with a high GL41 and high-fructose diets78 produce high levels of ROS and OS. In contrast, Paleolithic and Mediterranean diets are associated with low OS.46
In search of the “perfect maternal diet”, cohort studies indicate that dietary patterns which are mainly based on vegetables, fruits, whole grains, fish, chicken and lean meat have a positive effect on the formation of a normal placenta and the developing fetus and are associated with a reduced risk of pregnancy complications.187,189,199
Summary And Conclusions
- Over the course of 6–8 generations, but especially in the last 2–3 generations, there has been a pandemic of obesity and non-infectious degenerative diseases known as civilization diseases.
- Among a variety of factors that may be involved in this development, Western diet and lifestyle, as well as epigenetic changes and fetal programming, are believed to play key roles.
- The evidence presented shows that Western diets significantly distort the fine-tuned metabolism that has evolved over a very long period of human evolution in adaptation to Paleolithic nutrition, including an increased production of ROS, insulin hypersecretion and IR, low-grade inflammation and an abnormal activation of SNS and RAS (displayed in Figure 1), which may cause or contribute to the development of civilization diseases.
- The suggested pathomechanism is also well-suited to explain the well-known but not completely understood close relationship between obesity and the wide range of comorbidities like T2DM, cardiovascular disease, cancer, etc., as diseases of the same etiopathology.
- With the increasing spread of Western diet and lifestyle worldwide, overweight and lifestyle diseases are also rapidly increasing in developing countries.
- As rates of obesity increase worldwide, a significant percentage of embryos and fetuses will be exposed to an overweight or obese in utero environment and likely to maternal overnutrition during key periods of perinatal development, making fetuses vulnerable to obesity and degenerative diseases later in life - a self-reinforcing process that further fuels the pandemic.
- Changing our lifestyle in accordance with our genetic constitution, including diet and physical activity, can help prevent or limit the development of these diseases. This view is underpinned by several human and animal intervention studies which show that diets composed of Mediterranean/Paleolithic foods such as meat, fish, eggs, fresh fruits and vegetables, roots, tubers, nuts and seeds have highly beneficial effects on risk factors for diseases of civilization.2,141–143,181
- Probably more important than the foods contained in these diets are foods that are not included, especially high-glycemic carbohydrate foods, like refined grains, sugar, corn, refined fructose, and foods that are high in omega-6 PUFAs. Therefore, quality rather than the quantity of dietary fat and carbohydrates are important.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Fung J, Berger A. Hyperinsulinemia and insulin resistance: scope of the problem. J Insulin Resist. 2016;1(1):1–6. doi:10.4102/jir.v1i1.18
2. Carrera-Bastos P, Fontes-Villalba M, O’Keefe JH. The western diet and lifestyle and diseases of civilization. Res Rep Clin Cardiol. 2011;2:15–35.
3. Trnovec T, Cook T, Kahayová K, Nyulassy S. Civilization as a threat to human health? Cent Eur J Public Health. 2001;9(1):49–52.
4. Cordain L, Eaton SB, Sebastian A, et al. Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr. 2005;81(2):341–354. doi:10.1093/ajcn.81.2.341
5. O’Dea K, White NG, Sinclair AJ. An investigation of nutrition-related risk factors in an isolated Aboriginal community in northern Australia: advantages of a traditionally-orientated life-style. Med J Aust. 1988;148:177–180.
6. Lindeberg S, Eliasson M, Lindahl B, Ahrén B. Low serum insulin in traditional Pacific Islanders – the Kitava study. Metabolism. 1999;4:1216–1219. doi:10.1016/S0026-0495(99)90258-5
7. Eaton SB, Konner M, Shostak M. Stone agers in the fast lane: chronic degenerative diseases in evolutionary perspective. Am J Med. 1988;84:739–749. doi:10.1016/0002-9343(88)90113-1
8. O’Dea K. Marked improvement in carbohydrate and lipid metabolism in diabetic Australian aborigines after temporary reversion to traditional lifestyle. Diabetes. 1984;33:596. doi:10.2337/diab.33.6.596
9. Lindeberg S, Jönsson T, Granfeldt Y, et al. A Palaeolithic diet improves glucose tolerance more than a Mediterranean-like diet in individuals with ischaemic heart disease. Diabetologia. 2007;50(9):1795–1807. doi:10.1007/s00125-007-0716-y
10. Gurven M, Kaplan H. Longevity among hunter-gatherers: a cross-cultural examination. Popul Dev Rev. 2007;33:321–365. doi:10.1111/j.1728-4457.2007.00171.x
11. O’Keefe JH, Cordain L. Cardiovascular disease resulting from a diet and lifestyle at odds with our paleolithic genome: how to become a 21st-century hunter-gatherer. Mayo Clin Proc. 2004;79(1):101–108. doi:10.4065/79.1.101
12. Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med. 1985;312:283–289. doi:10.1056/NEJM198501313120505
13. Rendel JM. The time scale of genetic changes. In: Boyden SV, editor. The Impact of Civilization on the Biology of Man. Canberry: Australian National University Press; 1970:27–47.
14. Cordain L, Miller JB, Eaton SB, Mann N, Holt SAH, Speth JD. Plant-animal subsistence ratios and macronutrient energy estimations in worldwide hunter-gatherer diets. Am J Clin Nutr. 2000;71:682–692. doi:10.1093/ajcn/71.3.682
15. Garn SM. What did our ancestors eat? Nutr Rev. 1989;47:337–345. doi:10.1111/j.1753-4887.1989.tb02765.x
16. Wolever TM, Bolognesi C. Prediction of glucose and insulin responses of normal subjects after consuming mixed meals varying in energy, protein, fat, carbohydrate and glycemic index. J Nutr. 1996;126(11):2807–2812. doi:10.1093/jn/126.11.2807
17. Björk I, Liljeberg H, Östan E. Low-glycemic index foods. Br J Nutr. 2000;83(Suppl 1):S149–S155. doi:10.1017/s0007114500001094
18. Hoppe C, Mølgaard C, Vaag A, Barkholt V, Michaelsen KF. High intakes of milk, but not meat, increase s-insulin and insulin resistance in 8-year-old boys. Eur J Clin Nutr. 2005;59(3):393–398. doi:10.1038/sj.ejcn.1602086
19. Ostman EM, Liljeberg Elmståhl HG, Björck IM. Inconsistency between glycemic and insulinemic responses to regular and fermented milk products. Am J Clin Nutr. 2001;74:96–100. doi:10.1093/ajcn/74.1.96
20. Gómez Candela C, Bermejo López LM, Loria Kohen V. Importance of a balanced omega 6/omega 3 ratio for the maintenance of health: nutritional recommendations. Nutr Hosp. 2011;26(2):323–329. doi:10.1590/S0212-16112011000200013
21. Simopoulos AP. An increase in the omega-6/omega-3 fatty acid ratio increases the risk for obesity. Nutrients. 2016;8(3):128. doi:10.3390/nu8030128
22. DiNicolantonio JJ, O’Keefe JH. Omega-6 vegetable oils as a driver of coronary heart disease: the oxidized linoleic acid hypothesis. Open Heart. 2018;5(2):e000898. doi:10.1136/openhrt-2018-000898
23. Manson JE, Cook NR, Lee IM; VITAL Research Group, et al. Marine n-3 fatty acids and prevention of cardiovascular disease and cancer. N Engl J Med. 2019;380(1):23–32. doi:10.1056/NEJMoa1811403
24. Gough DR, Cotter TG. Hydrogen peroxide: a Jekyll and Hyde signaling molecule. Cell Death Dis. 2011;2:e213. doi:10.1038/cddis.2011.82
25. Henriksen E, Diamond-Stanic M, Marchionne E. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic Biol Med. 2011;51:993e9. doi:10.1016/j.freeradbiomed.2011.04.011
26. Rani V, Deep G, Singh RK, Palle K, Yadav UC. Oxidative stress and metabolic disorders: pathogenesis and therapeutic strategies. Life Sci. 2016;148:183–193. doi:10.1016/j.lfs.2016.02.002
27. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49(11):1603–1616. doi:10.1016/j.freeradbiomed.2010.09.006
28. Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res. 2015;116(3):531–549. doi:10.1161/CIRCRESAHA.116.303584
29. Alberts B, Bray D, Johnson A, et al. How cells obtain energy from food. In: Alberts B, Bray D, Johnson A, et al, editors. Essential Cell Biology. An Introduction to the Molecular Biology of the Cell. Vol. 4. New York: Garland Publishing Inc; 2014:419–466.
30. Muoio DM. Metabolic Inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell. 2014;159:1253–1262. doi:10.1016/j.cell.2014.09.052
31. Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi:10.1016/s0014-5793(97)01191-5
32. O’Keefe JH, Gheewala NM, O’Keefe JO. Dietary strategies for improving post-prandial glucose, lipids, inflammation, and cardiovascular health. J Am Coll Cardiol. 2008;51(3):249–255. doi:10.1016/j.jacc.2007.10.016
33. Tan BL, Norhaizan ME, Liew WPP. Nutrients and oxidative stress: friend or foe? Oxid Med Cell Longev. 2018;2018:9719584. doi:10.1155/2018/9719584
34. Esmaillzadeh A, Kimiagar M, Mehrabi Y, Azadbakht L, Hu FB, Willett WC. Dietary patterns, insulin resistance, and prevalence of the metabolic syndrome in women. Am J Clin Nutr. 2007;85:910–918. doi:10.1093/ajcn/85.3.910
35. Westman EC, Feinman RD, Mavropoulos JC, et al. Low-carbohydrate nutrition and metabolism. Am J Clin Nutr. 2007;86:276–284. doi:10.1093/ajcn/86.2.276
36. Rodin J, Wack J, Ferrannini E, DeFronzo RA. Effect of insulin and glucose on feeding behavior. Metabolism. 1985;34:826–831. doi:10.1016/0026-0495(85)90106-4
37. Holt SH, Miller JB. Increased insulin responses to ingested foods are associated with lessened satiety. Appetite. 1995;24:43–54. doi:10.1016/S0195-6663(95)80005-0
38. Drewnowski A. The role of energy density. Lipids. 2003;38(2):109–115. doi:10.1007/s11745-003-1039-3
39. DiMeglio DP, Mattes RD. Liquid versus solid carbohydrate: effects on food intake and body weight.Int. J Obes Relat Metab Disord. 2000;24:794–800. doi:10.1038/sj.ijo.0801229
40. Liesa M. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi:10.1016/j.cmet.2013.03.002
41. Limkunakul C, Sundell MB, Pouliot B, Graves AJ, Shintani A, Ikizler TA. Glycemic load is associated with oxidative stress among prevalent maintenance hemodialysis patients. Nephrol Dial Transplant. 2014;29(5):1047–1053. doi:10.1093/ndt/gft489
42. Busserolles J, Rock E, Gueux E, Mazur A, Grolier P, Rayssiguier Y. Short-term consumption of a high-sucrose diet has a pro-oxidant effect in rats. Br J Nutr. 2002;87(4):337–342. doi:10.1079/BJNBJN2002524
43. Sies H, Stahl W, Sevanian A. Nutritional, dietary and postprandial oxidative stress. J Nutr. 2005;135(5):969–972. doi:10.1093/jn/135.5.969
44. Erdelyi I, Levenkova N, Lin EY, et al. Western-Style diets induce oxidative stress and dysregulate immune responses in the colon in a mouse model of sporadic colon cancer. J Nutr. 2009;139(11):2072–2078. doi:10.3945/jn.108.104125
45. Monnier L, Mas E, Ginet C, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA. 2006;295(14):1681–1687. doi:10.1001/jama.295.14.1681
46. Whalen KA, McCullough ML, Flanders WD, Hartman TJ, Judd S, Bostick RM. Paleolithic and Mediterranean diet pattern scores are inversely associated with biomarkers of inflammation and oxidative balance in adults. J Nutr. 2016;146(6):1217–1226. doi:10.3945/jn.115.224048
47. Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes. 2011;60:2441–2449. doi:10.2337/db11-0425
48. Corkey BE. Banting lecture: hyperinsulinemia: cause or consequence? Diabetes. 2012;61:4–13. doi:10.2337/db11-1483
49. Mehran AE, Templeman NM, Brigidi GS, et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012;16(6):723–737. doi:10.1016/j.cmet.2012.10.019
50. Kopp W. High-insulinogenic nutrition - an etiologic factor for obesity and the metabolic syndrome? Metabolism. 2003;52(7):840–844. doi: 10.1016/s0026-0495(02)05294-0
51. Reaven G. All obese individuals are not created equal: insulin resistance is the major determinant of cardiovascular disease in overweight/obese individuals. Diab Vasc Dis Res. 2005;2:105–112. doi:10.3132/dvdr.2005.017
52. Nolan CJ, Prentki M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: time for a conceptual framework shift. Diab Vasc Dis Res. 2019;16(2):118–127. doi:10.1177/1479164119827611
53. Erion KA, Corkey BE. Hyperinsulinemia: a cause of obesity? Curr Obes Rep. 2017;6(2):178–186. doi:10.1007/s13679-017-0261-z
54. Templeman NM, Skovsø S, Page MM, Lim GE, Johnson JD. A causal role for hyperinsulinemia in obesity. J Endocrinol. 2017;232:R173–83. doi:10.1530/JOE-16-0449
55. Odeleye OE, de Courten M, Pettitt DJ, Ravussin E. Fasting hyperinsulinemia is a predictor of increased body weight gain and obesity in Pima Indian children. Diabetes. 1997;46(8):1341–1345. doi:10.2337/diab.46.8.1341
56. Cusin I, Rohner-Jeanrenaud F, Terrettaz J, Jeanrenaud B. Hyperinsulinemia and its impact on obesity and insulin resistance. Int J Obes. 1992;16(Suppl 4):S1–11.
57. Le Stunff C, Bougneres P. Early changes in postprandial insulin secretion, not in insulin sensitivity, characterize juvenile obesity. Diabetes. 1994;43:696–702. doi:10.2337/diab.43.5.696
58. Pénicaud L, Kinebanyan MF, Ferré P, et al. Development of VMH obesity: in vivo insulin secretion and tissue insulin sensitivity. Am J Physiol. 1989;257:E255–E260. doi:10.1152/ajpendo.1989.257.2.E255
59. Dankner R, Chetrit A, Shanik MH, Raz I, Roth J. Basal-state hyperinsulinemia in healthy normoglycemic adults is predictive of type 2 diabetes over a 24-year follow-up: a preliminary report. Diabetes Care. 2009;32(8):1464–1466. doi:10.2337/dc09-0153
60. Catalano KJ, Maddux BA, Szary J, Youngren JF, Goldfine ID, Schaufele F. Insulin resistance induced by hyperinsulinemia coincides with a persistent alteration at the insulin receptor tyrosine kinase domain. PLoS One. 2014;9(9):e108693. doi:10.1371/journal.pone.0108693
61. Juan CC, Fang VS, Kwok CF, Perng JC, Chou YC, Ho LT. Exogenous hyperinsulinemia causes insulin resistance, hyperendothelinemia and subsequent hypertension in rats. Metab Clin Exp. 1999;48:465–471. doi:10.1016/S0026-0495(99)90105-1
62. Ge X, Yu Q, Qi W, Shi X, Zhai Q. Chronic insulin treatment causes insulin resistance in 3T3-L1 adipocytes through oxidative stress. Free Radic Res. 2008;42:582–591. doi:10.1080/10715760802158448
63. Posa JK, Selvaraj S, Sangeetha KN, Baskaran SK, Lakshmi BS. p53 mediates impaired insulin signaling in 3T3-L1 adipocytes during hyperinsulinemia. Cell Biol Int. 2014;38:818–824. doi:10.1002/cbin.10275
64. Rowe JW, Young JB, Minaker KL, Stevens AL, Pallotta J, Landsberg L. Effect of insulin and glucose infusions on sympathetic nervous system activity in normal man. Diabetes. 1981;30:219–225. doi:10.2337/diab.30.3.219
65. Rooney DP, Edgar JD, Sheridan B, Atkinson AB, Bell PM. The effects of low dose insulin infusions on the renin-angiotensin and sympathetic nervous systems in normal man. Eur J Clin Invest. 1991;21:430–435. doi:10.1111/j.1365-2362.1991.tb01391.x
66. Haenni A, Reneland R, Lind L, Lithell H. Serum aldosterone changes during hyperinsulinemia are correlated to body mass index and insulin sensitivity in patients with essential hypertension. J Hypertens. 2001;19:107–112. doi:10.1097/00004872-200103000-00017
67. Pi J, Bai Y, Zhang Q, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56(7):1783–1791. doi:10.2337/db06-1601
68. Saadeh M, Ferrante TC, Kane A, et al. Reactive oxygen species stimulate insulin secretion in rat pancreatic islets: studies using mono-oleoyl-glycerol. PLoS One. 2012;7(1):e30200. doi:10.1371/journal.pone.0030200
69. Ning J, Hong T, Yang X, et al. Insulin and insulin signaling play a critical role in fat induction of insulin resistance in mouse. Am J Physiol Endocrinol Metab. 2011;301(2):E391–E401. doi:10.1152/ajpendo.00164.2011
70. Ramalingam L, Menikdiwela K, LeMieux M, et al. The renin angiotensin system, oxidative stress and mitochondrial function in obesity and insulin resistance. Biochim Biophys Acta. 2017;1863(5):1106–1114. doi:10.1016/j.bbadis.2016.07.019
71. Lembo G, Capaldo B, Rendina V, et al. Acute noradrenergic activation induces insulin resistance in human skeletal muscle. Am J Physiol Endo Met. 1994;266:E242–7.
72. Verdile G, Keane KN, Cruzat VF, et al. Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and Alzheimer’s Disease. Mediators Inflamm. 2015;2015:105828. doi:10.1155/2015/125380
73. Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440(7086):944–948. doi:10.1038/nature04634
74. Youn JY, Siu KL, Lob HE, Itani H, Harrison DG, Cai H. Role of vascular oxidative stress in obesity and metabolic syndrome. Diabetes. 2014;63(7):2344–2355. doi:10.2337/db13-0719
75. Schraml E, Quan P, Stelzer I, et al. Norepinephrine treatment and aging lead to systemic and intracellular oxidative stress in rats. Exp Gerontol. 2007;42(11):1072–1078. doi:10.1016/j.exger.2007.08.003
76. Heinonen I, Rinne P, Ruohonen ST, Ruohonen S, Ahotupa M, Savontaus E. The effects of equal caloric high fat and western diet on metabolic syndrome, oxidative stress and vascular endothelial function in mice. Acta Physiol (Oxf). 2014;211(3):515–527. doi:10.1111/apha.12253
77. Prasad K, Dhar I. Oxidative stress as a mechanism of added sugar-induced cardiovascular disease. Int J Angiol. 2014;23(4):217–226. doi:10.1055/s-0034-1387169
78. Castro MC, Massa ML, Arbeláez LG, Schinella G, Gagliardino JJ, Francini F. Fructose-induced inflammation, insulin resistance and oxidative stress: a liver pathological triad effectively disrupted by lipoic acid. Life Sci. 2015;137:1–6. doi:10.1016/j.lfs.2015.07.010
79. Matsuzawa-Nagata N, Takamura T, Ando H, et al. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism. 2008;57(8):1071–1077. doi:10.1016/j.metabol.2008.03.010
80. Schofield CJ, Sutherland C. Disordered insulin secretion in the development of insulin resistance and Type 2 diabetes. Diabet Med. 2012;29(8):972–979. doi:10.1111/j.1464-5491.2012.03655.x
81. Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A. Specific and combined effects of insulin and glucose on functional pancreatic beta-cell mass in vivo in adult rats. Endocrinology. 2003;144(6):2717–2727. doi:10.1210/en.2002-221112
82. Mahler RJ. The relationship between the hyperplastic pancreatic islet and insulin insensitivity in obesity. Acta Diabetol Lat. 1981;18:1–17.
83. Gonzalez A, Merino B, Marroquí L, et al. Insulin hypersecretion in islets from diet-induced hyperinsulinemic obese female mice is associated with several functional adaptations in individual β-cells. Endocrinology. 2013;154(10):3515–3524. doi:10.1210/en.2013-1424
84. Ferrannini E, Camastra S, Gastaldelli A, et al. Beta-cell function in obesity: effects of weight loss. Diabetes. 2004;53(Suppl 3):S26–S33. doi:10.2337/diabetes.53.suppl_3.s26
85. Preeyasombat C, Bacchetti P, Lazar AA, Lustig RH. Racial and etiopathologic dichotomies in insulin hypersecretion and resistance in obese children. J Pediatr. 2005;146(4):474–481. doi:10.1016/j.jpeds.2004.12.014
86. Patel MS, Srinivasan M. Metabolic programming in the immediate postnatal life. Ann Nutr Metab. 2011;58(Suppl 2):18–28. doi:10.1159/000328040
87. Pawlak DB, Bryson JM, Denyer GS, Brand-Miller JC. High glycemic index starch promotes hypersecretion of insulin and higher body fat in rats without affecting insulin sensitivity. J Nutr. 2001;131(1):99–104. doi:10.1093/jn/131.1.99
88. Gonsolin D, Couturier K, Garait B, et al. High dietary sucrose triggers hyperinsulinemia, increases myocardial beta-oxidation, reduces glycolytic flux and delays post-ischemic contractile recovery. Mol Cell Biochem. 2007;295(1–2):217–228. doi:10.1007/s11010-006-9291-7
89. Barnard RJ, Roberts CK, Varon SM, Berger JJ. Diet-induced insulin resistance precedes other aspects of the metabolic syndrome. J Appl Physiol. 1998;84:1311–1315. doi:10.1152/jappl.1998.84.6.1967
90. Grimditch GK, Barnard RJ, Hendricks L, Weitzman D. Peripheral insulin sensitivity as modified by diet and exercise training. Am J Clin Nutr. 1988;48:38–43. doi:10.1093/ajcn/48.1.38
91. Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr. 2004;79(4):537–543. doi:10.1093/ajcn/79.4.537
92. Sleder J, Chen YD, Cully MD, Reaven GM. Hyperinsulinemia in fructose-induced hypertriglyceridemia in the rat. Metabolism. 1980;29(4):303–305. doi:10.1016/0026-0495(80)90001-3
93. Baena M, Sangüesa G, Dávalos A, et al. Fructose, but not glucose, impairs insulin signaling in the three major insulin-sensitive tissues. Sci Rep. 2016;6:26149. doi:10.1038/srep26149
94. Grunfeld B, Balzareti M, Romo M, Gimenez M, Gutman R. Hyperinsulinemia in normotensive offspring of hypertensive parents. Hypertension. 1994;23:I12–5. doi:10.1161/01.HYP.23.1_Suppl.I12
95. Vlasáková Z, Pelikánová T, Karasová L, Skibová J. Insulin secretion, sensitivity, and metabolic profile of young healthy offspring of hypertensive parents. Metabolism. 2004;53:469–475. doi:10.1016/j.metabol.2003.10.030
96. Lambert EA, Straznicky NE, Dixon JB, Lambert GW. Should the sympathetic nervous system be a target to improve cardiometabolic risk in obesity? Am J Physiol Heart Circ Physiol. 2015;309(2):H244–H258. doi:10.1152/ajpheart.00096.2015
97. Scott EM, Greenwood JP, Vacca G, Stoker JB, Gilbey SG, Mary DA. Carbohydrate ingestion, with transient endogenous insulinemia, produces both sympathetic activation and vasodilatation in normal humans. Clin Sci. 2001;102:523–529. doi:10.1042/cs1020523
98. Welle S, Ulavivat U, Campell G. Thermic effect of feeding in men: increased plasma norepinephrine levels following glucose but not protein or fat consumption. Metabolism. 1981;30:953–958. doi:10.1016/0026-0495(81)90092-5
99. Heseltine D, Potter JF, Hartley G, Macdonald IA, James OF. Blood pressure, heart rate and neuroendocrine responses to a high carbohydrate and a high fat meal in healthy young subjects. Clin Sci. 1990;79(5):517–522. doi:10.1042/cs0790517
100. Tentolouris N, Tsigos C, Perea D, et al. Differential effect of high-fat and high-carbohydrate isoenergetic meals on cardiac autonomic nervous system activity in lean and obese women. Metabolism. 2003;52:1426–1432. doi:10.1016/s0026-0495(03)00322-6
101. Khoury N, McGill JB. Changes in insulin sensitivity following administration of the clinically used low-dose pressor, norepinephrine. Diabetes Metab Res Rev. 2011;27(6):604–608. doi:10.1002/dmrr.1212
102. Gamboa A, Okamoto LE, Arnold AC, et al. Autonomic blockade improves insulin sensitivity in obese subjects. Hypertension. 2014;64:867–874. doi:10.1161/HYPERTENSIONAHA.114.03738
103. Flaa A, Aksnes TA, Kjeldsen SE, Eide I, Rostrup M. Increased sympathetic reactivity may predict insulin resistance: an 18-year follow-up study. Metabolism. 2008;57(10):1422–1427. doi:10.1016/j.metabol.2008.05.012
104. Lavoie JL, Sigmund CD. Minireview: overview of the renin-angiotensin system – an endocrine and paracrine system. Endocrinology. 2003;144:2179–2183. doi:10.1210/en.2003-0150
105. Skov J, Persson F, Frøkiær J, Christiansen JS. Tissue renin-angiotensin systems: a unifying hypothesis of metabolic disease. Front Endocrinol (Lausanne). 2014;5:23. doi:10.3389/fendo.2014.00023
106. Grassi G. Renin-angiotensin-sympathetic cross talks in hypertension: reappraising the relevance of peripheral interactions. J Hypertens. 2001;19:1713–1716. doi:10.1097/00004872-200103000-00017
107. Saxena PR. Interaction between the renin–angiotensin–aldosterone and sympathetic nervous systems. J Cardiovasc Pharmacol. 1992;19:S80–8. doi:10.1097/00005344-199219006-00013
108. Olivares-Reyes JA, Arellano-Plancarte A, Castillo-Hernandez JR. Angiotensin II and the development of insulin resistance: implications for diabetes. Mol Cell Endocrinol. 2009;302(2):128–139. doi:10.1016/j.mce.2008.12.011
109. Pacurari M, Kafoury R, Tchounwou PB, Ndebele K. The renin-angiotensin-aldosterone system in vascular inflammation and remodeling. Int J Inflam. 2014;2014:689360. doi:10.1155/2014/689360
110. Hitomi H, Kiyomoto H, Nishiyama A. Angiotensin II and oxidative stress. Curr Opin Cardiol. 2007;22(4):311–315. doi:10.1097/HCO.0b013e3281532b53
111. Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 2012;95:194–204. doi:10.1093/cvr/cvs164
112. Chan SMH, Lau YS, Miller AA, et al. Angiotensin II causes β-cell dysfunction through an ER stress-induced proinflammatory response. Endocrinology. 2017;158(10):3162–3173. doi:10.1210/en.2016-1879
113. Tanga J, Lib Z, Lub L, Chob CH. β-Adrenergic system, a backstage manipulator regulating tumour progression and drug target in cancer therapy. Semin Cancer Biol. 2013;23:533–542. doi:10.1016/j.semcancer.2013.08.009
114. Calder PC, Ahluwalia N, Albers R, et al. A consideration of biomarkers to be used for evaluation of inflammation in human nutritional studies. Br J Nutr. 2013;109:S1–S34. doi:10.1017/S0007114512005119
115. Hunter P. The inflammation theory of disease. The growing realization that chronic inflammation is crucial in many diseases opens new avenues for treatment. EMBO Rep. 2012;13(11):968–970. doi:10.1038/embor.2012.142
116. Khansari N, Shakiba Y, Mahmoudi M. Chronic inflammation and oxidative stress as a major cause of age-related diseases and cancer. Recent Pat Inflamm Allergy Drug Discov. 2009;3(1):73–80.
117. Rufino AT, Ribeiro M, Ferreira JP, Judas F, Ferreira Mendes A. Hyperglycemia and hyperinsulinemia-like conditions independently induce inflammatory responses in human chondrocytes. J Funct Morphol Kinesiol. 2017;2(2):15. doi:10.3390/jfmk2020015
118. Pedersen DJ, Guilherme A, Danai LV, et al. A major role of insulin in promoting obesity-associated adipose tissue inflammation. Mol Metab. 2015;4(7):507–518. doi:10.1016/j.molmet.2015.04.003
119. Karakas M, Haase T, Zeller T. Linking the sympathetic nervous system to the inflammasome: towards new therapeutics for atherosclerotic cardiovascular disease. Eur Heart J. 2018;39(1):70–72. doi:10.1093/eurheartj/ehx374
120. Christ A, Günther P, Lauterbach MAR, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. 2018;172(1–2):162–175. doi:10.1016/j.cell.2017.12.013
121. Neuhouser ML, Schwarz Y, Wang C, et al. A low-glycemic load diet reduces serum C-reactive protein and modestly increases adiponectin in overweight and obese adults. J Nutr. 2011;142(2):369–374. doi:10.3945/jn.111.149807
122. James PT, Leach R, Kalamara E, Shayeghi M. The worldwide obesity epidemic. Obes Res. 2001;9(Suppl4):228S–233S. doi:10.1038/oby.2001.123
123. Strauss RS, Pollack HA. Epidemic increase in childhood overweight, 1986–1998. JAMA. 2001;286(22):2845–2848. doi:10.1001/jama.286.22.2845
124. GBD Obesity Collaborators, et al.Health effects of overweight and obesity in 195 countries over 25 years.N Engl J Med.2017;377(1):13–27. doi:10.1056/NEJMoa1614362
125. Ludwig DS, Ebbeling CB. The carbohydrate-insulin model of obesity: beyond “calories in, calories out”. JAMA Intern Med. 2018;178(8):1098–1103. doi:10.1001/jamainternmed.2018.2933
126. Choquet H, Meyre D. Genetics of obesity: what have we learned? Curr Genomics. 2011;12(3):169–179. doi:10.2174/138920211795677895
127. Hu F. Genetic predictors of obesity. In: Hu F, editor. Obesity Epidemiology. New York City: Oxford University Press; 2008:437–460.
128. Kopp W. Significant dietary changes during human evolution and the development of cancer: from cells in trouble to cells causing trouble. J Carcinog Mutagen. 2017;8:303. doi:10.4172/2157-2518
129. Campbell PJ, Carlson MG, Hill JO, Nurjhan N. Regulation of free fatty acid metabolism by insulin in humans: role of lipolysis and reesterification. Am J Physiol. 1992;263(6 Pt 1):E1063–E1069. doi:10.1152/ajpendo.2006.263.6.E1063
130. Sigal RJ, EI-Hashimy M, Martin BC, Soeldner S, Krolewski AS, Warram JH. Acute post-challenge hyperinsulinemia predicts weight gain: a prospective study. Diabetes. 1997;46(6):1025–1029. doi:10.2337/diab.46.6.1025
131. Chen YY, Wang JP, Jiang YY, et al. Fasting plasma insulin at 5 years of age predicted subsequent weight increase in early childhood over a 5-year period: the Da Qing children cohort study. PLoS One. 2015;10:e0127389. doi:10.1371/journal.pone.0127389
132. Lustig RH, Greenway F, Velasquez-Mieyer P, et al. A multicenter, randomized, double-blind, placebo-controlled, dose-finding trial of a long-acting formulation of octreotide in promoting weight loss in obese adults with insulin hypersecretion. Int J Obes (Lond). 2006;30:331–341. doi:10.1038/sj.ijo.0803074
133. Eckel RA. Insulin resistance: an adaptation for weight maintenance. Lancet. 1992;340:1452–1453. doi:10.1016/0140-6736(92)92633-q
134. Naja F, Hwalla N, Itani L, Karam S, Sibai AM, Nasreddine L. A Western dietary pattern is associated with overweight and obesity in a national sample of Lebanese adolescents (13–19 years): a cross-sectional study. Br J Nutr. 2015;114(11):1909–1919. doi:10.1017/S0007114515003657
135. Rasool S, Geetha T, Broderick TL, Babu JR. High fat with high sucrose diet leads to obesity and induces myodegeneration. Front Physiol. 2018;9:1054. doi:10.3389/fphys.2018.01054
136. Le Stunff C, Fallin D, Schork NJ, Bougnères P. The insulin gene VNTR is associated with fasting insulin levels and development of juvenile obesity. Nat Genet. 2000;26(4):444–446. doi:10.1038/82579
137. Coelho M, Oliveira T, Fernandes R. Biochemistry of adipose tissue: an endocrine organ. Arch Med Sci. 2013;9(2):191–200. doi:10.5114/aoms.2013.33181
138. Manna P, Jain SK. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: causes and therapeutic strategies. Metab Syndr Relat Disord. 2015;13(10):423–444. doi:10.1089/met.2015.0095
139. Seaman DR. Weight gain as a consequence of living a modern lifestyle: a discussion of barriers to effective weight control and how to overcome them. J Chiropr Humanit. 2013;20(1):27–35. doi:10.1016/j.echu.2013.08.001
140. Nordmann AJ, Nordmann A, Briel M, et al. Effects of low-carbohydrate vs low-fat diets on weight loss and cardiovascular risk factors: a meta-analysis of randomized controlled trials. Arch Intern Med. 2006;166(3):285–293. doi:10.1001/archinte.166.3.285
141. Lindeberg S, Jönsson T, Granfeldt Y, et al. A Palaeolithic diet improves glucose tolerance more than a Mediterranean-like diet in individuals with ischaemic heart disease. Diabetologia. 2007;50(9):1795–1807. doi:10.1007/s00125-007-0716-y
142. Jönsson T, Ahrén B, Pacini G, et al. A Paleolithic diet confers higher insulin sensitivity, lower C-reactive protein and lower blood pressure than a cereal-based diet in domestic pigs. Nutr Metab (Lond). 2006;3:39. doi:10.1186/1743-7075-3-39
143. Frassetto LA, Schloetter M, Mietus-Synder M, Morris RC
144. Drews G, Krippeit-Drews P, Düfer M. Oxidative stress and beta-cell dysfunction. Pflugers Arch. 2010;460(4):703–718. doi:10.1007/s00424-010-0862-9
145. Chawla T, Sharma D, Singh A. Role of the renin angiotensin system in diabetic nephropathy. World J Diabetes. 2010;1(5):141–145. doi:10.4239/wjd.v1.i5.141
146. Carnethon MR, Golden SH, Folsom AR, Haskell W, Liao D. Prospective investigation of autonomic nervous system function and the development of type 2 diabetes: the Atherosclerosis Risk In Communities study, 1987–1998. Circulation. 2003;107(17):2190–2195. doi:10.1161/01.CIR.0000066324.74807.95
147. Park KH, Park WJ. Endothelial dysfunction: clinical implications in cardiovascular disease and therapeutic approaches. J Korean Med Sci. 2015;30(9):1213–1225. doi:10.3346/jkms.2015.30.9.1213
148. Yang X, Li Y, Li Y, et al. Oxidative stress-mediated atherosclerosis: mechanisms and therapies. Front Physiol. 2017;8:600. doi:10.3389/fphys.2017.00600
149. Gomolak JR, Didion SP. Angiotensin II-induced endothelial dysfunction is temporally linked with increases in interleukin-6 and vascular macrophage accumulation. Front Physiol. 2014;5:396. doi:10.3389/fphys.2014.00268
150. Arcaro G, Cretti A, Balzano S, et al. Insulin causes endothelial dysfunction in humans: sites and mechanisms. Circulation. 2002;105(5):576–582. doi:10.1161/hc0502.103333
151. Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi:10.1161/CIRCULATIONAHA.105.563213
152. Libby P. Inflammation in Atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. doi:10.1161/ATVBAHA.108.179705
153. Grassi G, Arenare F, Pieruzzi F, Brambilla G, Mancia G. Sympathetic activation in cardiovascular and renal disease. J Nephrol. 2009;22:190–195.
154. Sarafidis PA, Bakris GL. Insulin and endothelin: an interplay contributing to hypertension development? J Clin Endocrinol Metab. 2007;92:379–385. doi:10.1210/jc.2006-1819
155. Kopp W. Diet-induced hyperinsulinemia as a key factor in the etiology of both benign prostatic hyperplasia and essential hypertension? Nutr Metab Insights. 2018;8(11):1178638818773072.
156. Hall JE, Granger JP, Do Carmo JM, et al. Hypertension: physiology and pathophysiology. Physiol. 2012;2:2393–2442.
157. Singh BM, Mehta JL. Interactions between the renin-angiotensin system and dyslipidemia: relevance in the therapy of hypertension and coronary heart disease. Arch Intern Med. 2003;163(11):1296–1304. doi:10.1001/archinte.163.11.1296
158. Reaven GM. Compensatory hyperinsulinemia and the development of an atherogenic lipoprotein profile: the price paid to maintain glucose homeostasis in insulin-resistant individuals. Endocrinol Metab Clin North Am. 2005;34(1):49–62. doi:10.1016/j.ecl.2004.12.001
159. Seyfried TN, Shelton LM. Cancer as a metabolic disease. Nutr Metab. 2010;7:7. doi:10.1186/1743-7075-7-7
160. Zuo T, Zhu M, Xu W. Roles of oxidative stress in polycystic ovary syndrome and cancers. Oxid Med Cell Longev. 2015;2016:8589318.
161. Vigneri R, Goldfine ID, Frittitta L. Insulin, insulin receptors, and cancer. J Endocrinol Invest. 2016;39(12):1365–1376. doi:10.1007/s40618-016-0508-7
162. George AJ, Thomas WG, Hannan RD. The renin-angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer. 2010;10:745–759. doi:10.1038/nrc2945
163. Todoric J, Antonucci L, Karin M. Targeting inflammation in cancer prevention and therapy. Cancer Prev Res (Phila). 2016;9(12):895–905. doi:10.1158/1940-6207.CAPR-16-0209
164. Cohen DH, LeRoith D. Obesity, type 2 diabetes, and cancer: the insulin and IGF connection. Endocr Relat Cancer. 2012;19(5):F27–F45. doi:10.1530/ERC-11-0374
165. Fishel MA, Watson GS, Montine TJ, et al. Hyperinsulinemia provokes synchronous increases in central inflammation and β-amyloid in normal adults. Arch Neurol. 2005;62(10):1539–1544. doi:10.1001/archneur.62.10.noc50112
166. Gebre AK, Altaye BM, Atey TM, Tuem KB, Berhe DF. Targeting renin-angiotensin system against Alzheimer’s disease. Front Pharmacol. 2018;9:440. doi:10.3389/fphar.2018.00440
167. Fitzgerald PJ. Is elevated norepinephrine an etiological factor in some cases of Alzheimer’s disease? Curr Alzheimer Res. 2010;7(6):506–516.
168. Shorakae S, Teede H, de Courten B, Lambert G, Boyle J, Moran LJ. The emerging role of chronic low-grade inflammation in the pathophysiology of polycystic ovary syndrome. Semin Reprod Med. 2015;33(4):257–269. doi:10.1055/s-0035-1556568
169. Lansdown A, Rees DA. The sympathetic nervous system in polycystic ovary syndrome: a novel therapeutic target? Clin Endocrinol (Oxf). 2012;77(6):791–801. doi:10.1111/cen.12003
170. Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract. 2014;105(2):141–150. doi:10.1016/j.diabres.2014.04.006
171. Almeida-Santos AF, Kangussu LM, Campagnole-Santos MJ. The renin-angiotensin system and the neurodegenerative diseases: a brief review. Protein Pept Lett. 2017;24(9):841–853. doi:10.2174/0929866524666170822120258
172. van Thiel BS, van der Pluijm I, Te Riet L, Essers J, Danser AH. The renin-angiotensin system and its involvement in vascular disease. Eur J Pharmacol. 2015;763(Pt A):3–14. doi:10.1016/j.ejphar.2015.03.090
173. Pasupathi P, Saravanan G, Farook J. Oxidative stress bio markers and antioxidant status in smokers compared to nonsmokers. Pharm Sci Res. 2009;1(2):55–62.
174. Facchini FS, Hollenbeck CB, Jeppesen J, Chen YD, Reaven GM. Insulin resistance and cigarette smoking. Lancet. 1992;339:1128–1130. doi:10.1016/0140-6736(92)91102-e
175. Narkiewicz K, van de Borne PJ, Hausberg M, et al. Cigarette smoking increases sympathetic outflow in humans. Circulation. 1998;98(6):528–534. doi:10.1161/01.cir.98.6.528
176. Oakes JM, Fuchs RM, Gardner JD, Lazartigues E, Yue X. Nicotine and the renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol. 2018;315(5):R895–R906. doi:10.1152/ajpregu.00099.2018
177. Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res. 2012;91(2):142–149. doi:10.1177/0022034511421200
178. Rehm J. The risks associated with alcohol use and alcoholism. Alcohol Res Health. 2011;34(2):135–143.
179. Shield KD, Rylett M, Gmel G, Gmel G, Kehoe-Chan TA, Rehm J. Global alcohol exposure estimates by country, territory and region for 2005–a contribution to the comparative risk assessment for the 2010 Global Burden of Disease Study. Addiction. 2013;108(5):912–922. doi:10.1111/add.12112
180. González K, Fuentes J, Márquez JL. Physical inactivity, sedentary behavior and chronic diseases. Korean J Fam Med. 2017;38(3):111–115. doi:10.4082/kjfm.2017.38.3.111
181. Martinez KB, Leone V, Chang EB. Western diets, gut dysbiosis, and metabolic diseases: are they linked? Gut Microbes. 2017;8(2):130–142. doi:10.1080/19490976.2016.1270811
182. De Filippis F, Pellegrini N, Vannini L, et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut. 2016;65(11):1812–1821. doi:10.1136/gutjnl-2015-309957
183. Calkins K, Devaskar SU. Fetal origins of adult disease. Curr Probl Pediatr Adolesc Health Care. 2011;41(6):158–176. doi:10.1016/j.cppeds.2011.01.001
184. Moosavi A, Motevalizadeh Ardekani A. Role of epigenetics in biology and human diseases. Iran Biomed J. 2016;20(5):246–258. doi:10.22045/ibj.2016.01
185. Kwon EJ, Kim YJ. What is fetal programming?: a lifetime health is under the control of in utero health. Obstet Gynecol Sci. 2017;60(6):506–519. doi:10.5468/ogs.2017.60.6.506
186. Alfaradhi MZ, Ozanne SE. Developmental programming in response to maternal overnutrition. Front Genet. 2011;2:27. doi:10.3389/fgene.2011.00027
187. Paknahad Z, Fallah A, Moravejolahkami AR. Maternal dietary patterns and their association with pregnancy outcomes. Clin Nutr Res. 2019;8(1):64–73. doi:10.7762/cnr.2019.8.1.64
188. Damasceno DC, Dallaqua B, Lovizutto Iessi I, Volpato GT, Campos KE. Impact of maternal over-nutrition during pregnancy on maternal oxidative stress and fetal skeletal/visceral anomalies of the rats. J Nutr Disorders Ther. 2016;6:1. doi:10.4172/2161-0509.1000185
189. Tzanetakou IP, Mikhailidis DP, Perrea DN. Nutrition during pregnancy and the effect of carbohydrates on the offspring’s metabolic profile: in search of the “perfect maternal diet”. Open Cardiovasc Med J. 2011;5:103–109. doi:10.2174/1874192401105010103
190. Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. 2001;60(5–20):26. doi:10.1093/bmb/60.1.5
191. Hay WWW
192. Desai M, Jellyman JK, Han G, Beall M, Lane RH, Ross MG. Maternal obesity and high-fat diet program offspring metabolic syndrome. Am J Obstet Gynecol. 2014;211(3):
193. Rkhzay-Jaf J, O’Dowd JF, Stocker CJ. Maternal obesity and the fetal origins of the metabolic syndrome. Curr Cardiovasc Risk Rep. 2012;6(5):487–495. doi:10.1007/s12170-012-0257-x
194. Perrone S, Tataranno ML, Santacroce A, et al. Fetal stress, maternal nutrition and oxidative stress hypothesis. J Pediatr Biochem. 2016;6(02):96–102. doi:10.1055/s-0036-1593811
195. Rodríguez-Rodríguez P, Ramiro-Cortijo D, Reyes-Hernández CG, López de Pablo AL, González MC, Arribas SM. Implication of oxidative stress in fetal programming of cardiovascular disease. Front Physiol. 2018;9:602. doi:10.3389/fphys.2018.00602
196. Thompson LP, Al-Hasan Y. Impact of oxidative stress in fetal programming. J Pregnancy. 2012;2012:582748. doi:10.1155/2012/582748
197. Mathers JC, McKay JA. Epigenetics - potential contribution to fetal programming. Adv Exp Med Biol. 2009;646:119–123. doi:10.1007/978-1-4020-9173-5_13
198. Malti N, Merzouk H, Merzouk SA, et al. Oxidative stress and maternal obesity: feto-placental unit interaction. Placenta. 2014;35(6):411–416. doi:10.1016/j.placenta.2014.03.010
199. Cetin I, Laoreti A. The importance of maternal nutrition for health. J Pediatr Neonat Individual Med. 2015;4(2):e040220.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.