Back to Journals » Biologics: Targets and Therapy » Volume 15
How Can We Engineer CAR T Cells to Overcome Resistance?
Authors Glover M ![]() , Avraamides S, Maher J
, Avraamides S, Maher J ![]()
Received 10 February 2021
Accepted for publication 19 April 2021
Published 19 May 2021 Volume 2021:15 Pages 175—198
DOI https://doi.org/10.2147/BTT.S252568
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Shein-Chung Chow
Maya Glover,1 Stephanie Avraamides,2 John Maher1– 4
1Leucid Bio Ltd., Guy’s Hospital, London, SE1 9RT, UK; 2King’s College London, School of Cancer and Pharmaceutical Sciences, Guy’s Hospital, London, SE1 9RT, UK; 3Department of Clinical Immunology and Allergy, King’s College Hospital NHS Foundation Trust, London, SE5 9RS, UK; 4Department of Immunology, Eastbourne Hospital, Eastbourne, East Sussex, BN21 2UD, UK
Correspondence: John Maher Tel +44 207 188 1468
Fax +44 207 188 0919
Email [email protected]
Abstract: Chimeric antigen receptor (CAR) T cell therapy has achieved unrivalled success in the treatment of B cell and plasma cell malignancies, with five CAR T cell products now approved by the US Food and Drug Administration (FDA). However, CAR T cell therapies for solid tumours have not been nearly as successful, owing to several additional challenges. Here, we discuss mechanisms of tumour resistance in CAR T cell therapy and the emerging strategies that are under development to engineer CAR T cells to overcome resistance.
Keywords: chimeric antigen receptor, cancer, immunotherapy, T-cell, resistance
Introduction
Chimeric antigen receptors (CARs) were first described over 30 years ago in which the variable regions of an immunoglobulin were fused into the constant regions of an αβ T cell receptor (TCR).1 Eshhar simplified this design through the use of a single-chain antibody fragment to direct target specificity, while an activating moiety such as CD3ξ was employed to deliver a cytotoxic signal.2 A key advance that resulted in clinical impact was the incorporation of co-stimulatory modules such as CD28 or 4–1BB within this basic framework.3 These so-called second generation (2G) CAR T cells achieved dramatic efficacy in relapsed/refractory patients with selected haematological malignancies. As a result, the US Food and Drug Administration (FDA) has approved four CD19-specific and one B cell maturation antigen (BCMA)-specific CAR T cell products for the treatment of B cell and plasma cell malignancies, respectively.4–8 Despite this success and continued research, the development of CAR T cell therapies for solid tumours continues to prove challenging with only sub-optimal efficacy achieved to date. Solid tumours deploy a unique set of obstacles that enable them to resist CAR T cell function and anti-tumour response. To address this, it is necessary to consider how we can engineer CAR T cells to overcome the hostile tumour microenvironment (TME) whilst avoiding immune pressure induced resistance, summarised in Table 1. In the sections that follow and are illustrated in Figure 1, we will consider a variety of strategies that have demonstrated promise in the quest for more resistant CAR T-cells.
 |
Table 1 Summary of Mechanisms of Tumour Resistance and How CAR T Cells Can Be Engineered to Overcome Resistance |
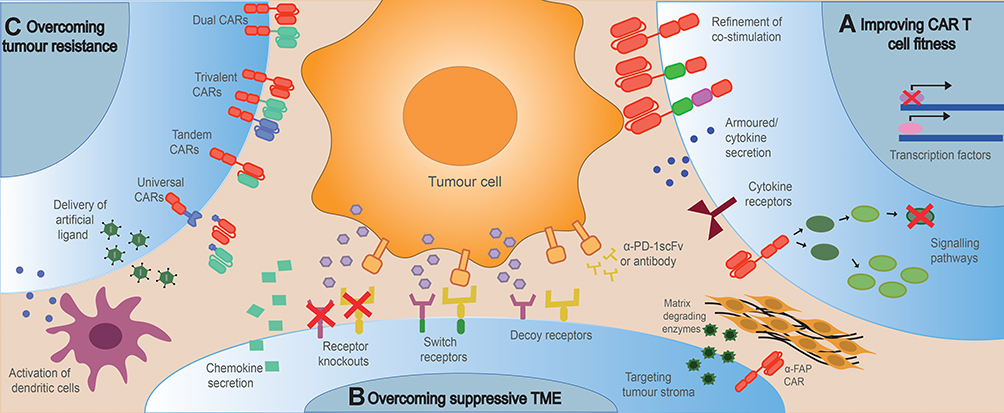 |
Figure 1 High level overview of strategies to engineer resistance in CAR T-cells. Tumour resistance to CAR T cell therapy can be overcome by (A) improving CAR T cell fitness to enhance proliferation, persistence and cytotoxicity; (B) engineering CAR T cells to resist the suppressive TME including checkpoint inhibitors, immunosuppressive cells and cytokines and the tumour stroma, whilst enhancing tumour homing and infiltration; (C) Engineering CAR T cells to overcome tumour resistance caused by antigen loss or downregulation by promoting epitope spreading, targeting multiple antigens or introducing artificial ligands to the tumour. |
Refinement of CAR Binding and Spacer Domain
Many CARs bind to antigen using single-chain antibody fragments (scFvs), comprising a variable heavy and variable light chain joined by a flexible linker. Selection of scFvs with appropriate affinity is important to avoid on-target off-tumour toxicities. Low-affinity ErbB2 (HER2) or epidermal growth factor receptor (EGFR) targeting CAR T cells can distinguish between healthy cells expressing low level of antigen and tumour cells expressing high level of antigen without compromising T cell activation.9–11 Similarly, a humanised folate receptor-alpha (αFR) specific scFv with reduced affinity for target antigen compared to the parental murine scFv could distinguish between high and low-density tumour antigen in vitro and in vivo.12 Selection of low-affinity scFvs has also improved CAR T cell function in some cases.13,14 Lower affinity was associated with a faster off-rate and increased serial killing of tumour cells, leading to enhanced function in vitro and in vivo.13 In addition to optimisation of affinity, the selection of humanised or fully human scFvs (rather than traditionally used murine variants) is preferred in order to prevent anti-CAR immune responses, as discussed in later sections.15 One concern with the use of scFvs as CAR binding domains is their frequent propensity to oligomerise. As a result, CAR clustering may occur, leading to tonic signalling and early exhaustion.16 Nanobodies represent an alternative to scFvs which may reduce the risk of unwanted oligomerisation, as they consist of a single heavy chain only.17 Furthermore, due to their small size, nanobodies can engage with some epitopes that are inaccessible to scFvs.18 Alternative binding domains include naturally occurring tumour-sensing receptors such as natural killer (NK) cell receptors,19 modified cytokines such as IL-13 mutein20 or the T1E panErbB ligand,21 and integrin-targeting peptides such as A2022
The spacer/hinge domain connects the binding domain and the transmembrane domain within a chimeric antigen receptor. Many CAR constructs utilise spacer/hinge regions derived from one of the four human IgG subclasses, or from CD8α and CD28. In the case of IgG-derived sequences, mutation of residues involved in Fc receptor engagement may be desirable to mitigate unwanted interactions.23 The selection of an appropriate spacer domain of optimal length is important for efficient binding of ligand, and is dependent on the position of the target epitope on the tumour surface.24 For example, incorporation of the highly flexible, elongated IgD hinge improved CAR-mediated recognition of the aberrantly glycosylated tumour target, Muc1.25 The use of different spacer domains may also affect the intensity of CAR T cell activation. Illustrating this, CD19 CARs containing the spacer and transmembrane domain from CD28 secreted lower cytokine levels and underwent reduced activation-induced cell death (AICD) compared with those containing the spacer and transmembrane domain of CD8α,26 although the influence of changing the spacer domain only was not evaluated in this study.26
Refinement of Co-Stimulation
Approved 2G CAR T cells therapies contain the co-stimulatory domains CD28 (Yescarta, Tecartus) or 41BB (Kymriah, Breyanzi, Abecma).4,5 Although these therapies provide comparable efficacy, the cell products demonstrate quite different kinetics of anti-tumour activity.27,28 When compared to 41BB-containing counterparts, CD28 CAR T cells undergo early expansion and mediate rapid initial tumour cell killing, exemplified by in vivo stress test models.27 Faster activation kinetics of CD28 containing CAR T cells was associated with increased phosphorylation of CAR CD3ζ, Lck, ZAP-70 and LAT following in vitro activation.29 Early phosphorylation events ultimately lead to higher Ca2+ influx, CD69 expression and increased IL-2 and interferon (IFN)-γ secretion, resulting in faster cytotoxic responses.27,29 Rapid activation following antigen exposure of CD28 CAR T cells may be dependent on basal phosphorylation of Lck in these T cells.29,30 A consequence of the superior initial activation of CD28 CARs is early exhaustion leading to poor persistence and diminished long-term anti-tumour function.31 Guedan et al31 reversed excessive activation and improved the persistence of CD28 CAR T cells by mutating a single amino acid within the CD28 endodomain. The YMNM motif of CD28 binds to SH2 containing proteins, including Grb2 and phospholipase C (PLC)γ1, activating downstream signalling pathways. Mutation of the asparagine (N) in this motif to phenylalanine (F) disrupts this interaction, reducing Ca2+ influx and NFAT activation, protecting cells from premature exhaustion, yet maintaining lytic capacity.31 These authors confirmed improved function following a single acid mutation by using mesothelin targeting CARs which demonstrated superior efficacy in two in vivo pancreatic cancer models.31 CD28 also contains a PRRP motif, which binds ITK, activating PLC-γ and ERK signalling pathways, and a PYAP motif, which binds Lck.32 Boucher et al32 also investigated the effect of mutating these signalling motifs in order to reduce CAR T cell exhaustion. CD19 targeting CAR T cells in which both the CD28 YMNM and PRRP motifs were mutated while the PYAP motif remained intact demonstrated superior persistence and reduced exhaustion in a B-ALL mouse model, demonstrating that reduction of CD28 signalling was beneficial in this model.32 In an alternative approach, Sun et al29 proposed that recruitment of phosphatases to the CAR would decrease early phosphorylation events, preventing excessive activation and the resulting early exhaustion. To achieve this, a FK506 binding protein (FKBP) rapamycin binding (FRB) domain was introduced into a CD28 CAR and was expressed alongside SHP1 phosphatase containing a FKBP domain. Following the addition of an inert rapalog, SHP1 was recruited to the CAR via FRB-FKBP binding, reducing CAR phosphorylation upon activation without compromising anti-tumour activity of a CD19 targeting CAR against an in vivo lymphoma xenograft model.29 Reduced exhaustion of CD28 CAR T cells was also achieved by integrating the CAR cDNA into the T-cell receptor α constant (TRAC) locus via CRISPR/Cas9 editing. This allowed TRAC promoter-regulated CAR expression, internalisation and re-expression following antigen encounter and resulted in CAR T cells with a less differentiated phenotype and reduced exhaustion.33
In contrast to CD28 variants, 41BB-containing CAR T cells demonstrate slower anti-tumour kinetics, yet improved persistence.27,28,34 Furthermore, 41BB CAR T cells are generally more resistant to exhaustion due to reduced tonic signalling, and exhibit a more favourable metabolic profile and less differentiated state.34 Modification of 41BB CAR T cells has been attempted to improve their initial activation kinetics but still maintain their longevity. Overexpression of Lck in 41BB CAR T cells counteracts the recruitment of SHP1 phosphatase, leading to increased CAR CD3ζ phosphorylation and Ca2+ influx.29 Lck overexpression improved initial expansion following antigen exposure without inducing an exhausted phenotype and improved the efficacy of GD2 targeting CAR T cells against an in vivo neuroblastoma model.29 Recently, it has been shown that 41BB-containing CARs continue to signal in endosomes following antigen encounter and internalisation.35 Mutation of intracellular lysine residues within the CAR prevents ubiquitination and 41BB signalling is thereby enhanced via colocalization with TRAF2 in endosomes, increasing signalling by mammalian target of rapamycin (mTOR), mitochondrial oxidative phosphorylation and memory cell differentiation.35
Harnessing the signalling capacities of both CD28 and 41BB has been attempted by placing both costimulatory domains upstream of CD3ζ in third generation (3G) CAR T cells. Some studies have demonstrated the superior efficacy of CD28 and 41BB containing 3G CAR T cells compared to their 2G counterparts with improved expansion, cytokine production and anti-tumour function.36–39 However some studies only show borderline superiority and even inferior activity compared to 2G CARs.28,40,41 Furthermore, clinical success of 3G CAR T cells has been limited. Despite this, the addition of a further costimulatory domain has been attempted with 4G CAR T cells consisting of 41BB, CD28 and CD27.42 Combining CD28 and 41BB signalling in other configurations has also been attempted. For example, the expression of 41BB or 41BBL alongside a CD28 2G CAR improves proliferation, persistence and anti-tumour efficacy compared to their 2G and 3G counterparts in both in vitro and in vivo models.27,43 Alternatively, in an attempt to improve CAR safety and specificity, a construct containing a first generation (1G) CAR expressed alongside a chimeric co-stimulatory receptor (CCR) containing CD28 and 41BB was engineered.44 Although this CAR construct provides a safety mechanism, as both CAR and CCR engagements are necessary for T cell activation, signalling by both CD28 and 41BB elements within the fused CCR was not demonstrated.
Second-generation CARs utilising co-stimulatory molecules other than CD28 and 41BB have also been investigated. Incorporation of inducible T cell co-stimulator (ICOS) resulted in Th1/Th17 polarisation of CAR T cells with increased IFN-γ, IL-17 and IL-22 secretion compared to both 41BB and CD28 CAR T cells.45 Furthermore, ICOS CAR T cells demonstrated enhanced persistence in an in vivo model of non-small cell lung cancer.45 Unlike CD28 CAR T cells, CAR T cells containing OX40 co-stimulation do not secrete IL-10 and yet secrete comparable levels of pro-inflammatory cytokines.46 Functional studies demonstrating enhanced function of 2G OX40 CAR T cells are lacking and studies investigating 3G OX40/CD28 CAR T cells have shown contrasting results of either enhanced or diminished cytokine secretion.47,48 Similar to 41BB, incorporation of CD27 in CAR designs led to enhanced persistence and survival in vivo compared to CD28 CAR T cells.49 The addition of either inducible or constitutively active MyD88 and CD40 has also resulted in enhanced performance of CAR T cells.50–52 More recently, Prinzing et al53 found that MyD88/CD40 CAR T cells demonstrated improved in vitro proliferation and restimulation ability while remaining in a less differentiated state compared to both CD28 and 41BB containing CAR T cells.53 The structural components of CAR T cells that can be refined to enhance function are illustrated in Figure 2.
 |
Figure 2 Structural components of CAR T cells. The structure of a first generation CAR consists of an antigen binding domain attached to a transmembrane domain via a hinge/spacer, followed by a CD3ξ signalling domain. Second generation CARs have an additional co-stimulatory domain and third generation CARs contain two co-stimulatory domains. Fourth generation CARs are armoured to secrete cytokines. All structural components of CAR T cells illustrated can be refined to enhance function. |
Armoured (Cytokine-Expressing) CAR T Cells
Engineering CAR T cells to secrete cytokines that enhance proliferation and effector function (known variously as “armoured CARs” or “TRUCKs”) represents another strategy that may be used to boost resistance of these cells (Figure 2). Several cytokines have been used in this manner as summarised below and in Table 2. Interleukin (IL)-12 was one of the first cytokines to be constitutively expressed alongside a CAR, in order to promote resistance to the immunosuppressive TME.54–56 Concerns over toxicity related to constitutive cytokine production led to the engineering of CAR T cells in which IL-12 transcription was placed under the control of the NFAT promoter. By this means, IL-12 production was rendered dependent on CAR T cell activation, meaning that this toxic cytokine should remain localised to the TME.57,58 Although studies on both immunodeficient and immunocompetent mouse models demonstrated control of IL-12 secretion, toxicity was seen in a clinical trial in which ex vivo expanded tumour-infiltrating lymphocytes (TILs) were armoured with inducible IL-12, suggesting possible leakiness of NFAT promoter activity.59 In an alternate approach, exemplified by Sachdeva et al,60 the IL-12 gene was placed under the IL-2Rα or PDCD1 regulatory elements in order to couple cytokine secretion to CAR activation.60 Armouring CAR T cells with IL-12 can also induce accumulation of activated macrophages, as demonstrated in an immune competent mouse model, resulting in the ability to target antigen-negative tumour cells.57,61
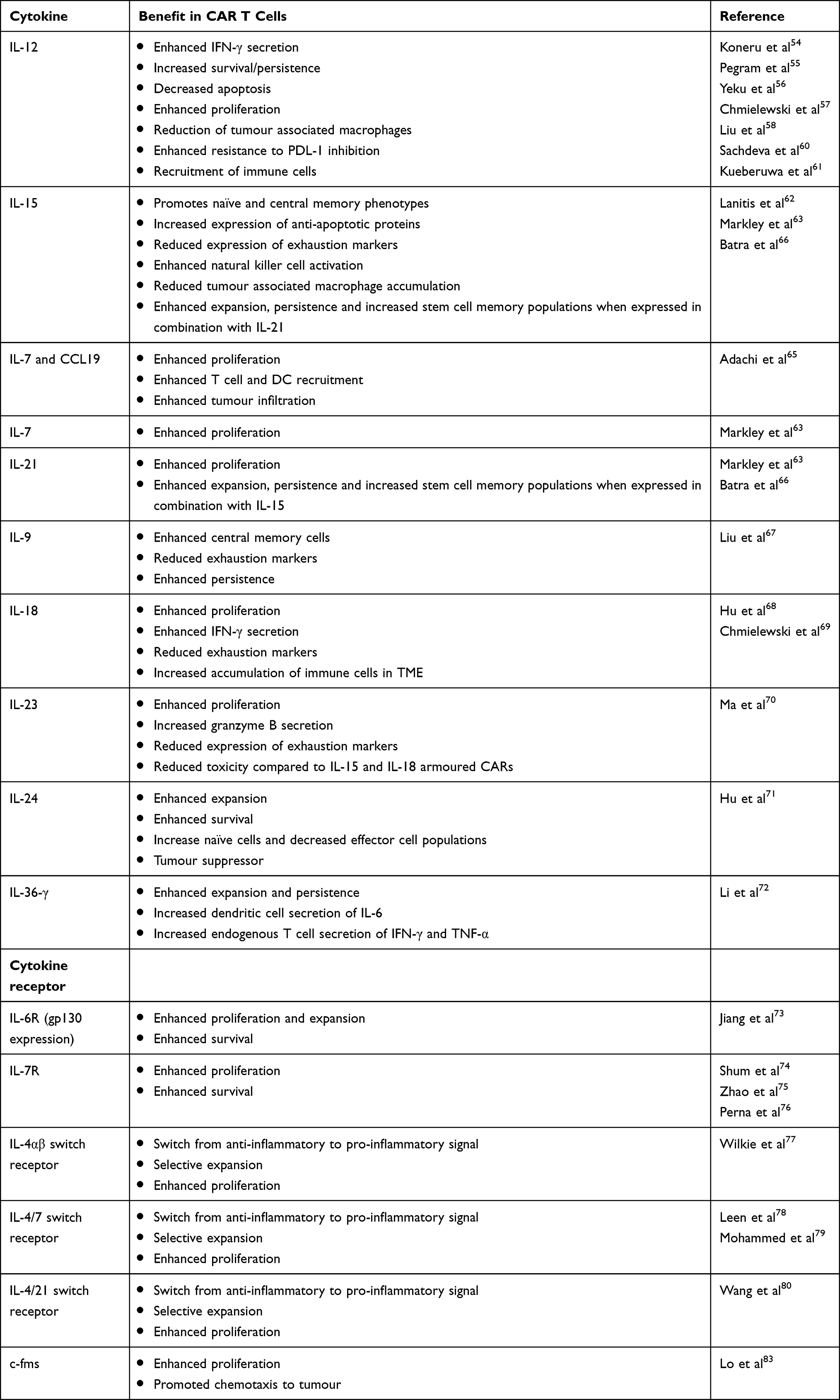 |
Table 2 Summary of Armoured CAR T Cells |
Armouring CARs with constitutively expressed IL-15 can also augment effector functions and enhance fitness. When tested in vitro and in immunocompetent mouse models, IL-15 maintained naïve and central memory T-cell phenotype, with increased expression of the anti-apoptotic protein Bcl-2 and reduced PD-1 expression.62,63 Similar results were observed when IL-15 is tethered to the CAR T cell membrane, an approach that also prevents unwanted bystander cell activation.64 Adachi et al65 engineered CAR T cells to co-express both IL-7 and CCL19 to promote T cell and dendritic cell (DC) recruitment and tumour infiltration in an immunocompetent mouse model. Secretion of IL-7 and CCL19 from CAR T cells improved anti-tumour responses in solid tumour mouse models with increased overall survival. Furthermore, mice treated with IL-7 and CCL19 secreting CAR T cells could eradicate both antigen positive and antigen-negative tumour rechallenge, indicating the development of a robust memory response and epitope spread.65 Markley et al63 set out to identify the optimal common gamma cytokine support for CD19 targeting CAR T cells by the constitutive co-expression of IL-2, IL-7, IL-15 or IL-21 in these cells. Evaluation was performed in a systemic B-cell lymphoma model in immunodeficient mice. They demonstrated that IL-7 and IL-21 armoured CAR T cells achieved superior anti-tumour responses compared to those producing IL-15 and IL-2, a finding that was suggested to depend on enhanced proliferation but not enhanced lytic activity of CAR T cells. Furthermore, no long-lived cells could be identified following IL-7 armoured CAR T cell treatment and only T effector memory cells could be identified in IL-21 armoured CARs in the mouse models with complete responses.63 When tested on a hepatocellular carcinoma xenograft model in immunodeficient mice, the combined constitutive co-expression of both IL-15 and IL-21 in CAR T cells resulted in superior expansion, persistence and anti-tumour efficacy, associated with increased stem cell memory and central memory populations, when compared to single cytokine expression.66 Similarly, IL-9 expression in CAR T cells also enhances the pool of central memory cells, with lower expression of exhaustion markers and enhanced in vivo persistence.67
The addition of constitutively active or inducible IL-18 to CAR T cells can also improve anti-tumour function with enhanced proliferation, IFN-γ production and a reduction in markers of exhaustion compared to IL-12 secreting CAR T cells.68,69 Furthermore, IL-18 secretion from CAR T cells engaged the local immune response in immunocompetent tumor-bearing mice by inducing an increase in M1-polarised macrophages and NK cells and a decrease in T regulatory cells (Tregs), suppressive DCs and M2-polarised macrophages within the TME.69 Alternatively, engineering CAR T cells to secrete IL-23 demonstrated reduced toxicity compared to constitutive expression of IL-15 and IL-18, with enhanced effector function demonstrated in both xenograft and syngeneic immunocompetent solid tumour mouse models.70 To induce CAR T cells to secrete IL-23, cells were engineered to express the IL-23 p40 subunit, which unlike the IL-23α p19 subunit and the IL-23R, is not upregulated upon T cell activation. The secretion of IL-23 was therefore rendered dependent on CAR T cell activation.70 In another approach, armouring CAR T cells with IL-24 has recently been shown to improve cytotoxicity, expansion and survival, with increased naïve cell and reduced effector cell populations in vitro with no signs of toxicity in immunodeficient mice.71 Furthermore, IL-24 acts as a tumour suppressor in many cancer types, promoting the therapeutic activity of IL-24 armoured CAR T cells.71 IL-36-γ armoured CARs have also been shown to mediate enhanced anti-tumour activity by engaging endogenous immune responses. IL-36-γ CAR T cells increase IL-6 production by DCs and IFN-γ and tumour necrosis factor (TNF)-α release by endogenous T cells, which could then eradicate a secondary challenge with antigen-negative tumour cells.72
In summary, many cytokines have now been tested for their ability to enhance CAR T cell anti-tumour responses. Further studies are necessary to compare and identify the best cytokine or combination of cytokines to armour CAR T cells.
Cytokine Receptor Expression in CAR T Cells
An alternative strategy to improve CAR T cell expansion and function involves the introduction of cytokine receptors into these cells. Jiang et al73 engineered CAR T cells to express a constitutively active form of gp130, a membrane protein which induces IL-6 trans signalling following the binding of IL-6/soluble IL-6 receptor (sIL-6R). Compared to a synthetic IL-6/sIL-6R complex, termed hyper IL-6, the constitutively active gp130 similarly enhanced CAR T cell function but without induction of GvHD in immunodeficient mouse models.73 Expression of constitutively active IL-7R enhanced CAR T cell proliferation, survival and anti-tumour response in immunodeficient mouse models, even when Tregs were co-infused.74–76 The concern of autonomous cell expansion with constitutively active cytokine receptor expression can in principle be circumvented by using switch chimeric cytokine receptors.77,78 CAR T cells have been engineered to respond to the immunosuppressive cytokine IL-4 by fusing the IL-4Rα ectodomain to the βc subunit of IL-2 and IL-15 receptor,77 the IL-7 receptor endodomain,78,79 or IL-21 receptor endodomain.80 Using these chimeric receptors, tumour derived IL-4 is harnessed to deliver a positive signal, increasing proliferation and anti-tumour response. Chimeric cytokine receptors are also useful for enhancing the expansion and selective proliferation of CAR T cells during the manufacture of cell products.77,81,82 Similarly, CAR T cells have been engineered to respond to tumour derived colony-stimulating factor (CSF)-1.83 Expression of c-fms, the CSF-1 receptor, in CAR T cells not only enhanced proliferation but also promoted chemotaxis in response to CSF-1.83
Manipulation of Signalling Pathways in CAR T Cells
Targeting distinct pathways in T cell signalling can influence effector function and differentiation of CAR T-cells. Cytokine signalling pathways can be enhanced by incorporating signalling domains directly into the CAR structure. Kagoya et al84 incorporated the IL-2Rβ domain which mediates STAT5 activation, alongside the YXXQ STAT-3 motif into 2G CARs.84 The addition of these domains resulted in superior persistence, proliferation and anti-tumour efficacy compared to matched 2G counterparts due to enhanced JAK/STAT signalling, in both liquid and solid tumour models in NSG mice.84 In a related approach, the expression of constitutively active STAT5 improved anti-tumour activity of CD19 CAR T cells, accompanied by enhanced persistence demonstrated in a mouse B cell lymphoma model.85 The phosphatase PTPN2 dephosphorylates multiple components of the JAK/STAT pathway inhibiting cytokine signalling. Engineering of PTPN2-deficient CAR T cells targeting ErbB2 resulted in enhanced cytotoxicity, tumour homing and tumour eradication when infused into lymphodepleted mice bearing ErbB2+ mammary tumours.86
The phosphoinositide 3-kinase (PI3K)/Akt signalling pathway is activated following engagement of the TCR and its coreceptors and promotes cell survival, glycolysis and cytokine synthesis.87 Within the TME, however, suppressive cytokines and molecules activate phosphatases which counteract Akt signalling.88 To overcome this, Akt has been overexpressed in CAR T cells which increased tumour cell killing both in vitro and in vivo models.89 Similar results were observed when a constitutively active Akt protein was expressed in CAR T cells.90 However, the sustained activation of Akt in CD8 T cells promotes terminal differentiation.91 Inhibiting Akt during ex vivo expansion of CAR T cells improved anti-tumour responses of CD19 CAR T cells in tumour-bearing immunodeficient mice, without reducing cell yield.91–93 Furthermore, Akt inhibition increases memory cell populations by promoting localisation of the transcriptional regulator of T cell memory, FOXO1, to the nucleus.93 Similarly, manipulating the PI3K/Akt signalling pathway further upstream by pre-treatment of CD33 targeting CAR T cells with a PI3K inhibitor prevented differentiation ex vivo, which enabled enhanced in vivo persistence in immunodeficient mice bearing CD33+ tumour cells.94 Careful manipulation of the PI3K/Akt pathway is necessary to engineer CAR T cells with favourable characteristics. Inhibition of Akt during manufacturing helps to prevent terminal differentiation. However, Akt expression by CAR T-cells is important when they are located within the TME to enhance effector responses.
The complexity of T cell signalling makes it challenging to identify targets to engineer CAR T cells with enhanced fitness and anti-tumour immunity. More recently CRISPR-Cas9 technology has been used to thoroughly screen potential therapeutic targets to improve T cell therapy. Gurusamy et al95 genetically edited 25 T cell receptor-driven kinases and monitored phenotypes related to T cell fitness, including cell expansion, differentiation, oxidative stress and genomic stress.95 The MAP kinase p38α was identified as a target which, when inhibited in CAR T cells, led to improved anti-tumour response in immune competent mouse tumour models.95 In an independent study, a CRISPR-Cas9 screen was combined with a screen of >500 small molecule drugs to identify mechanisms to improve CAR T cell cytotoxicity.96 FADD and TRAIL-R2 were identified as important components for the induction of cancer cell apoptosis which could be sensitised through treatment with SMAC mimetics.96 Using these methods to screen for important components of CAR T cell signalling to improve fitness and cytotoxicity will allow us to identify ways to engineer more efficacious CAR T cells to overcome resistance.
Manipulation of Transcription Factors in CAR T Cells
Targeting transcription factors in CAR T cells has proven a useful tool to improve fitness. Lynn et al97 demonstrated that tonic signalling from a GD2 targeting CAR caused dysregulation of AP-1 transcription factor family members with increased expression of immunoregulatory components, BATF, JunB and IRF4.97 Engineering CAR T cells to overexpress c-Jun shifts the balance in favour of the formation of proinflammatory c-Jun/c-Fos heterodimers, resulting in reduced terminal differentiation, increased expansion and improved anti-tumour responses against both solid tumour and leukemic xenograft models.97 NFAT is a transcription factor associated with T cell activation and effector responses, but also contributes to T cell exhaustion and tolerance.98 Only when NFAT is bound to AP-1 can it promote the expression of activating genes, which is likely the mechanism of improved function when CAR T cells are engineered to overexpress c-Jun.97,98 Furthermore, exhaustion of CD19 targeting CAR T cells has been associated with the activity of three NR4A family transcription factors (NR4A1, NR4A2, NR4A3) which are regulated by NFAT.99 Specifically, NR4A1 promotes T cell exhaustion by not only inducing tolerance-related genes but also by competing for AP-1 binding sites, thereby preventing transcription of activating genes.100 Triple knockout of the NR4A transcription factors in CAR T cells eliminated exhausted phenotypes and enhanced anti-tumour responses in a solid tumour xenograft model.99 Another NFAT driven transcription factor family, TOX, has similar functions to NR4A transcription factors in promoting T cell exhaustion, and these two transcription factor families can positively regulate the expression of each other.101,102 CAR TILs engineered to no longer express two TOX transcription factors (TOX and TOX2) also achieved improved in vivo anti-tumour responses with reduced expression of inhibitory receptors.101
Improving T Cell Fitness During Manufacturing of CAR T Cell Therapy
Most therapeutic CAR T cells are manufactured as autologous products using patient-derived leukapheresis. Despite the success of autologous CAR T cell therapy in the clinic, the manufacture of these products has encountered many difficulties.103 An adequate number of T cells must be harvested to ensure sufficient starting material for manufacture. However, the quantity and quality of patient-derived cells are both often compromised due to disease burden and/or prior treatment.103–105 To ensure sufficient CAR T cell expansion, cytokines are used during the manufacturing process, most commonly IL-2. However, supplementation with IL-7/IL-15 has been shown to induce superior activation and expansion with increased populations of CAR T cells with naïve and stem cell memory phenotypes.106–108 Alternatively, CAR T cells have been engineered to secrete cytokines to promote their expansion and survival, as discussed above. Similarly, CAR T cells can be engineered to co-express switch receptors that also promote selective expansion during manufacture in response to cognate ligand.58,62,63
Manufacture of autologous CAR T cells is often lengthy, costly and can be subject to compromise by patient T cell dysfunction. Moreover, products are not immediately available at the time of clinical need. To address these shortcomings, allogeneic “off the shelf” CAR T cell products are under active development, whereby a single cryopreserved batch of drug contains multiple dosing units.103 Indeed, allogeneic CD19 CAR T cells have been tested in clinical trials in patients with B cell malignancies with some success.109,110 However, the development of graft-versus-host disease (GVHD) remains of concern when using allogeneic cell products. CAR constructs containing 41BB co-stimulatory modules have elicited increased GVHD compared to CD28-containing counterparts in lymphoma mouse models, possibly due to excessive activation of CD28 CAR T cells resulting in cell death, whereas 41BB CAR T cells have superior persistence.111 To prevent GVHD, allogeneic CAR T cells can be edited to knock out αβ TCR chains without compromising CAR T cell efficacy.33,112–115 Alternatively, allogeneic CAR engineered cell products can be derived from virus-specific T-cells (www.atarabio.com, accessed April 2nd, 2021), NK cells, γδ T cells, invariant NK T cells, macrophages or induced pluripotent stem cells.116–122
Improving CAR T Cell Homing and Infiltration of Solid Tumours
The success of CAR T cell therapy in liquid tumours can partially be attributed to the high probability of tumour cell-CAR T cell interaction in circulation. By contrast, CAR T cells must migrate to and infiltrate solid tumours in order to achieve therapeutic success in that setting. In selected cases, CAR T cell therapy can be delivered intratumorally or using a regional delivery approach.123,124 However, many solid tumours are inaccessible to intratumoral injection, owing to location, proximity to vital structures and/or metastatic spread. To direct homing of CAR T cells to specific tumour types, a suitable chemokine receptor may be co-expressed in these cells. The chemokine CXCL8 is produced by tumour cells and chemoattracts neutrophils and myeloid-derived suppressor cells which naturally express the receptors, CXCR1 and 2.22,100,125 CAR T cells engineered to express these chemokine receptors show improved homing to tumours and enhanced tumour regression in solid tumour models in immunodeficient mice.22,125,126 CAR T cells engineered to express CCR2b or CCR4 also demonstrate improved trafficking to CCL2-expressing or CCL17- and CCL22-expressing tumour cells, respectively.127,128 However, it should be noted that excessive secretion of some chemokines (eg CXCL12) can inhibit T cell migration.129–131 Pharmacological inhibition of the chemokine receptor CXCR4 in combination with checkpoint inhibition led to improved T cell homing to CXCL12 secreting tumours in a mouse model of pancreatic ductal adenocarcinoma.132 Furthermore, in a proof-of-concept study, patients who previously resisted immunotherapy were treated with CXCR4 inhibitor and showed increased immune cell infiltration into tumours.131 Engineering CAR T cells to be deficient in CXCR4 may also enhance trafficking to tumours.
The tumour stroma consists of fibroblasts, immune cells, extracellular matrix and vasculature and forms a physical, chemical and biological barrier to immune cell infiltration whilst supporting tumour growth. Fibroblast activation protein (FAP) is expressed on cancer-associated fibroblasts (CAFs) and has been targeted in an attempt to improve CAR T cell infiltration. FAP targeting CAR T cells inhibit the growth of subcutaneous solid tumours in immunocompetent mouse models by targeting FAP+ stromal cells, whilst increasing endogenous CD8 T cell infiltration.133 Administration of anti-FAP CAR T cells in combination with CAR T cells that target tumour-associated EphA2 achieved improved therapeutic efficacy in immunodeficient mice bearing a lung tumour xenograft, when compared to treatment with either CAR T cell population alone.134 Production of a dual antigen targeted CAR that incorporates specificity for FAP and an additional target could circumvent the need for multiple infusions. One concern with CARs targeting FAP is the potential for on-target/off-tumour toxicity as FAP is widely expressed in healthy tissues and has important functions in wound healing. In keeping with this, some but not all studies have shown that anti-FAP CAR T cells can cause bone marrow toxicity in mouse models.135
The tumour extracellular matrix (ECM) has also been targeted to improve CAR T cell penetration in solid tumours. Illustrating this, CAR T cells that have been engineered to secrete heparanase induces degradation of heparan sulfate proteoglycans (HSPG) in the ECM. Improved infiltration and anti-tumour response by heparinase-expressing CAR T cells was demonstrated in immunodeficient mice implanted with xenograft models of neuroblastoma.136
Overcoming Immune Checkpoints in CAR T Cells
Tumours and associated stromal cells express immune checkpoints that decrease CAR T cell function.137,138 The checkpoint inhibitors, ipilimumab (anti-CTLA4), nivolumab and pembrolizumab (anti-PD-1) are frequently used to treat cancer patients and are under investigation for use in combination with CAR T cell therapy.139–142 To avoid the need for separate dosing, CAR T cells can be engineered to circumvent immune checkpoints as summarised in Table 3. Secretion of anti-PD-L1 antibodies or scFvs by CAR T cells improved anti-tumour responses with reduced expression of exhaustion markers in multiple mouse models including a humanised mouse model of renal cell carcinoma and a patient-derived xenograft (PDX) model of gastric cancer.143–147 In an alternative strategy, a dominant negative PD-1 receptor has been designed, consisting of PD-1 lacking the intracellular domain, and expressed in CAR T cells. The dominant negative receptor can outcompete endogenous receptors for PD-L1 on tumours.148 Genetic elimination of PD-1 has been achieved by CRISPR/Cas9 technology in CAR T cells, which were engineered alongside endogenous TCR and β2 microglobulin (B2M) knockdown to produce checkpoint resistant, allogeneic CAR T cells.149–151 PD-1 has also been downregulated using small hairpin (sh)RNA in CAR T cells to improve anti-tumour responses.148,152 Combined knockdown of immune checkpoints has been investigated by Zou et al,153 where PD-1 as well as Tim-3 and Lag-3 were downregulated by shRNA in CAR T cells. This triple knockdown approach led to enhanced tumour infiltration and superior efficacy in a xenograft model that was dependent on increased CD56 expression.153 PD-1 knockdown combined with CTLA-4 knockdown using small interfering (si)RNAs however, has not shown much improvement compared to PD-1 knockdown alone.154 Additionally, shRNA knockdown of CTLA-4 alone has shown no significant effect in CD28 2G CAR T cells.155 CAR T cells secreting anti-CTLA-4 minibodies, however, improved control of tumour growth in a glioma xenograft model compared to control CAR T cells, whereas CAR T cells secreting anti-PD-1 or anti-Tim-3 minibodies showed no improvement.156
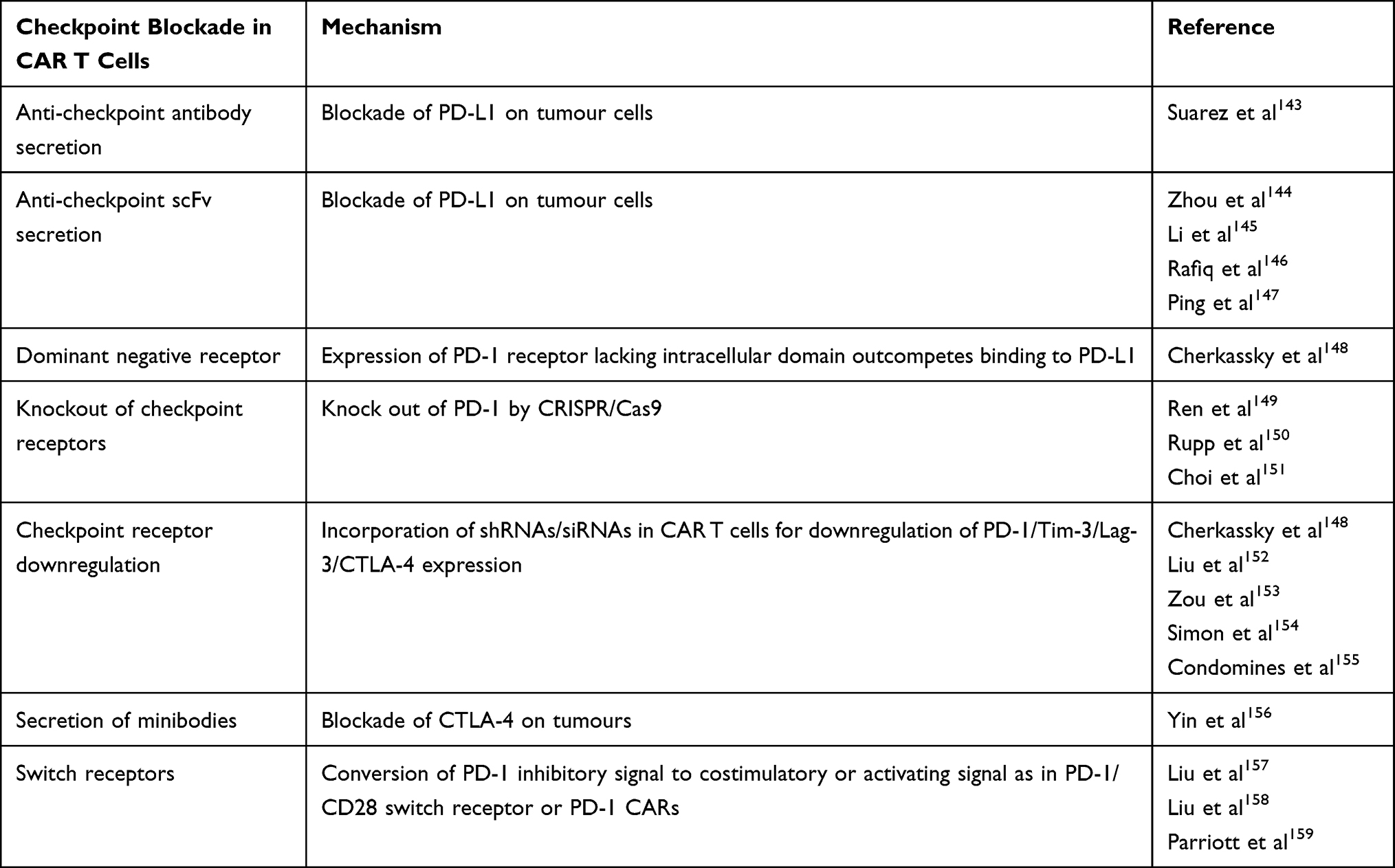 |
Table 3 Summary of Techniques to Overcome Checkpoint Blockade by CAR T Cells |
An alternative approach to overcome immune checkpoints involves the construction of a switch receptor. Illustrating this, the extracellular domain of PD-1 was fused to the intracellular domain of CD28, thereby converting a potentially inhibitory signal to an activating signal. Such a PD-1 switch receptor was co-expressed with 1G and 2G CARs, leading to improved cytokine secretion and tumour killing of solid tumour xenograft model and in a Phase 1 clinical trial treating B cell lymphoma.157,158 Similarly, sources of either signal one (eg CD3ξ) or signal two (eg DAP10) have been fused into the extracellular domain of PD-1 producing an anti-PD-1 CAR which can target a range of tumour types.159
Overcoming Immunosuppressive Cells and Cytokines Within the TME
The tumour microenvironment includes a complex population of immunoinhibitory cells including Tregs, tumour associated macrophages (TAMs), myeloid-derived suppressor cells (MDSC), and DCs. These cells inhibit optimal CAR T cell function by producing inhibitory cytokines, enzymes and immunoinhibitory proteins. Patients are generally lymphodepleted before CAR T cell treatment to support expansion of the infused cells by removing inhibitory cell populations and increasing the pool of available activating cytokines.160 Without lymphodepletion, CAR T cell production of IL-2 can enhance Treg function and expansion. Suryadevara et al161 produced a CAR T cell with decreased IL-2 secretion potential by mutating the PYAP motif in CD28, preventing Lck binding. These CAR T cells were able to resist the inhibitory effects of Tregs in the TME of non-lymphodepleted mice; however, anti-tumour function was dependent on additional 41BB signalling.161
The inhibitory cytokine transforming growth factor (TGF)-β is not only produced by Tregs in the tumour microenvironment but also by tumour cells and stromal cells. 2G CARs containing CD28 rather than 41BB are more resistant to the inhibitory effects of TGF-β in a manner that is dependent on increased IL-2 signalling, in contrast to Treg resistance.162 The introduction of cytokine autocrine loops into CAR T cells can confer resistance to TGF-β. This was elegantly demonstrated in CD28 2G CAR T-cells that lacked an Lck binding motif and therefore did not produce IL-2, but in which a hybrid IL-7 receptor delivered IL-2 signalling.162 The functionality of a dominant negative TGF-β receptor, consisting of a receptor lacking intracellular signalling domains, has also been demonstrated to enhance anti-tumour responses of both tumour-specific CTLs and CAR T cells163–165 Introduction of the dominant negative receptor in prostate-specific membrane antigen targeting CAR T cells can enhance proliferation, cytokine secretion, persistence and tumour eradication in a metastatic prostate cancer mouse model.166 In a proof-of-principle study demonstrating CAR activation by soluble antigen, a second generation anti-TGF-β CAR was produced using scFvs with specificity for this cytokine.167 The CAR not only functions as a switch receptor by converting an inhibitory signal into an activating signal but also works as a dominant negative receptor by inhibiting endogenous TGF-β signalling.167 Subsequently, it was demonstrated that CD4+ T cells transduced with the TGF-β CAR enhanced in vitro anti-tumour responses of anti-CD20 CAR T cells in the presence of TGF-β.168 The investigators demonstrate that the expression of the anti-TGF-β CAR in Tregs did not enhance their suppressive effects or inhibit CD19-CAR T cell effector response. However, a concern is that enrichment of transduced Tregs in a CAR T cell product may occur via the induction of a TGF-β autocrine loop.168 More recently, CRISPR/Cas9 technology has been used to knockout the TGF-β receptor II in CAR T cells, resulting in enhanced anti-tumour responses in cell line-derived and PDX solid tumour models in immunodeficient mice.169 Furthermore, knockdown of TGF-β receptor II reduced exhaustion marker expression and not only decreased regulatory cell differentiation but also increased the presence of memory cell populations.169 Mechanisms of conferring resistance to TGF-β could in principle be translated to other inhibitory cytokines in order to improve CAR T cell function within the TME.
Overcoming Immunosuppressive Molecules Within the TME
Adenosine is produced in the TME from ATP by ectonucleases expressed on tumour cells.170 Adenosine binds to adenosine receptors on T cells, leading to the activation of protein kinase A (PKA) and the initiation of immunosuppressive effects. Illustrating this, PKA interferes with T cell signalling pathways, resulting in impaired T cell proliferation and effector function.171 Pharmacological inhibition of the adenosine receptor A2A in CAR T cells has been achieved by incorporating small molecule inhibitors into nanoparticles.172 Nanoparticle loaded CD19 CAR T cells accumulated within the TME and elicited anti-tumour responses in CD19 positive solid tumour xenograft models.172 Alternatively, CAR T cells deficient in the adenosine receptor A2A showed improved in vivo anti-tumour responses associated with increased cytokine production.173–175
Prostaglandin E2 (PGE2) is also produced within the TME by tumour-derived cyclooxygenase 2 (COX2)-mediated conversion of arachidonic acid. Similar to adenosine, PGE2 binds receptors that are found on T cells, resulting in PKA activation. Manipulation of the PKA pathway in CAR T cells can circumvent the negative effects of adenosine and PGE2 in the TME. PKA activation is dependent on its recruitment to the immune synapse by the anchor protein ezrin.176 Disruption of this binding by the introduction of small peptides in CAR T cells improved tumour infiltration and anti-tumour responses against both human and mouse mesothelioma cell lines in vivo and rendered CAR T cells resistant to tumour derived PGE2 and adenosine.176
Overcoming Disruptive Effects of Tumour Metabolism
Activation of oncogenes in tumour cells increases their metabolic capacity leading to increased tumour growth, deprivation of microenvironmental nutrients, accumulation of harmful metabolites and increased tumour resistance to immune cell attack.
Tumour cells outcompete immune cells for glucose, causing insufficient glycolysis and reduced effector functions of TIL cells. High expression of glycolysis-related genes in tumours is associated with reduced infiltration and sensitivity to cytotoxicity mediated by adoptively infused T cells. Furthermore, highly glycolytic tumour cells render themselves resistant to immunotherapy by downregulating the IFN-γ pathway and expression of chemokines.177 Incorporating mechanisms to enable CAR T cells to resist these effects may help to overcome tumour resistance to immunotherapy. Checkpoint blockade can disrupt mTOR signalling in tumour cells, resulting in increased glucose availability and T cell function, as demonstrated in an in vivo mouse sarcoma model.178 Tumour cells increase glucose metabolism by upregulating glycolytic enzymes including lactate dehydrogenase A (LDHA). Lactate dehydrogenase A converts pyruvate to lactic acid, which is exported out into the TME, reducing pH. Glycolytically active immune cells rely on a gradient for lactic acid export. Increased lactic acid in the TME compromises this export process and can lead to net lactic acid uptake by immune cells. Inefficient lactic acid export prevents sufficient glycolysis and causes poor proliferation and cytokine production associated with reduced NFAT upregulation and translocation to the nucleus.179,180 Levels of LDHA and lactic acid accumulation in the TME are both associated with poor prognosis and tumour metastasis.180 Inhibition of LDHA reduced tumour cell growth when undertaken in combination with tumour reactive T cell immunotherapy in a mouse model of melanoma; an approach that could be translated to CAR T cell therapy.177 Neutralisation of the acidic TME can also improve T cell infiltration into solid tumours and improves tumour regression in response to checkpoint inhibitors, as demonstrated in an in vivo model of melanoma.181
Similar to glucose, tumour cells outcompete T cells for tryptophan in the TME, leading to reduced T cell proliferation. Tumour cells produce indoleamine 2,3-dioxygenase (IDO), an enzyme that metabolises tryptophan to generate harmful metabolites. Tryptophan metabolites including kynurenine and 3-hydroxyanthranilic acid (3-HAA) directly inhibit CAR T cell expansion, cytotoxicity, cytokine secretion and promote apoptosis.182 One potential beneficial effect of lymphodepletion prior to adoptive cell therapy entails downregulated IDO expression in malignant cells.183 Furthermore, the combination of CAR T cell therapy with small molecule inhibitors of IDO can improve in vivo anti-tumour activity.184,185
Tumour cells and tumour-associated myeloid cells produce arginase which catabolises arginine, an amino acid that supports T cell proliferation and function. Depletion of arginine in the TME is detrimental to the function of CAR T cells, reducing proliferation and cytotoxicity.186 Expression of two arginine resynthesis enzymes, argininosuccinate synthase (ASS) and ornithine transcarbamylase (OTC) in CAR T cells enables cells to utilise low levels of arginine in the tumour microenvironment maintaining CAR T cell function and proliferation, exemplified in both leukaemia and solid tumour mouse models.187
Finding alternative nutrients for T cell metabolism is one method which has been explored to avoid nutrient competition in the TME to support T cell proliferation and function. Inosine has been identified as an alternative T cell substrate which many tumour cell lines are unable to utilise. Supplementation of inosine in both in vitro and in vivo models enhances the ability of CAR T cells to control tumour growth.188
Manipulating CAR T cells to utilise specific metabolism pathways may foster resistance within the nutrient depleted TME. Naïve T cells rely upon oxidation of fatty acids.189,190 However, effector T cells upregulate glycolytic pathways after antigen exposure. Conversely, memory populations increase oxidative phosphorylation as a source of energy.189,190 Incorporation of alternative costimulatory domains within CARs can promote different metabolic fates.191 41BB containing CAR T cells demonstrate improved persistence due to central memory differentiation, associated with enhanced mitochondrial biogenesis and oxidative metabolism.191 In contrast, CD28 containing CARs promote enhanced glycolysis associated with differentiation of effector phenotypes.191 Manipulation of CAR T cells during expansion can also improve metabolic potential. Sukumar et al192 demonstrated that highly glycolytic T cells have reduced capacity to develop into memory T cells.192 Inhibiting glycolysis during expansion increased differentiation of central memory and stem cell memory phenotypes, improving anti-tumour function in a in vivo melanoma model.192 Methods to isolate T cells with a desired metabolic profile for adoptive cell therapy (ACT) have also been investigated. Selecting T cells based on their mitochondrial membrane potential (using a lipophilic catatonic dye) selects memory T cell precursors that have superior persistence and anti-tumour ability in the pmel-1 mouse model system.193 Applying this to CAR T cells may improve their function, although the required incorporation of additional manufacturing steps is not ideal.
Overcoming Antigen Negative Tumour Resistance
Relapse due to antigen loss following CAR T cell treatment has been observed with multiple targets including CD19,194 BCMA,195 CD22,196 EGFRvIII,197 IL-13Rα2124,198 and ErbB2.199 Antigen loss can occur due to frameshift and missense mutations resulting in deletion of the target antigen and outgrowth of antigen-negative cancer cells that are resistant to CAR T cell therapy.194,200 Lineage switch following CD19 CAR T cell therapy has also been observed due to immune pressure inducing a drastic reprogramming of malignant cells, switching from lymphoid to myeloid phenotypes.201,202 In the case of CD19, alternative splicing can lead to the production of a mutant protein lacking the transmembrane domain, preventing cell surface expression. Skipping of exon 2 results in the expression of a truncated CD19 lacking the extracellular epitope recognised by the commonly used FMC63 or SJ25C1 scFvs, rendering cancer cells undetectable by derived CARs.194 Fischer et al203 demonstrated that these spliced variants exist pre-CAR T cell treatment in B-ALL patients and therefore could be used as predictors for antigen escape.203 More recently it has been shown that CAR T cells can induce reversible tumour antigen loss via trogocytosis whereby CAR T cells actively remove antigen from the tumour cell surface. Not only does this result in antigen escape but also induces fratricide and exhaustion of the CAR T cells.204
Resistance to CAR T cell therapy can also occur due to downregulation of tumour antigen and low target density on tumour cells.196,205 The threshold for CAR T cell activation is dependent on target density and therefore antigen downregulation prevents successful CAR T cell activation and anti-tumour responses.206–208 Majzner et al208 investigated how CAR T cell structure affects the threshold level for activation. Comparison of the two FDA approved CD19 targeting CAR T cell products revealed that CD28 co-stimulation provided a lower activation threshold as compared to 41BB. The activation threshold of 41BB containing CARs could be reduced by the addition of a further CD3ζ domain, enabling improved response to low antigen density tumour cells in vitro and in vivo.208 Activation threshold is also dependent on hinge and transmembrane domain of CD19 targeting CAR T cells.208 Both 1G and 2G CAR T cells containing the hinge and transmembrane domains from CD28 have reduced activation threshold compared to CARs containing hinge and transmembrane domains from CD8. This was shown to be dependent on improved receptor clustering within the immunological synapse and recruitment of ZAP70.208
To avoid relapse due to antigen escape, targeting of multiple antigens has been attempted. This has been demonstrated following multiple infusions of distinctly targeted CAR T cells, dual targeting CARs, trivalent CARs, tandem CARs and universal CARs (Figure 3). Dual targeting CARs, where two distinct 2G CARs are expressed on a single cell, has been exemplified for many target combinations.204,209–213 Dual targeting CD19 and CD22 CAR T cells have demonstrated the ability to overcome antigen escape induced by trogocytosis.204 Using mathematical models, Hedge et al199 analysed the expression pattern of antigens on glioblastoma cells and proposed that targeting of both ErbB2 and IL-13Rα2 would be the most efficient way to prevent therapeutic failure of CAR T cell immunotherapy due to antigen loss and/or heterogeneity. The benefit of targeting two cancer-associated antigens was demonstrated using pooled ErbB2 and IL-13Rα2 CAR T cells and dual targeting CAR T cells, expressing the two distinct second-generation CARs on a single cell.199 In this study, the authors showed that targeting three antigens did not enhance the probability of recognising the majority of tumour cells compared to targeting two antigens in their cohort of primary tumours. Nonetheless, trivalent CAR T cells have been developed for glioblastoma in which three 2G CARs targeting ErbB2, IL-13Rα2 and EphA2 are expressed on a single cell. Trivalent CARs in this setting were able to overcome tumour antigenic heterogeneity, variability between patients and demonstrated increased cytotoxicity and cytokine release compared to dual targeting CAR T cells.214 Additionally, trispecific CAR T cells targeting CD19, CD20 and CD22 have been developed to overcome CD19 antigen loss and demonstrated the ability to target and kill CD19 negative blasts from relapsed patients that received CD19 CAR T cell therapy.215 Tandem CARs are an alternative strategy to target multiple antigens, whereby two distinct scFvs are fused together by a flexible linker.216,217 Tandem CARs can be activated via one antigen but activation is enhanced when both targets are engaged, as demonstrated in a proof-of-concept study targeting CD19 and ErbB2216 and CD20 and ErbB2.218
 |
Figure 3 Structure of CARs to target multiple antigens. Tumour antigen loss can be overcome by targeting multiple antigens by various CAR T cell structures. Dual CAR T cells contain two distinct 2G CARs expressed on a single cell which recognise two different target antigens. Trivalent CAR T cells contain three distinct CARs able to recognise three different target antigens. Tandem CARs contain two distinct scFvs fused together by a flexible linker. Universal CARs consist of universal receptor and an antigen binding adaptor molecule, of which different target binders can be utilised. |
Universal CARs consist of a universal receptor and an antigen-binding adaptor molecule. Multiple antigens can be targeted by infusing multiple adaptor molecules with specificity for different targets.219 Importantly, in the case of antigen escape, a different antigen can be targeted without the need for additional CAR T cell production and infusion. In a related concept, CAR T cell immunotherapy can be combined with the infusion of bispecific antibodies (BiTEs) which bind a target antigen and CD3, thereby inducing an immunological synapse and T cell activation.220 Co-treatment with BiTEs and CAR T cells allow for dual-antigen targeting, avoiding the outgrowth of antigen-negative tumours. Combining αFR- targeting CAR T cells with an oncolytic virus secreting an EGFR-targeting BiTE enabled activation of CAR T cells in response to αFR positive as well as αFR negative but EGFR positive solid tumour mouse models.221 Furthermore, CAR negative T cells found in the CAR T cell product could also be activated by the EGFR-BiTEs, enhancing the anti-tumour response.221 CAR T cells have also been engineered to secrete BiTEs themselves. For example, secretion of an EGFR-targeting BiTEs by EGFRvIII targeting CAR T cells enhanced antitumour immunity against heterogenous glioblastoma tumour xenografts in mouse models.222
An alternative strategy to avoid antigen escape is to induce expression of artificial antigens which CAR T cells can recognise. Glycometabolic labelling of tumour cells with unnatural sugar residues can be targeted by CARs in which specificity is directed by the artificial ligand – so-called Click CAR T cells. Incorporation of artificial receptor and ligand into CD19 CAR T cells and target tumour cells improved selectivity, infiltration and homing.223 The introduction of artificial targets also has the potential to avoid off-tumour/on-target toxicity. Park et al224 used an oncolytic virus to deliver a target antigen, specifically truncated CD19, into solid tumours and demonstrated successful tumour eradication in both human xenograft models and a mouse immunocompetent model. Rechallenge of mice previously treated with oncolytic viruses and CAR T cells inhibited the growth of new tumours, suggesting induction of endogenous immune response and epitope spreading.224 Epitope spreading following CAR T cell therapy has been demonstrated with a 3G EGFRvIII targeting CAR which was able to control antigen-negative tumour rechallenge in immunocompetent mouse models.225 Promoting epitope spreading can maintain tumour control even when the CAR T cell target is lost. The cross-presenting DC growth factor FMS-like tyrosine kinase 3 ligand (Flt3L) has recently been expressed by CAR T cells leading to the facilitation of an endogenous anti-tumour immune response, with increased epitope spreading.226 In vivo experiments with Flt3L CAR T cells demonstrated increased DC and T cell activation which led to control of both antigen positive and negative tumour when used in combination with an additional adjuvant.226
Overcoming Resistance in Tumours That Retain Target Antigen
Acquired resistance to CAR T cell therapy can also occur without antigen loss. An in vitro model of CAR T cell resistance suggests that one mechanism by which such resistance can be acquired is disruption of TRAIL signalling.227 Sensitising resistant tumour cells with histone deacetylase inhibitors can partially reverse resistance by restoring the TRAIL apoptotic pathway.227 Kearney et al228 performed a CRISPR screen to identify additional mechanisms of acquired tumour immune resistance. Following immune pressure, tumour cells can lose their sensitivity to TNF-mediated killing, which is associated with caspase-8 downregulation.228 SMAC mimetics can sensitise cells to TNF-mediated killing and could be used in combination with CAR T cells to avoid acquired resistance.229
Alternatively, it is notable that certain oncogenic pathways can render tumour cells intrinsically resistant to adoptive T cell therapy. For example, loss of phosphatase and tensin homolog (PTEN) in tumour cells is associated with reduced T cell trafficking, increased production of inhibitory cytokines and resistance to apoptosis, as demonstrated in in vitro and in vivo melanoma models.230 Furthermore, PTEN loss is associated with increased vascular endothelial growth factor expression, resulting in increased trafficking of immunosuppressive cells into the TME.230
Enhancing CAR T Cell Efficacy by Refining Immunogenicity of Target Binders
Anti-CAR T cell immune responses may be pre-existent or may develop following CAR T cell immunotherapy. Clinical evaluation of the anti-CD19 CAR T cell therapy, Tisagenlecleucel, demonstrated that the majority of patients had detectable anti-CAR antibodies before treatment, which increased in a small group of patients post-CAR T cell therapy.231,232 The existence of anti-CAR antibodies, however, did not appear to affect expansion or efficacy in this study.231,232 In contrast, patients who develop cellular immune responses to CAR T cells did not respond to initial or subsequent doses of CD19 CAR T cells.233 Unlike the situation in CD19 CAR T cell immunotherapy, CAR-specific antibody responses appear to disrupt the efficacy of CAR T cells targeting different antigens. A study investigating bispecific CAR T cells targeted against two epitopes of BCMA correlated the presence of anti-CAR antibodies to disease relapse and reduced number of circulating CAR T cells.234 Furthermore, in a Phase I clinical trial, CAR T cells targeting αFR in patients with ovarian cancer developed anti-CAR antibody responses, reducing CAR T cell efficacy and persistence.235 CAR T cell therapy has even induced anaphylaxis, an event thought to reflect the induction of an IgE response against murine scFv epitopes.236 Methods to reduce CAR immunogenicity have been extensively reviewed by Wagner et al237 and encompasses the removal of immunogenic CAR components, exchange of mouse-derived scFvs with humanised/human scFvs,238 eliminating immunogenic linkers through the use of heavy chain only CARs including nanobodies239 and replacement of scFvs with receptor-ligand binding moieties.240–242
Conclusions
Despite intensive efforts, no successful CAR T cell therapy for solid tumours has been developed as yet. It is apparent that engineering CAR T cells to overcome just one aspect of tumour resistance may not be enough. The emerging picture of a highly successful CAR T cell seems to be one that is less differentiated from the stem/central memory cell phenotype, allowing long-term persistence in patients. Furthermore, CAR T cells must be able to successfully home to and infiltrate highly complex tumours and, once within the hostile TME, they must resist immunosuppression to maintain full functionality. It remains to be seen whether the use of one or more of the strategies described in this review can enable CAR T-cells to resist the panoply of defence mechanisms commonly deployed in solid tumours.
Acknowledgments
Research in JM’s laboratory is supported by Leucid Bio, Breast Cancer Now, British Lung Foundation, the Wellcome Trust, the J P Moulton Charitable Foundation, the Medical Research Council, the King’s Health Partners Research and Development Fund, the Experimental Cancer Medicine Centre at King’s College London, the Cancer Research UK Centre at King’s Health Partners and by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Disclosure
JM is founder, chief scientific officer and shareholder in Leucid Bio. MG is an employee and shareholder in Leucid Bio. The authors report no other conflicts of interest in this work.
References
1. Kuwana Y, Asakura Y, Utsunomiya N, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149(3):960–968. doi:10.1016/0006-291X(87)90502-X
2. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720–724. doi:10.1073/pnas.90.2.720
3. Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161(6):2791–2797.
4. Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31–42. doi:10.1016/S1470-2045(18)30864-7
5. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56.
6. Wang M, Munoz J, Goy A, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–1342. doi:10.1056/NEJMoa1914347
7. Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839–852. doi:10.1016/S0140-6736(20)31366-0
8. Munshi NC, Anderson LD
9. Liu X, Jiang S, Fang C, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015;75(17):3596–3607. doi:10.1158/0008-5472.CAN-15-0159
10. Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol. 2004;173(12):7647–7653. doi:10.4049/jimmunol.173.12.7647
11. Caruso HG, Hurton LV, Najjar A, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015;75(17):3505–3518. doi:10.1158/0008-5472.CAN-15-0139
12. Song DG, Ye Q, Poussin M, Liu L, Figini M, Powell DJ
13. Ghorashian S, Kramer AM, Onuoha S, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. 2019;25(9):1408–1414. doi:10.1038/s41591-019-0549-5
14. Park S, Shevlin E, Vedvyas Y, et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep. 2017;7(1):14366. doi:10.1038/s41598-017-14749-3
15. Zhao Y, Liu Z, Wang X, et al. Treatment with humanized selective CD19CAR-T cells shows efficacy in highly treated B-ALL patients who have relapsed after receiving murine-based CD19CAR-T therapies. Clin Cancer Res. 2019;25(18):5595–5607. doi:10.1158/1078-0432.CCR-19-0916
16. Ajina A, Maher J. Synergistic combination of oncolytic virotherapy with CAR T-cell therapy. Prog Mol Biol Transl Sci. 2019;164:217–292.
17. Xie YJ, Dougan M, Jailkhani N, et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci. 2019;116(16):7624–7631. doi:10.1073/pnas.1817147116
18. Wesolowski J, Alzogaray V, Reyelt J, et al. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med Microbiol Immunol. 2009;198(3):157–174. doi:10.1007/s00430-009-0116-7
19. Baumeister SH, Murad J, Werner L, et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res. 2019;7(1):100–112. doi:10.1158/2326-6066.CIR-18-0307
20. Brown CE, Aguilar B, Starr R, et al. Optimization of IL13Ralpha2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol Ther. 2018;26(1):31–44. doi:10.1016/j.ymthe.2017.10.002
21. Davies DM, Foster J, Van Der Stegen SJ, et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol Med. 2012;18:565–576. doi:10.2119/molmed.2011.00493
22. Whilding LM, Halim L, Draper B, et al. CAR T-cells targeting the integrin alphavbeta6 and co-expressing the chemokine receptor CXCR2 demonstrate enhanced homing and efficacy against several solid malignancies. Cancers. 2019;11:5. doi:10.3390/cancers11050674
23. Hombach A, Hombach AA, Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc “spacer” domain in the extracellular moiety of chimeric antigen receptors avoids “off-target” activation and unintended initiation of an innate immune response. Gene Ther. 2010;17(10):1206–1213. doi:10.1038/gt.2010.91
24. Guest RD, Hawkins RE, Kirillova N, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005;28(3):203–211. doi:10.1097/01.cji.0000161397.96582.59
25. Wilkie S, Picco G, Foster J, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180(7):4901–4909. doi:10.4049/jimmunol.180.7.4901
26. Alabanza L, Pegues M, Geldres C, et al. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. 2017;25(11):2452–2465. doi:10.1016/j.ymthe.2017.07.013
27. Zhao Z, Condomines M, van der Stegen SJC, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415–428. doi:10.1016/j.ccell.2015.09.004
28. Carpenito C, Milone MC, Hassan R, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106(9):3360–3365. doi:10.1073/pnas.0813101106
29. Sun C, Shou P, Du H, et al. THEMIS-SHP1 recruitment by 4-1BB tunes LCK-mediated priming of chimeric antigen receptor-redirected T cells. Cancer Cell. 2020;37(2):216–225 e216. doi:10.1016/j.ccell.2019.12.014
30. Frigault MJ, Lee J, Basil MC, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. 2015;3(4):356–367. doi:10.1158/2326-6066.CIR-14-0186
31. Guedan S, Madar A, Casado-Medrano V, et al. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J Clin Invest. 2020;130(6):3087–3097. doi:10.1172/JCI133215
32. Boucher JC, Li G, Kotani H, et al. CD28 costimulatory domain-targeted mutations enhance chimeric antigen receptor T-cell function. Cancer Immunol Res. 2021;9(1):62–74. doi:10.1158/2326-6066.CIR-20-0253
33. Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–117. doi:10.1038/nature21405
34. Long AH, Haso WM, Shern JF, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–590. doi:10.1038/nm.3838
35. Li W, Qiu S, Chen J, et al. Chimeric antigen receptor designed to prevent ubiquitination and downregulation showed durable antitumor efficacy. Immunity. 2020;53(2):456–470 e456. doi:10.1016/j.immuni.2020.07.011
36. Wang J, Jensen M, Lin Y, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007;18(8):712–725. doi:10.1089/hum.2007.028
37. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18(2):413–420. doi:10.1038/mt.2009.210
38. Tammana S, Huang X, Wong M, et al. 4-1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum Gene Ther. 2010;21(1):75–86. doi:10.1089/hum.2009.122
39. Karlsson H, Svensson E, Gigg C, et al. Evaluation of intracellular signaling downstream chimeric antigen receptors. PLoS One. 2015;10(12):e0144787. doi:10.1371/journal.pone.0144787
40. Abate-Daga D, Lagisetty KH, Tran E, et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum Gene Ther. 2014;25(12):1003–1012. doi:10.1089/hum.2013.209
41. Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–1464. doi:10.1038/mt.2009.83
42. Sangsuwannukul T, Supimon K, Sujjitjoon J, et al. Anti-tumour effect of the fourth-generation chimeric antigen receptor T cells targeting CD133 against cholangiocarcinoma cells. Int Immunopharmacol. 2020;89(Pt B):107069. doi:10.1016/j.intimp.2020.107069
43. Dai Q, Han P, Qi X, et al. 4-1BB signaling boosts the anti-tumor activity of CD28-incorporated 2(nd) generation chimeric antigen receptor-modified T cells. Front Immunol. 2020;11:539654. doi:10.3389/fimmu.2020.539654
44. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71–75. doi:10.1038/nbt.2459
45. Guedan S, Chen X, Madar A, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 2014;124(7):1070–1080. doi:10.1182/blood-2013-10-535245
46. Hombach AA, Heiders J, Foppe M, Chmielewski M, Abken H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4(+) T cells. Oncoimmunology. 2012;1(4):458–466. doi:10.4161/onci.19855
47. Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12(5):933–941. doi:10.1016/j.ymthe.2005.04.016
48. Quintarelli C, Orlando D, Boffa I, et al. Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology. 2018;7(6):e1433518. doi:10.1080/2162402X.2018.1433518
49. Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ
50. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible activation of MyD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov. 2017;7(11):1306–1319. doi:10.1158/2159-8290.CD-17-0263
51. Foster AE, Mahendravada A, Shinners NP, et al. Regulated expansion and survival of chimeric antigen receptor-modified T cells using small molecule-dependent inducible MyD88/CD40. Mol Ther. 2017;25(9):2176–2188. doi:10.1016/j.ymthe.2017.06.014
52. Collinson-Pautz MR, Chang WC, Lu A, et al. Constitutively active MyD88/CD40 costimulation enhances expansion and efficacy of chimeric antigen receptor T cells targeting hematological malignancies. Leukemia. 2019;33(9):2195–2207. doi:10.1038/s41375-019-0417-9
53. Prinzing B, Schreiner P, Bell M, Fan Y, Krenciute G, Gottschalk S. MyD88/CD40 signaling retains CAR T cells in a less differentiated state. JCI Insight. 2020;5(21):21. doi:10.1172/jci.insight.136093
54. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 2015;4(3):e994446.
55. Pegram HJ, Purdon TJ, van Leeuwen DG, et al. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia. 2015;29(2):415–422. doi:10.1038/leu.2014.215
56. Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep. 2017;7(1):10541. doi:10.1038/s41598-017-10940-8
57. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71(17):5697–5706. doi:10.1158/0008-5472.CAN-11-0103
58. Liu Y, Di S, Shi B, et al. Armored inducible expression of IL-12 enhances antitumor activity of glypican-3-targeted chimeric antigen receptor-engineered T cells in hepatocellular carcinoma. J Immunol. 2019;203(1):198–207. doi:10.4049/jimmunol.1800033
59. Zhang L, Morgan RA, Beane JD, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res. 2015;21(10):2278–2288. doi:10.1158/1078-0432.CCR-14-2085
60. Sachdeva M, Busser BW, Temburni S, et al. Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat Commun. 2019;10(1):5100. doi:10.1038/s41467-019-13088-3
61. Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol Ther Oncolytics. 2018;8:41–51. doi:10.1016/j.omto.2017.12.003
62. Lanitis E, Rota G, Kosti P, et al. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. J Exp Med. 2021;218(2). doi:10.1084/jem.20192203
63. Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115(17):3508–3519. doi:10.1182/blood-2009-09-241398
64. Hurton LV, Singh H, Najjar AM, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A. 2016;113(48):E7788–E7797. doi:10.1073/pnas.1610544113
65. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36(4):346–351. doi:10.1038/nbt.4086
66. Batra SA, Rathi P, Guo L, et al. Glypican-3-specific CAR T cells coexpressing IL15 and IL21 have superior expansion and antitumor activity against hepatocellular carcinoma. Cancer Immunol Res. 2020;8(3):309–320. doi:10.1158/2326-6066.CIR-19-0293
67. Liu L, Bi E, Ma X, et al. Enhanced CAR-T activity against established tumors by polarizing human T cells to secrete interleukin-9. Nat Commun. 2020;11(1):5902. doi:10.1038/s41467-020-19672-2
68. Hu B, Ren J, Luo Y, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 2017;20(13):3025–3033. doi:10.1016/j.celrep.2017.09.002
69. Chmielewski M, Abken H. CAR T cells releasing IL-18 convert to T-Bet(high) FoxO1(low) effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. 2017;21(11):3205–3219. doi:10.1016/j.celrep.2017.11.063
70. Ma X, Shou P, Smith C, et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat Biotechnol. 2020;38(4):448–459. doi:10.1038/s41587-019-0398-2
71. Hu Q, Zhang Y, Wang P, et al. IL-24 armored CAR19-T cells show enhanced antitumor activity and persistence. Signal Transduct Target Ther. 2021;6(1):14. doi:10.1038/s41392-020-00380-8
72. Li X, Daniyan AF, Lopez AV, Purdon TJ, Brentjens RJ. Cytokine IL-36gamma improves CAR T-cell functionality and induces endogenous antitumor response. Leukemia. 2021;35(2):506–521.
73. Jiang Z, Liao R, Lv J, et al. IL-6 trans-signaling promotes the expansion and anti-tumor activity of CAR T cells. Leukemia. 2020. doi:10.1038/s41375-020-01085-1
74. Shum T, Omer B, Tashiro H, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017;7(11):1238–1247. doi:10.1158/2159-8290.CD-17-0538
75. Zhao Z, Li Y, Liu W, Li X. Engineered IL-7 receptor enhances the therapeutic effect of AXL-CAR-T cells on triple-negative breast cancer. Biomed Res Int. 2020;2020:4795171.
76. Perna SK, Pagliara D, Mahendravada A, et al. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin Cancer Res. 2014;20(1):131–139. doi:10.1158/1078-0432.CCR-13-1016
77. Wilkie S, Burbridge SE, Chiapero-Stanke L, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. 2010;285(33):25538–25544. doi:10.1074/jbc.M110.127951
78. Leen AM, Sukumaran S, Watanabe N, et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol Ther. 2014;22(6):1211–1220. doi:10.1038/mt.2014.47
79. Mohammed S, Sukumaran S, Bajgain P, et al. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther. 2017;25(1):249–258. doi:10.1016/j.ymthe.2016.10.016
80. Wang Y, Jiang H, Luo H, et al. An IL-4/21 inverted cytokine receptor improving CAR-T cell potency in immunosuppressive solid-tumor microenvironment. Front Immunol. 2019;10:1691. doi:10.3389/fimmu.2019.01691
81. Klampatsa A, Achkova DY, Davies DM, et al. Intracavitary “T4 immunotherapy” of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. 2017;393:52–59. doi:10.1016/j.canlet.2017.02.015
82. van Schalkwyk MC, Papa SE, Jeannon JP, Guerrero Urbano T, Spicer JF, Maher J. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum Gene Ther Clin Dev. 2013;24(3):134–142. doi:10.1089/humc.2013.144
83. Lo AS, Taylor JR, Farzaneh F, Kemeny DM, Dibb NJ, Maher J. Harnessing the tumour-derived cytokine, CSF-1, to co-stimulate T-cell growth and activation. Mol Immunol. 2008;45(5):1276–1287. doi:10.1016/j.molimm.2007.09.010
84. Kagoya Y, Tanaka S, Guo T, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. 2018;24(3):352–359. doi:10.1038/nm.4478
85. Ding ZC, Shi H, Aboelella NS, et al. Persistent STAT5 activation reprograms the epigenetic landscape in CD4(+) T cells to drive polyfunctionality and antitumor immunity. Sci Immunol. 2020;5(52). doi:10.1126/sciimmunol.aba5962
86. Wiede F, Lu K-H, Du X, et al. PTPN 2 phosphatase deletion in T cells promotes anti-tumour immunity and CAR T-cell efficacy in solid tumours. EMBO J. 2020;39(2):e103637. doi:10.15252/embj.2019103637
87. Kim EH, Suresh M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front Immunol. 2013;4:20. doi:10.3389/fimmu.2013.00020
88. Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–9553. doi:10.1128/MCB.25.21.9543-9553.2005
89. Wu Y, Deng Z, Tang Y, Zhang S, Zhang YQ. Over-expressing Akt in T cells to resist tumor immunosuppression and increase anti-tumor activity. BMC Cancer. 2015;15:603. doi:10.1186/s12885-015-1611-4
90. Sun J, Dotti G, Huye LE, et al. T cells expressing constitutively active Akt resist multiple tumor-associated inhibitory mechanisms. Mol Ther. 2010;18(11):2006–2017. doi:10.1038/mt.2010.185
91. Kim EH, Sullivan JA, Plisch EH, et al. Signal integration by Akt regulates CD8 T cell effector and memory differentiation. J Immunol. 2012;188(9):4305–4314. doi:10.4049/jimmunol.1103568
92. Urak R, Walter M, Lim L, et al. Ex vivo Akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J Immunother Cancer. 2017;5:26. doi:10.1186/s40425-017-0227-4
93. Klebanoff CA, Crompton JG, Leonardi AJ, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight. 2017;2(23). doi:10.1172/jci.insight.95103
94. Zheng W, O’Hear CE, Alli R, et al. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia. 2018;32(5):1157–1167. doi:10.1038/s41375-017-0008-6
95. Gurusamy D, Henning AN, Yamamoto TN, et al. Multi-phenotype CRISPR-Cas9 screen identifies p38 kinase as a target for adoptive immunotherapies. Cancer Cell. 2020;37(6):818–833 e819. doi:10.1016/j.ccell.2020.05.004
96. Dufva O, Koski J, Maliniemi P, et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood. 2020;135(9):597–609. doi:10.1182/blood.2019002121
97. Lynn RC, Weber EW, Sotillo E, et al. c-jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576(7786):293–300. doi:10.1038/s41586-019-1805-z
98. Martinez GJ, Pereira RM, Aijo T, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity. 2015;42(2):265–278. doi:10.1016/j.immuni.2015.01.006
99. Chen J, Lopez-Moyado IF, Seo H, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 2019;567(7749):530–534. doi:10.1038/s41586-019-0985-x
100. Liu X, Wang Y, Lu H, et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature. 2019;567(7749):525–529. doi:10.1038/s41586-019-0979-8
101. Seo H, Chen J, Gonzalez-Avalos E, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A. 2019;116(25):12410–12415. doi:10.1073/pnas.1905675116
102. Khan O, Giles JR, McDonald S, et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019;571(7764):211–218. doi:10.1038/s41586-019-1325-x
103. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. “Off-the-shelf” allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19(3):185–199. doi:10.1038/s41573-019-0051-2
104. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–167.
105. Thommen DS, Schumacher TN, Cell T. Dysfunction in cancer. Cancer Cell. 2018;33(4):547–562. doi:10.1016/j.ccell.2018.03.012
106. Gargett T, Brown MP. Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy. 2015;17(4):487–495. doi:10.1016/j.jcyt.2014.12.002
107. Cieri N, Camisa B, Cocchiarella F, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121(4):573–584. doi:10.1182/blood-2012-05-431718
108. Hoffmann JM, Schubert ML, Wang L, et al. Differences in expansion potential of naive chimeric antigen receptor T cells from healthy donors and untreated chronic lymphocytic leukemia patients. Front Immunol. 2017;8:1956. doi:10.3389/fimmu.2017.01956
109. Brudno JN, Somerville RP, Shi V, et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol. 2016;34(10):1112–1121. doi:10.1200/JCO.2015.64.5929
110. Kochenderfer JN, Dudley ME, Carpenter RO, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122(25):4129–4139. doi:10.1182/blood-2013-08-519413
111. Ghosh A, Smith M, James SE, et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat Med. 2017;23(2):242–249. doi:10.1038/nm.4258
112. Osborn MJ, Webber BR, Knipping F, et al. Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9, and megaTAL nucleases. Mol Ther. 2016;24(3):570–581. doi:10.1038/mt.2015.197
113. Torikai H, Reik A, Liu PQ, et al. A foundation for universal T-cell based immunotherapy: t cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119(24):5697–5705. doi:10.1182/blood-2012-01-405365
114. Torikai H, Reik A, Soldner F, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 2013;122(8):1341–1349. doi:10.1182/blood-2013-03-478255
115. Poirot L, Philip B, Schiffer-Mannioui C, et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015;75(18):3853–3864. doi:10.1158/0008-5472.CAN-14-3321
116. Han J, Chu J, Keung Chan W, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. 2015;5:11483. doi:10.1038/srep11483
117. Liu E, Tong Y, Dotti G, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–531. doi:10.1038/leu.2017.226
118. Mehta RS, Rezvani K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front Immunol. 2018;9:283. doi:10.3389/fimmu.2018.00283
119. Capsomidis A, Benthall G, Van Acker HH, et al. Chimeric antigen receptor-engineered human gamma delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol Ther. 2018;26(2):354–365. doi:10.1016/j.ymthe.2017.12.001
120. Themeli M, Kloss CC, Ciriello G, et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol. 2013;31(10):928–933. doi:10.1038/nbt.2678
121. Rotolo A, Caputo VS, Holubova M, et al. Enhanced anti-lymphoma activity of CAR19-iNKT cells underpinned by dual CD19 and CD1d targeting. Cancer Cell. 2018;34(4):596–610 e511. doi:10.1016/j.ccell.2018.08.017
122. Klichinsky M, Ruella M, Shestova O, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38(8):947–953. doi:10.1038/s41587-020-0462-y
123. Papa S, van Schalkwyk M, Maher J. Clinical evaluation of ErbB-targeted CAR T-cells, following intracavity delivery in patients with ErbB-expressing solid tumors. Methods Mol Biol. 2015;1317:365–382.
124. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi:10.1056/NEJMoa1610497
125. Liu G, Rui W, Zheng H, et al. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur J Immunol. 2020;50(5):712–724. doi:10.1002/eji.201948457
126. Jin L, Tao H, Karachi A, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019;10(1):4016. doi:10.1038/s41467-019-11869-4
127. Craddock JA, Lu A, Bear A, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33(8):780–788. doi:10.1097/CJI.0b013e3181ee6675
128. Di Stasi A, De Angelis B, Rooney CM, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392–6402. doi:10.1182/blood-2009-03-209650
129. Poznansky MC, Olszak IT, Foxall R, Evans RH, Luster AD, Scadden DT. Active movement of T cells away from a chemokine. Nat Med. 2000;6(5):543–548. doi:10.1038/75022
130. Vianello F, Papeta N, Chen T, et al. Murine B16 melanomas expressing high levels of the chemokine stromal-derived factor-1/CXCL12 induce tumor-specific T cell chemorepulsion and escape from immune control. J Immunol. 2006;176(5):2902–2914. doi:10.4049/jimmunol.176.5.2902
131. Biasci D, Smoragiewicz M, Connell CM, et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc Natl Acad Sci U S A. 2020;117(46):28960–28970. doi:10.1073/pnas.2013644117
132. Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–20217. doi:10.1073/pnas.1320318110
133. Wang LC, Lo A, Scholler J, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2(2):154–166. doi:10.1158/2326-6066.CIR-13-0027
134. Kakarla S, Chow KK, Mata M, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21(8):1611–1620. doi:10.1038/mt.2013.110
135. Tran E, Chinnasamy D, Yu Z, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. 2013;210(6):1125–1135. doi:10.1084/jem.20130110
136. Caruana I, Savoldo B, Hoyos V, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21(5):524–529. doi:10.1038/nm.3833
137. Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. 2016;9:5023–5039. doi:10.2147/OTT.S105862
138. Oh SA, Wu D-C, Cheung J, et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nature Cancer. 2020;1(7):681–691. doi:10.1038/s43018-020-0075-x
139. John LB, Devaud C, Duong CP, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19(20):5636–5646. doi:10.1158/1078-0432.CCR-13-0458
140. Gargett T, Yu W, Dotti G, et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther. 2016;24(6):1135–1149. doi:10.1038/mt.2016.63
141. Chong EA, Melenhorst JJ, Lacey SF, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. 2017;129(8):1039–1041. doi:10.1182/blood-2016-09-738245
142. Heczey A, Louis CU, Savoldo B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. 2017;25(9):2214–2224. doi:10.1016/j.ymthe.2017.05.012
143. Suarez ER, Chang de K, Sun J, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7(23):34341–34355. doi:10.18632/oncotarget.9114
144. Zhou JT, Liu JH, Song TT, Ma B, Amidula N, Bai C. EGLIF-CAR-T cells secreting PD-1 blocking antibodies significantly mediate the elimination of gastric cancer. Cancer Manag Res. 2020;12:8893–8902. doi:10.2147/CMAR.S260915
145. Li S, Siriwon N, Zhang X, et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin Cancer Res. 2017;23(22):6982–6992. doi:10.1158/1078-0432.CCR-17-0867
146. Rafiq S, Yeku OO, Jackson HJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. 2018;36(9):847–856. doi:10.1038/nbt.4195
147. Ping Y, Li F, Nan S, et al. Augmenting the effectiveness of CAR-T cells by enhanced self-delivery of PD-1-neutralizing scFv. Front Cell Dev Biol. 2020;8:803. doi:10.3389/fcell.2020.00803
148. Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130–3144. doi:10.1172/JCI83092
149. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23(9):2255–2266. doi:10.1158/1078-0432.CCR-16-1300
150. Rupp LJ, Schumann K, Roybal KT, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7(1):737. doi:10.1038/s41598-017-00462-8
151. Choi BD, Yu X, Castano AP, et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J Immunother Cancer. 2019;7(1):304. doi:10.1186/s40425-019-0806-7
152. Liu G, Zhang Q, Li D, et al. PD-1 silencing improves anti-tumor activities of human mesothelin-targeted CAR T cells. Hum Immunol. 2021;82(2):130–138. doi:10.1016/j.humimm.2020.12.002
153. Zou F, Lu L, Liu J, et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat Commun. 2019;10(1):4109. doi:10.1038/s41467-019-11893-4
154. Simon B, Harrer DC, Schuler-Thurner B, et al. The siRNA-mediated downregulation of PD-1 alone or simultaneously with CTLA-4 shows enhanced in vitro CAR-T-cell functionality for further clinical development towards the potential use in immunotherapy of melanoma. Exp Dermatol. 2018;27(7):769–778. doi:10.1111/exd.13678
155. Condomines M, Arnason J, Benjamin R, Gunset G, Plotkin J, Sadelain M. Tumor-targeted human T cells expressing CD28-based chimeric antigen receptors circumvent CTLA-4 inhibition. PLoS One. 2015;10(6):e0130518. doi:10.1371/journal.pone.0130518
156. Yin Y, Boesteanu AC, Binder ZA, et al. Checkpoint blockade reverses anergy in IL-13Ralpha2 humanized scFv-based CAR T cells to treat murine and canine gliomas. Mol Ther Oncolytics. 2018;11:20–38. doi:10.1016/j.omto.2018.08.002
157. Liu X, Ranganathan R, Jiang S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016;76(6):1578–1590. doi:10.1158/0008-5472.CAN-15-2524
158. Liu H, Lei W, Zhang C, et al. CD19-specific CAR T cells that express a PD-1/CD28 chimeric switch-receptor are effective in patients with PD-L1-positive B-cell lymphoma. Clin Cancer Res. 2020.
159. Parriott G, Deal K, Crean S, Richardson E, Nylen E, Barber A. T-cells expressing a chimeric-PD1-Dap10-CD3zeta receptor reduce tumour burden in multiple murine syngeneic models of solid cancer. Immunology. 2020;160(3):280–294. doi:10.1111/imm.13187
160. Bechman N, Maher J. Lymphodepletion strategies to potentiate adoptive T-cell immunotherapy - what are we doing; where are we going? Expert Opin Biol Ther. 2020;1–11.
161. Suryadevara CM, Desai R, Farber SH, et al. Preventing Lck activation in CAR T cells confers treg resistance but requires 4-1BB signaling for them to persist and treat solid tumors in nonlymphodepleted hosts. Clin Cancer Res. 2019;25(1):358–368. doi:10.1158/1078-0432.CCR-18-1211
162. Golumba-Nagy V, Kuehle J, Hombach AA, Abken H. CD28-zeta CAR T cells resist TGF-beta repression through IL-2 signaling, which can be mimicked by an engineered IL-7 autocrine loop. Mol Ther. 2018;26(9):2218–2230. doi:10.1016/j.ymthe.2018.07.005
163. Foster AE, Dotti G, Lu A, et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother. 2008;31(5):500–505. doi:10.1097/CJI.0b013e318177092b
164. Zhang L, Yu Z, Muranski P, et al. Inhibition of TGF-beta signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. 2013;20(5):575–580. doi:10.1038/gt.2012.75
165. Bollard CM, Rossig C, Calonge MJ, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99(9):3179–3187. doi:10.1182/blood.V99.9.3179
166. Kloss CC, Lee J, Zhang A, et al. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26(7):1855–1866. doi:10.1016/j.ymthe.2018.05.003
167. Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. 2018;14(3):317–324. doi:10.1038/nchembio.2565
168. Hou AJ, Chang ZL, Lorenzini MH, Zah E, Chen YY. TGF-beta-responsive CAR-T cells promote anti-tumor immune function. Bioeng Transl Med. 2018;3(2):75–86. doi:10.1002/btm2.10097
169. Tang N, Cheng C, Zhang X, et al. TGF-beta inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020;5(4). doi:10.1172/jci.insight.133977
170. Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;29(39):5346–5358. doi:10.1038/onc.2010.292
171. Aandahl EM, Moretto WJ, Haslett PA, et al. Inhibition of antigen-specific T cell proliferation and cytokine production by protein kinase A type I. J Immunol. 2002;169(2):802–808. doi:10.4049/jimmunol.169.2.802
172. Siriwon N, Kim YJ, Siegler E, et al. CAR-T cells surface-engineered with drug-encapsulated nanoparticles can ameliorate intratumoral T-cell hypofunction. Cancer Immunol Res. 2018;6(7):812–824. doi:10.1158/2326-6066.CIR-17-0502
173. Beavis PA, Henderson MA, Giuffrida L, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. 2017;127(3):929–941. doi:10.1172/JCI89455
174. Masoumi E, Jafarzadeh L, Mirzaei HR, et al. Genetic and pharmacological targeting of A2a receptor improves function of anti-mesothelin CAR T cells. J Exp Clin Cancer Res. 2020;39(1):49. doi:10.1186/s13046-020-01546-6
175. Li N, Tang N, Cheng C, et al. Improving the anti-solid tumor efficacy of CAR-T cells by inhibiting adenosine signaling pathway. Oncoimmunology. 2020;9(1):1824643. doi:10.1080/2162402X.2020.1824643
176. Newick K, O’Brien S, Sun J, et al. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol Res. 2016;4(6):541–551. doi:10.1158/2326-6066.CIR-15-0263
177. Cascone T, McKenzie JA, Mbofung RM, et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 2018;27(5):977–987 e974. doi:10.1016/j.cmet.2018.02.024
178. Chang CH, Qiu J, O’Sullivan D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229–1241. doi:10.1016/j.cell.2015.08.016
179. Fischer K, Hoffmann P, Voelkl S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109(9):3812–3819. doi:10.1182/blood-2006-07-035972
180. Brand A, Singer K, Koehl GE, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016;24(5):657–671. doi:10.1016/j.cmet.2016.08.011
181. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016;76(6):1381–1390. doi:10.1158/0008-5472.CAN-15-1743
182. Fallarino F, Grohmann U, Vacca C, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9(10):1069–1077. doi:10.1038/sj.cdd.4401073
183. Ninomiya S, Narala N, Huye L, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. 2015;125(25):3905–3916. doi:10.1182/blood-2015-01-621474
184. Hu M, Zhou W, Wang Y, et al. Discovery of the first potent proteolysis targeting chimera (PROTAC) degrader of indoleamine 2,3-dioxygenase 1. Acta Pharm Sin B. 2020;10(10):1943–1953. doi:10.1016/j.apsb.2020.02.010
185. Yazdanifar Y, Zhou Z, Grover G, et al. Overcoming immunological resistance enhances the efficacy of a novel anti-tMUC1-CAR T cell treatment against pancreatic ductal adenocarcinoma. Cells. 2019;8(9):1070. doi:10.3390/cells8091070
186. Mussai F, Egan S, Hunter S, et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res. 2015;75(15):3043–3053. doi:10.1158/0008-5472.CAN-14-3443
187. Fultang L, Booth S, Yogev O, et al. Metabolic engineering against the arginine microenvironment enhances CAR-T cell proliferation and therapeutic activity. Blood. 2020;136(10):1155–1160. doi:10.1182/blood.2019004500
188. Wang T, Gnanaprakasam JNR, Chen X, et al. Inosine is an alternative carbon source for CD8(+)-T-cell function under glucose restriction. Nat Metab. 2020;2(7):635–647. doi:10.1038/s42255-020-0219-4
189. Shyer JA, Flavell RA, Bailis W. Metabolic signaling in T cells. Cell Res. 2020;30(8):649–659. doi:10.1038/s41422-020-0379-5
190. Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345–1360. doi:10.1084/jem.20151159
191. Kawalekar OU, O’Connor RS, Fraietta JA, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(3):712. doi:10.1016/j.immuni.2016.02.023
192. Sukumar M, Liu J, Ji Y, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123(10):4479–4488. doi:10.1172/JCI69589
193. Sukumar M, Liu J, Mehta GU, et al. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 2016;23(1):63–76. doi:10.1016/j.cmet.2015.11.002
194. Sotillo E, Barrett DM, Black KL, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282–1295. doi:10.1158/2159-8290.CD-15-1020
195. Brudno JN, Maric I, Hartman SD, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267–2280. doi:10.1200/JCO.2018.77.8084
196. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–28. doi:10.1038/nm.4441
197. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399). doi:10.1126/scitranslmed.aaa0984
198. Krenciute G, Prinzing BL, Yi Z, et al. Transgenic expression of IL15 improves antiglioma activity of IL13Ralpha2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. 2017;5(7):571–581. doi:10.1158/2326-6066.CIR-16-0376
199. Hegde M, Corder A, Chow KK, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. 2013;21(11):2087–2101. doi:10.1038/mt.2013.185
200. Orlando EJ, Han X, Tribouley C, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24(10):1504–1506. doi:10.1038/s41591-018-0146-z
201. Jacoby E, Nguyen SM, Fountaine TJ, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun. 2016;7(1):12320. doi:10.1038/ncomms12320
202. Gardner R, Wu D, Cherian S, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127(20):2406–2410. doi:10.1182/blood-2015-08-665547
203. Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187–195. doi:10.1097/CJI.0000000000000169
204. Hamieh M, Dobrin A, Cabriolu A, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568(7750):112–116. doi:10.1038/s41586-019-1054-1
205. Cohen AD, Garfall AL, Stadtmauer EA, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. 2019;129(6):2210–2221. doi:10.1172/JCI126397
206. Ramakrishna S, Highfill SL, Walsh Z, et al. Modulation of target antigen density improves CAR T-cell functionality and persistence. Clin Cancer Res. 2019;25(17):5329–5341. doi:10.1158/1078-0432.CCR-18-3784
207. Walker AJ, Majzner RG, Zhang L, et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol Ther. 2017;25(9):2189–2201. doi:10.1016/j.ymthe.2017.06.008
208. Majzner RG, Rietberg SP, Sotillo E, et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. 2020;10(5):702–723. doi:10.1158/2159-8290.CD-19-0945
209. Dai H, Wu Z, Jia H, et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J Hematol Oncol. 2020;13(1):30. doi:10.1186/s13045-020-00856-8
210. Chen KH, Wada M, Pinz KG, et al. A compound chimeric antigen receptor strategy for targeting multiple myeloma. Leukemia. 2018;32(2):402–412. doi:10.1038/leu.2017.302
211. Wei X, Lai Y, Li J, et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology. 2017;6(3):e1284722. doi:10.1080/2162402X.2017.1284722
212. Wilkie S, van Schalkwyk MC, Hobbs S, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32(5):1059–1070. doi:10.1007/s10875-012-9689-9
213. de Larrea CF, Staehr M, Lopez AV, et al. Defining an optimal dual-targeted CAR T-cell therapy approach simultaneously targeting BCMA and GPRC5D to prevent BCMA escape-driven relapse in multiple myeloma. Blood Cancer Discov. 2020;1(2):146–154. doi:10.1158/2643-3230.BCD-20-0020
214. Bielamowicz K, Fousek K, Byrd TT, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018;20(4):506–518. doi:10.1093/neuonc/nox182
215. Fousek K, Watanabe J, Joseph SK, et al. CAR T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia. 2021;35(1):75–89. doi:10.1038/s41375-020-0792-2
216. Grada Z, Hegde M, Byrd T, et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids. 2013;2:e105. doi:10.1038/mtna.2013.32
217. Schneider D, Xiong Y, Wu D, et al. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J Immunother Cancer. 2017;5:42. doi:10.1186/s40425-017-0246-1
218. De Munter S, Ingels J, Goetgeluk G, et al. Nanobody based dual specific CARs. Int J Mol Sci. 2018;19(2):403. doi:10.3390/ijms19020403
219. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173(6):1426–1438 e1411. doi:10.1016/j.cell.2018.03.038
220. Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol. 2006;43(6):763–771. doi:10.1016/j.molimm.2005.03.007
221. Wing A, Fajardo CA, Posey AD
222. Choi BD, Yu X, Castano AP, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. 2019;37(9):1049–1058. doi:10.1038/s41587-019-0192-1
223. Pan H, Li W, Chen Z, et al. Click CAR-T cell engineering for robustly boosting cell immunotherapy in blood and subcutaneous xenograft tumor. Bioact Mater. 2021;6(4):951–962. doi:10.1016/j.bioactmat.2020.09.025
224. Park AK, Fong Y, Kim SI, et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci Transl Med. 2020;12(559):559. doi:10.1126/scitranslmed.aaz1863
225. Sampson JH, Choi BD, Sanchez-Perez L, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20(4):972–984. doi:10.1158/1078-0432.CCR-13-0709
226. Lai J, Mardiana S, House IG, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol. 2020;21(8):914–926. doi:10.1038/s41590-020-0676-7
227. Torres-Collado AX, Jazirehi AR. Overcoming resistance of human non-hodgkin’s lymphoma to CD19-CAR CTL therapy by celecoxib and histone deacetylase inhibitors. Cancers. 2018;10(6):6. doi:10.3390/cancers10060200
228. Kearney CJ, Vervoort SJ, Hogg SJ, et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci Immunol. 2018;3(23):eaar3451. doi:10.1126/sciimmunol.aar3451
229. Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305(5689):1471–1474. doi:10.1126/science.1098231
230. Peng W, Chen JQ, Liu C, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6(2):202–216. doi:10.1158/2159-8290.CD-15-0283
231. Awasthi R, Pacaud L, Waldron E, et al. Tisagenlecleucel cellular kinetics, dose, and immunogenicity in relation to clinical factors in relapsed/refractory DLBCL. Blood Adv. 2020;4(3):560–572. doi:10.1182/bloodadvances.2019000525
232. Mueller KT, Waldron E, Grupp SA, et al. Clinical pharmacology of tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin Cancer Res. 2018;24(24):6175–6184. doi:10.1158/1078-0432.CCR-18-0758
233. Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+: CD8+composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–2138. doi:10.1172/JCI85309
234. Xu J, Chen LJ, Yang SS, et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci U S A. 2019;116(19):9543–9551. doi:10.1073/pnas.1819745116
235. Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–6115. doi:10.1158/1078-0432.CCR-06-1183
236. Maus MV, Haas AR, Beatty GL, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. 2013;1(1):26–31. doi:10.1158/2326-6066.CIR-13-0006
237. Wagner DL, Fritsche E, Pulsipher MA, et al. Immunogenicity of CAR T cells in cancer therapy. Nat Rev Clin Oncol. 2021. doi:10.1038/s41571-021-00476-2
238. Brudno JN, Lam N, Vanasse D, et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med. 2020;26(2):270–280. doi:10.1038/s41591-019-0737-3
239. Mikkilineni L, Manasanch EE, Lam N, et al. T cells expressing an anti-B-Cell Maturation Antigen (BCMA) chimeric antigen receptor with a fully-human heavy-chain-only antigen recognition domain induce remissions in patients with relapsed multiple myeloma. Blood. 2019;134(Supplement_1):3230. doi:10.1182/blood-2019-129088
240. Zhang T, Barber A, Sentman CL. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res. 2006;66(11):5927–5933. doi:10.1158/0008-5472.CAN-06-0130
241. Sengupta S, Thaci B, Crawford AC, Sampath P. Interleukin-13 receptor alpha 2-targeted glioblastoma immunotherapy. Biomed Res Int. 2014;2014:952128. doi:10.1155/2014/952128
242. Whilding LM, Parente-Pereira AC, Zabinski T, et al. Targeting of aberrant alphavbeta6 integrin expression in solid tumors using chimeric antigen receptor-engineered T cells. Mol Ther. 2017;25(10):2427. doi:10.1016/j.ymthe.2017.09.018
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.