Back to Journals » Cancer Management and Research » Volume 12
HNRNPCL1, PRAMEF1, CFAP74, and DFFB: Common Potential Biomarkers for Sporadic and Suspected Lynch Syndrome Endometrial Cancer
Authors Gao Y ![]() , Zhang X, Wang T, Zhang Y, Wang Q, Hu Y
, Zhang X, Wang T, Zhang Y, Wang Q, Hu Y ![]()
Received 5 June 2020
Accepted for publication 22 September 2020
Published 4 November 2020 Volume 2020:12 Pages 11231—11241
DOI https://doi.org/10.2147/CMAR.S262421
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Yuan Gao,1,* Xiuping Zhang,2,* Tian Wang,1 Ye Zhang,1 Qingxuan Wang,1 Yuanjing Hu1
1Department of Gynecological Oncology, Tianjin Central Hospital of Gynecology & Obstetrics, Tianjin, People’s Republic of China; 2Reproductive Center, Shanxi Maternal and Child Health Hospital, Taiyuan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yuanjing Hu Department of Gynecological Oncology
Tianjin Central Hospital of Gynecology & Obstetrics, Tianjin 300100, People’s Republic of China
Tel +86-18920196053
Email [email protected]
Purpose: To investigate the genes of patients with sporadic endometrial cancer (EC) and suspected Lynch syndrome (LS)-related EC in the Chinese population. Identification of meaningful mutation sites can provide theoretical basis for molecular targeted therapy, aiming to improve the prognosis of patients with EC.
Methods: We recruited 388 patients with EC for mismatch repair (MMR) immunohistochemistry and MLH1 methylation analysis. Based on the results, they were divided into four groups: MMR without deletion group (sporadic EC group 1); MLH1&PMS2 deletion and MLH1 methylation group (sporadic EC group 2); MSH2 and/or MSH6 deletion group (suspected LS group); and unclassified group (remainder cases). Patients from each group were randomly screened for whole-exome sequencing detection. Genome Analysis Toolkit, VarScant, MuTect, and CONTRA were used to detect the insertions/deletions, single nucleotide polymorphisms, and copy number variations. Gene Ontology term and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses were performed with Database for Annotation, Visualization and Integrated Discovery. Protein–protein interaction analysis was accomplished through the STRING database.
Results: The MMR immunohistochemistry results were positive (without MMR deletion) and negative in 299 patients and 89 patients, respectively. The 32, 10, 13, and 7 patients in the sporadic EC group 1, sporadic EC group 2, suspected LS group, and unclassified group were randomly selected for whole-exome sequencing, respectively. These three groups had a total of 86 common mutation sites, which were distributed on 26 genes. Among the top 30 common high-frequency mutation sites, 12, 5, 4, and 3 mutation sites were located on HNRNPCL1, PRAMEF1, HNRNPCL2, and CFAP74, respectively. Protein–protein interaction analysis showed that DFFB was associated with the most genes. There were some differences in the number of specific mutations in the families of different LS-related EC proband.
Conclusion: HNRNPCL1, PRAMEF1, CFAP74, and DFFB may be potential biomarkers for EC or LS-related EC.
Keywords: endometrial cancer, Lynch syndrome, whole-exome sequencing, WES, mismatch repair gene, gene mutation
Introduction
Endometrial cancer (EC) is one of the most common tumors of the female reproductive system, with nearly 200,000 new cases recorded each year.1 Following ovarian and cervical cancers, EC is the third most common gynecological malignancy that can cause death.2 In China, with the improvement of economic conditions and dietary habits, the incidence of EC has also increased annually.3 It is currently second only to cervical cancer and ranks second among malignant tumors of the female reproductive system. Lynch syndrome (LS), also termed hereditary non-polyposis colorectal cancer, is an autosomal dominant hereditary disease caused by mutations in the DNA mismatch repair (MMR) gene.4 Previous studies had mostly focused on LS-related colorectal cancer, and great progress had been made in gene levels and preventive research studies. In recent years, a growing body of evidences have shown that the risk of EC in females with LS is 40–60%, reaching or even exceeding that of colorectal cancer, and has become the first tumor to receive clinical attention.5 With the increasing incidence of EC, hereditary endometrial malignancies have received more research attention. EC can be divided into sporadic EC and LS-related EC according to the deletion of the MMR gene.
There is a certain relationship between the occurrence and development of EC and a variety of genes. For example, studies have shown that >80% of EC and >50% of paracancer lesions have the PTEN gene mutation.6 POLE mutational status is an independent prognostic factor for patients with EC.7 In addition, genes including MDC1, KIAA1109, and CTNNB1 have also been associated with the occurrence and development of EC.8–10 In recent years, with the continuous understanding of the role of genes in various diseases and the development of sequencing technology, next-generation sequencing technologies have been generally applied to investigate genes that play an important role in the development of a variety of cancers and related mechanisms.11 Among them, whole-exome sequencing (WES) is the most frequently applied genome sequencing method.12 Exons contain the information required to encode proteins and their DNA can be captured and enriched using sequence capture technology. Although the exon region only accounts for approximately 1% of the entire genome,13 it contains 85% of the pathogenic mutations. Previous studies on the gene mutation of EC were mostly systematic or single. This study aims to reveal the common mutation genes between sporadic EC and suspected LS-related EC as the potential biomarkers, which may be beneficial to the comprehensive screening and treatment of EC.
The present study compared the mutation characteristics of patients with sporadic EC and suspected LS-related EC by WES, and found mutation genes that are closely related to EC. This will provide a theoretical basis for disclosing the molecular mechanism of EC and facilitate targeted therapy.
Patients and Methods
Patients and Sample Collection
A total of 388 patients with EC who underwent surgical treatment in Tianjin Central Hospital of Gynecology & Obstetrics (Tianjin, China) from October 2017 to February 2019 were collected. Hysterectomy specimens were collected for immunohistochemical detection of four MMR proteins and grouped according to the results of immunohistochemistry. Furthermore, the tumor formalin-fixed paraffin-embedded samples of the patients and blood plasma samples of families of two probands were collected. Written informed consent was provided by each patient that enrolled in this study. This study was approved by The Ethics Committee of Tianjin Central Hospital of Gynecology & Obstetrics.
Immunohistochemistry of MMR Proteins
All specimens were routinely pathologically sectioned and stained with hematoxylin and eosin. The paraffin sections with normal tissues as internal controls were selected for immunohistochemical stained. Immunohistochemical streptavidin-peroxidase method was used for labeling, and 3,3ʹ,4,4ʹ-Biphenyltetramine tetrahydrochloride was used for color development. Antibodies mutL homolog 1 (MLH1), mutS homolog 2 (MSH2), MSH6, and PMS1 homolog 2, mismatch repair system component (PMS2) were purchased from Beijing ZSGB-Bio Co., Ltd. (Beijing, China). The expression of MMR protein is localized in the nucleus. Positive staining of the nuclei of tumor cells nuclei indicates that the MMR protein is positively expressed. In contrast, negative staining indicates the absence of MMR protein expression. When interpreting the results, attention should be paid to the selection of internal controls. Positive staining of non-tumorous endometrial stroma, glands, and lymphocyte nuclei is shown by a brown-yellow color. Only complete absence of expression in the tumor cell nucleus in an internal control-positive background can be considered effective.
Methylation Analysis of the MLH1 Promoter
Cases with loss of MLH1 expression were tested for MLH1 methylation using tumor DNA. The bisulfite modified sequencing technique (EZ DNA Methylation-Gold kit; Zymo Research, Irvine, CA, USA) was used to detect the methylation status of MLH1. The whole experimental procedure was performed strictly according to the instructions provided by the manufacturer.
WES
DNA from the paraffin sections was extracted using a Genomic DNA extraction kit (Qiagen, Hilden, Germany). The blood plasma samples were used to extract circulating free DNA (ccfDNA) with the QIAamp circulating nucleic acid kit (Qiagen) based on the instructions provided by the manufacturer.
Library preparation for each sample was carried out according to the instructions provided by the manufacturer. Briefly, 3 μg of high-quality genomic DNA or ccfDNA was applied to construct the library. Subsequently, the DNA was fragmented (150–200 bp fragments) using the SureSelectXT Library Prep Kit (Agilent Technologies, Santa Clara, CA, USA), followed by end-repair and adapter ligation. The adaptor-ligated libraries were hybridized using the SureSelectXT Reagent Kit and the hybridized DNA was captured using streptavidin-coated magnetic beads (Agilent Technologies). The captured exomes were pooled and sequenced using the Illumina Hiseq Xten platform (Illumina, Inc., San Diego, CA, USA). The Genome Analysis Toolkit (GATK, version 2.3.9) software (Broad Institute, Cambridge, MA, USA) was applied to analyze the raw data. The average sequencing depth reached 100×, and it was >20× in up to 96% of the regions. The QC30 of the raw data for all samples was >85%, and the allele frequency exceeded 40% for all mutations.
Mutation Analysis
The data were aligned to the hg19 reference genome using the Burrows–Wheeler Aligner. The GATK (version 2.3.9; Broad Institute) was applied to re-compare the reads in the interval, calibrate and rearrange the alkali matrix quality values, and determine the sequence depth and coverage. The statistics of sequencing results are listed in Table 1. Insertions/deletions were detected by GATK (version 2.3.9; Broad Institute) and VarScant (version 2.4.3; McDonnell Genome Institute, Washington University, St. Louis, MO, USA), single nucleotide polymorphisms were identified by MuTect (version 1.1.4; Broad Institute) and VarScan (version 2.4.3; McDonnell Genome Institute), copy number variations were determined by CONTRA (http://jtjli.users.sourceforge.net), and fusions were identified with a self-developed fusion program.
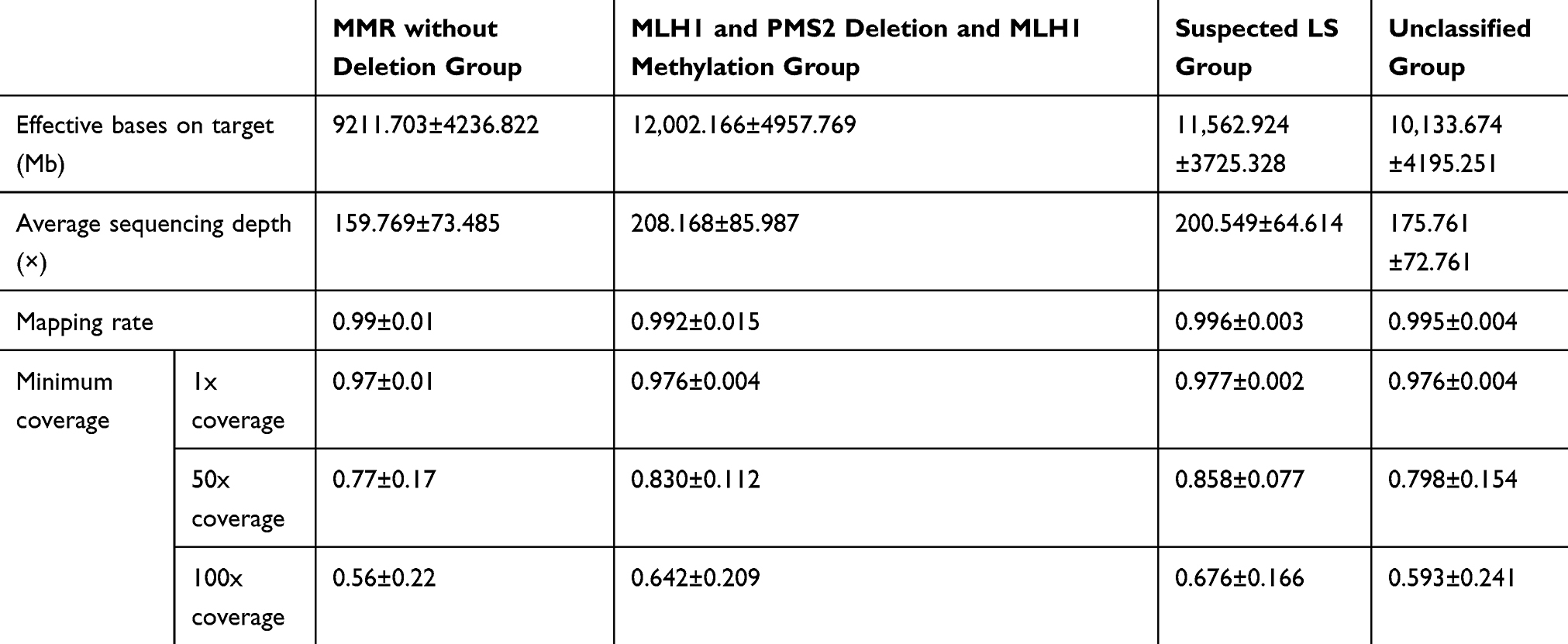 |
Table 1 Statistics of Sequencing Data |
Bioinformatics Analysis
The mutation sites were annotated to the COSMIC (Catalogue of Somatic Mutations in Cancer) database (https://cancer.sanger.ac.uk/cosmic). Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed with the Database for Annotation, Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/). The results of KEGG, GO, mutation type statistics, and high-frequency mutation gene statistics were plotted using ggplot2 (version 3.2.1; Hadley Wickham, RStudio, Boston, MA, USA). Protein–protein interaction (PPI) analysis was conducted using the STRING database (https://string-db.org/). Various Venn diagrams were drawn by VennDiagram (version 1.6.20; R package), Vennerable (version 3.0; R package), and Venn (version 1.9; R package). P<0.05 was the cut-off criterion for statistical significance.
Results
MMR Immunohistochemical Analysis
MMR immunohistochemical analysis revealed that 299 of the 388 patients with EC were positive for MMR immunohistochemistry, whereas 89 patients were negative. Patients without MMR deletion were classified into the sporadic EC group 1. According to the results of MMR immunohistochemistry and MLH1 methylation analyses, we divided the negative patients into the MLH1&PMS2 deletion and MLH1 methylation group (sporadic EC group 2), MSH2 and/or MSH6 deletion group (suspected LS group), and unclassified group (with those not satisfying the conditions of any of the other three groups). Of note, 32 patients in the sporadic EC group 1, 10 patients in the sporadic EC group 2, 13 suspected LS patients, and seven unclassified patients were randomly selected for WES. The clinical characteristics and immunohistochemical results of the patients are shown in Supplementary Tables S1 and S2, respectively.
Analysis of Common Mutation Sites
In this study, the most common mutation type in any group of samples was the missense mutation, followed by coding sequence insertion/deletion and frameshift (Figure 1). The sporadic EC group 1, sporadic EC group 2, and suspected LS-related group had a total of 86 common mutation sites, which were distributed on 26 genes (Figure 2).
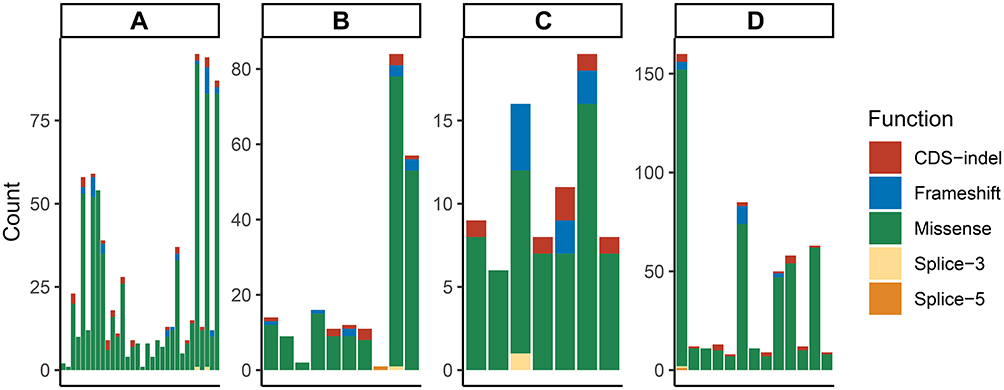 |
Figure 1 Distribution of mutation types in each group. A represents the without MMR deletion group; B represents the MLH1&PMS2 deletion and MLH1 methylation group; C represents unclassified patients; and D represents the MSH2 and/or MSH6 deletion group. Each column represents different mutation types and counts in one patient. |
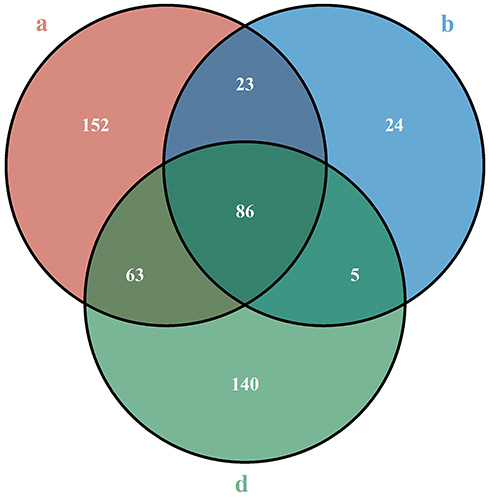 |
Figure 2 Venn diagrams of mutation sites of the (a, b, and d groups). a represents the without MMR deletion group; b represents the MLH1&PMS2 deletion and MLH1 methylation group; and d represents the MSH2 and/or MSH6 deletion group. |
The top 30 common high-frequency mutation sites mainly belonged to the missense mutation. These mutation sites were located at the ATPase family AAA domain containing 3B (ATAD3B), solute carrier family 35 member E2B (SLC35E2B), heterogeneous nuclear ribonucleoprotein C-like 1 (HNRNPCL1), PRAME family member 1 (PRAMEF1), cilia and flagella associated protein 74 (CFAP74), HNRNPCL2, PRAMEF7, family with sequence similarity 131 member C (FAM131C), DNA fragmentation factor subunit beta (DFFB), and PRAMEF18 (Figure 3). Among these genes, HNRNPCL1, PRAMEF1, HNRNPCL2, and CFAP74 contained 12, 5, 4, and 3 mutation sites, respectively, and the residual six genes had only one mutation site (ATAD3B c.[1915C>T], SLC35E2B c.[686G>A], PRAMEF7 c.[1037T>C], FAM131C c.[776C>T], DFFB c.[587G>A], PRAMEF18 c.[313T>A]). In addition to CFAP74 c.[1212_1213insGAG], HNRNPCL2 c.[674G>C], HNRNPCL2 c.[685A>T], HNRNPCL2 c.[686T>C], and HNRNPCL2 c.[697G>A], the other 25 high-frequency common mutation sites had annotation information in the COSMIC database (Table 2).
 |
Table 2 The Annotation Results of Top 30 High-Frequency Mutation Sites in COSMIC Database |
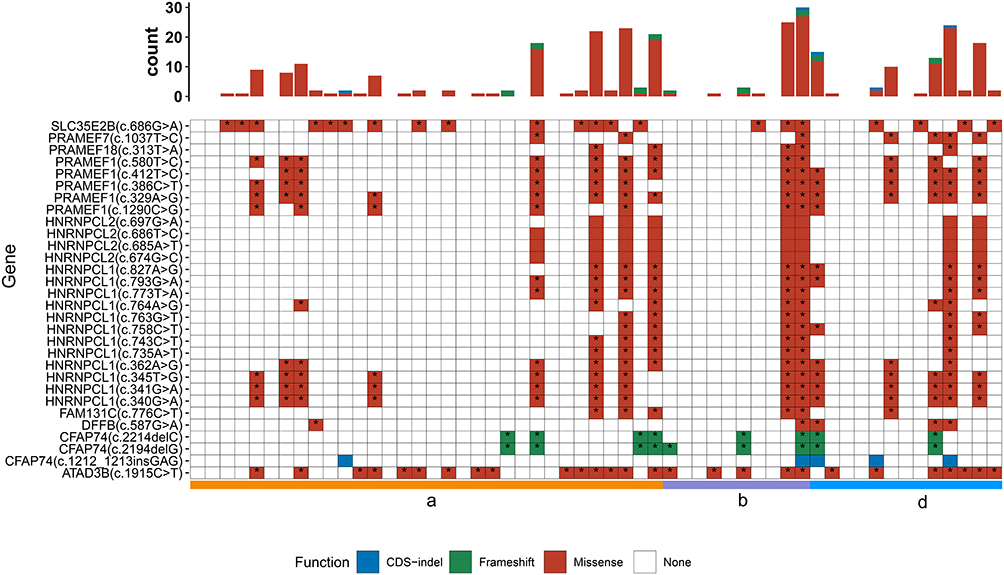 |
Figure 3 Mutation types, distribution in each sample, and annotation information in the COSMIC database of the top 30 common high-frequency mutation sites. a represents the without MMR deletion group; b represents the MLH1&PMS2 deletion and MLH1 methylation group; and d represents the MSH2 and/or MSH6 deletion group. * represents that the mutation is recorded in COSMIC database. Each column represents mutations in one patient. |
Enrichment GO and Pathway Terms
KEGG pathway and GO enrichment analyses were performed on the above 10 genes in which a high-frequency mutation site was detected. These genes were found to be enriched in five GO terms, including one cellular component and four biological processes. In cellular components, the intracellular ribonucleoprotein complex was the only term which was highly enriched. The highly enriched biological processes were negative regulation of cell differentiation, negative regulation of the apoptotic process, positive regulation of cell proliferation and negative regulation of transcription, DNA-templated (Figure 4A).
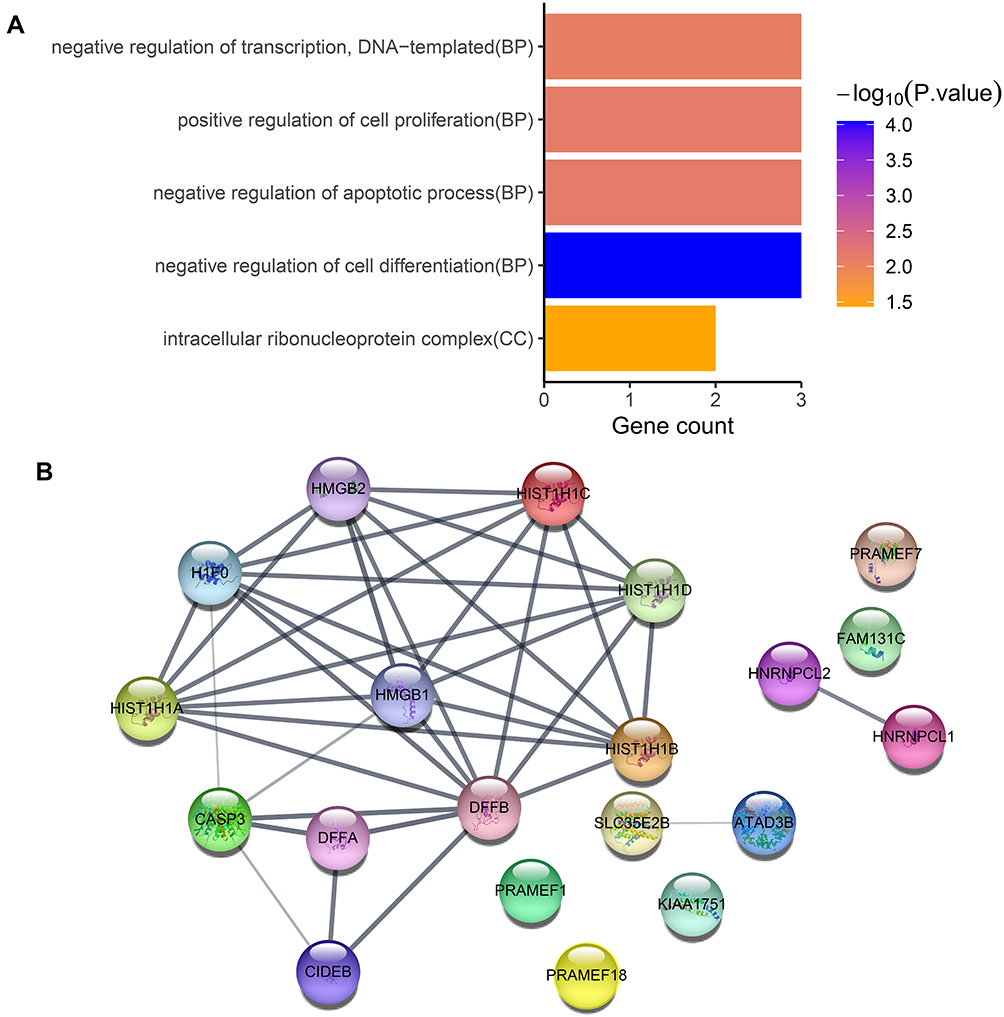 |
Figure 4 (A) GO enrichment analysis of genes in which the top 30 common high-frequency mutation sites are located. (B) The PPI network of these genes in which the top 30 common high-frequency mutation sites are located. |
PPI Network
The lowest interaction score in the STRING analysis was 0.4. To investigate the presence of an indirect relationship between genes, the maximum number of interactions was set to ≤10. Among these genes, DFFB was associated with most genes, such as histone cluster 1 H1 family member b (HIST1H1B), HIST1H1D, HIST1H1C, high mobility group box 2 (HMGB2), HMGB1, H1 histone family member 0 (H1F0), HIST1H1A, caspase 3 (CASP3), DNA fragmentation factor subunit alpha (DFFA), and cell death-inducing DFFA like effector b (CIDEB) (Figure 4B).
Genetic Analysis of Families of Suspected LS-Related EC Patients with MSH2/MSH6 Deletion
We further analyzed the mutation characteristics of the family members of two suspected LS-related EC patient with MSH2/MSH6 deletion through WES sequencing of ccfDNA. The first proband was a 34-year-old woman, and the International Federation of Gynecology and Obstetrics stage was IA. The proband, her cousin, and her two aunts formed the first family. The second 48-year-old proband who was also diagnosed with IA-stage EC and her sister constituted the second family. There were 752 common mutation sites in the first family (Figure 5A), accounting for 25.84%, 26.88%, 26.91%, and 27.74% of the mutations in samples of the proband, her cousin, and two aunts, respectively. After removing common genes with the second family, the first family had 321 specific mutation sites. This number was lower than that of the proband (408), her cousin (323), and two aunts (467 and 517, respectively) (Figure 6). The second family had a total of 1,759 common mutation sites, which accounted for 65.61% and 62.05% of the number of mutations of the proband and her sister, respectively (Figure 5B). After removing the genes shared with the first family, there were 846 specific mutation sites in the second family. This number exceeded that of the proband (615) and her sister (638) (Figure 6).
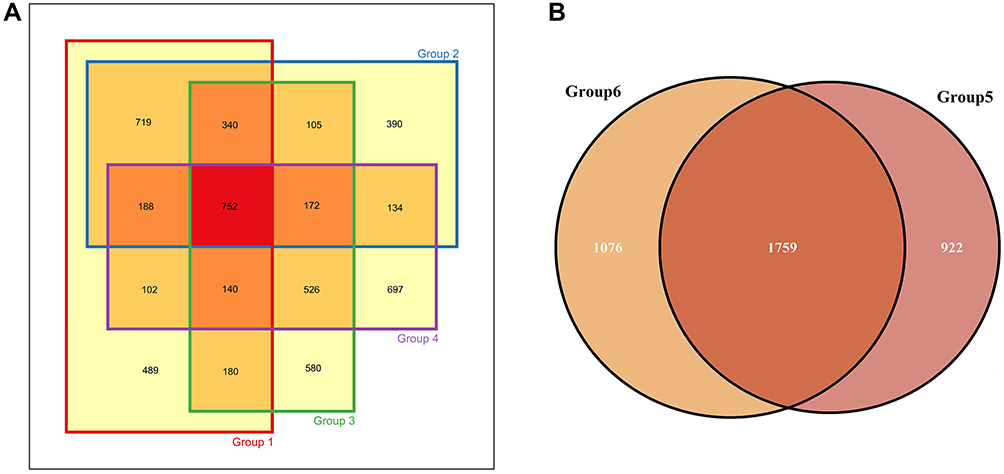 |
Figure 5 Venn diagrams of mutation sites in different families. (A) The family of the first proband. (B) The family of the second proband. Group 1 represents the first proband; Group 2 represents her cousin; Group 3 and Group 4 represents her aunts. Group 5 and Group 6 represent the second proband and her sister. |
 |
Figure 6 Statistics of mutation sites in the members of the first and second families. The dots and links under the histogram represent different combinations of six members in two families, and columns represent specific mutation site numbers in different combinations of members. The orange column represents the number of specific mutation sites in the second family; the blue column represents the number of specific mutation sites in the first family. |
Discussion
In previous studies that searched for genes related to EC, there was no clear distinction between sporadic EC and LS-related EC,14 or only sporadic EC15 or LS-related EC16 was studied independently. However, research studies have shown differences in genetic mutation information among patients with sporadic EC and LS-related EC.17 Accordingly, during the screening of representative genes related to the occurrence and development of EC, the difference in mutation genes between patients should be eliminated, and the genes that are prevalent in all patients with EC should be identified. These common genes have more research significance and application value. Therefore, in the present study, according to the results of MMR immunohistochemistry and MLH1 methylation analyses, patients with EC were divided into the four aforementioned groups. Patients with suspected LS-related EC were classified based on the immunohistochemistry results as MSH2 and/or MSH6 deletion and MLH1&PMS2 without deletion (Supplementary Table S2). Subsequently, we used the WES technology and bioinformatics analysis to obtain the common mutated genes of the three groups of patients and analyzed their roles in the occurrence and development of EC. There were fewer patients with LS-related EC, rendering the collection of samples difficult; hence, this study is limited by an insufficient sample size. However, to the best of our knowledge, this was the first study to investigate the mutational characteristics common to sporadic EC and LS-related EC in the Chinese population, which has certain innovation and research significance. In addition, this was a preliminary study; thus, the results are exploratory, and further research is warranted to verify and advance these findings.
This study found that 12, 5, 4, and 3 of the top 30 common high-frequency mutation sites were located in HNRNPCL1, PRAMEF1, HNRNPCL2, and CFAP74, respectively. In addition to CFAP74 c. [1212_1213insGAG] and all mutations in HNRNPCL2, these mutation sites had annotation information in the COSMIC database. Although only one common high-frequency mutation site was located on DFFB, this gene was related to multiple genes in the PPI analysis. The HNRNPCL1 gene is located on 1p36.21, also termed HNRPCL1. HNRNPCL1 is a protein-coding gene and may play a role in nucleosome assembly by neutralizing basic proteins.18 GO annotations related to this gene include nucleic acid binding and nucleotide binding. The PRAMEF1 gene is a member of the PRAME (preferentially expressed antigen of melanoma) gene family which is expressed in numerous types of cancer but may function in reproductive tissues during development.19 PRAMEF1 is associated with four GO terms, namely negative regulation of cell differentiation, negative regulation of the apoptotic process, positive regulation of cell proliferation, and negative regulation of transcription, DNA-templated. CFAP74 is also known as C1orf222 or KIAA1751. CFAP74 is a protein that contains the ASPM-SPD-2-Hydin domain. The proteins containing this domain are commonly associated with cilia, flagella, centrosomes, and Golgi bodies, and they bind microtubules. DFFB participates in nucleic acid metabolism and is mainly involved in DNA replication and repair, transcription regulation, etc.20 DFFB is also a crucial protein in the apoptotic pathway, triggering both DNA fragmentation and chromatin condensation during this process. Although there are relatively few studies on the correlation between HNRNPCL1, PRAMEF1, CFAP74, and DFFB and tumors, these mutation sites on the four genes identified in this study had annotation information in the COSMIC database. These results indicated that they are associated with the occurrence and development of tumors. Consequently, HNRNPCL1, PRAMEF1, CFAP74, and DFFB may be regarded as common potential biomarkers for EC and LS-related EC, and warrant further investigation.
LS-related EC is different from sporadic EC in that it is an autosomal dominant hereditary disease, and its etiology and pathogenesis are linked to mutations or abnormal expressions of MMR genes, such as MLH1, MSH2, MSH6, and PMS2.21 From a genetic point of view, LS-related EC is difficult to be completely classified into type I (hormonal-dependent) or type II (non-hormonal-dependent) EC, and is often ignored by clinicians or pathologists.22 However, strengthening the screening and detection of LS-related EC has certain clinical significance. In at least 50% of patients with LS, EC appears as the first malignant tumor prior to colorectal cancer.23 Therefore, the significance of early screening, diagnosis, and treatment of LS-related EC lies in the detection of other LS-related tumors with poorer prognosis in these patients and high-risk members of their families. This approach prevents the occurrence of other tumors and improves the survival rate.24 This study analyzed the characteristics of gene mutations in two families of suspected LS-related EC patients with MSH2/MSH6 deletion through ccfDNA sequencing with WES. Differences in the number of specific mutations were found in different families, and these specific mutations may reflect their genetic characteristics, which has certain research significance. Studies with larger sample sizes are necessary for more in-depth research.
However, LS carriers could not be diagnosed, as there were no MMR gene mutation detections performed for suspected LS-related patients and their families. A reason for this limitation is that testing of parents or siblings was not possible because of the age of the patients, and some family members of the patients refused to undergo genetic testing due to insufficient understanding. Another objective reason is the high cost of sequencing; thus, the patients and their family members were unwilling to pay for this examination. In addition, since this study is a retrospective analysis, only formalin-fixed paraffin-embedded samples of tumor tissues were obtained in all enrolled patients; normal tissue or blood samples were not used to determine whether the patients were Lynch carriers. Therefore, the present study employed only immunohistochemistry to identify patients with suspected LS-related EC. Nevertheless, in terms of the rationality of the entire study, genetic testing would be necessary to improve the completeness and accuracy of our research; this will be addressed in the follow-up studies. In future research, we will expand the sample size, and conduct family study to identify patients with LS-related and sporadic EC. Moreover, we will further analyze the differences of MMR gene mutation and their impact on protein expression.
Conclusion
HNRNPCL1, PRAMEF1, CFAP74, and DFFB may be common potential biomarkers for EC and LS-related EC, providing new insights for research on the diagnosis and treatment of EC in the future.
Abbreviations
EC, endometrial cancer; LS, Lynch syndrome; MMR, mismatch repair; WES, whole-exome sequencing; ccfDNA, circulating free DNA; GATK, Genome Analysis Toolkit; COSMIC, Catalogue of Somatic Mutations in Cancer; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DAVID, Database for Annotation, Visualization and Integrated Discovery; PPI, protein–protein interaction.
Data Availability
We have presented all our main data in the form of tables and figures. The data supporting the conclusions of this article are included within the article.
Ethics Approval and Consent to Participate
All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional committee. The collection and analysis of samples has met the guidelines outlined in the Declaration of Helsinki. Ethical approval of the ethics committee of Tianjin Central Hospital of Gynecology & Obstetrics.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed on the journal to which the article will be submitted; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
The work was supported by Tianjin Science and Technology Plan Project (17ZXMFSY00160). The sponsor had no involvement in the project design, experiment, data processing and article writing, etc.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
2. Lortet-Tieulent J, Ferlay J, Bray F, Jemai A. International patterns and trends in endometrial cancer incidence, 1978–2013. J Nati Cancer Inst. 2018;110(4):354–361. doi:10.1093/jnci/djx214
3. Gao Y, Zhao M, Dai X, et al. The prevalence of endometrial cancer in pre- and postmenopausal Chinese women. Menopause. 2016;23(8):884–887. doi:10.1097/GME.0000000000000684
4. Chapelle ADL. The incidence of lynch syndrome. Fam Cancer. 2005;4(3):233–237. doi:10.1007/s10689-004-5811-3
5. Mills AM, Liou S, Ford JM, et al. Lynch syndrome screening should be considered for all patients with newly diagnosed endometrial cancer. Am J Surg Pathol. 2014;38(11):1501–1509. doi:10.1097/PAS.0000000000000321
6. Che Y, Yao Q, Dai S, et al. Study of the mutation and expression of PTEN gene in endometrial carcinoma and epithelial ovarian cancer. Zhonghua Fu Chan Ke Za Zhi. 2002;37(10):608–611.
7. Li Y, Bian Y, Wang K, et al. POLE mutations improve the prognosis of endometrial cancer via regulating cellular metabolism through AMF/AMFR signal transduction. BMC Med Genet. 2019;20(1):202. doi:10.1186/s12881-019-0936-2
8. Merentitis D, Nguyen BD, Samartzis EP, Noske A, Brandt S, Dedes KJ. Loss of MDC1 in endometrial carcinoma is associated with loss of MRN complex and MMR deficiency. Anticancer Res. 2019;39(12):6547–6553. doi:10.21873/anticanres.13870
9. Qiao Z, Jiang Y, Wang L, et al. Mutations in KIAA1109, CACNA1C, BSN, AKAP13, CELSR2, and HELZ2 are associated with the prognosis in endometrial cancer. Front Genet. 2019;10:909. doi:10.3389/fgene.2019.00909
10. Imboden S, Tapia C, Scheiwiller N, et al. Early-stage endometrial cancer, CTNNB1 mutations, and the relation between lymphovascular space invasion and recurrence. Acta Obstet Gynecol Scand. 2020;99(2):196–203.
11. Avila M, Meric-Bernstam F. Next-generation sequencing for the general cancer patient. Clin Adv Hematol Oncol. 2019;17(8):447–454.
12. Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Nati Acad Sci USA. 2009;106(45):19096–19101. doi:10.1073/pnas.0910672106
13. Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272–276. doi:10.1038/nature08250
14. Chang YS, Huang HD, Yeh KT, et al. Identification of novel mutations in endometrial cancer patients by whole-exome sequencing. Int J Oncol. 2017;50(5):1778–1784. doi:10.3892/ijo.2017.3919
15. Temko D, Van Gool IC, Rayner E, et al. Somatic POLE exonuclease domain mutations are early events in sporadic endometrial and colorectal carcinogenesis, determining driver mutational landscape, clonal neoantigen burden and immune response. J Pathol. 2018;245(3):283–296. doi:10.1002/path.5081
16. Yokoyama T, Takehara K, Sugimoto N, et al. Lynch syndrome-associated endometrial carcinoma with MLH1 germline mutation and MLH1 promoter hypermethylation: a case report and literature review. BMC Cancer. 2018;18(1):576. doi:10.1186/s12885-018-4489-0
17. Libera L, Craparotta I, Sahnane N, et al. Targeted gene sequencing of Lynch syndrome–related and sporadic endometrial carcinomas. Hum Pathol. 2018;81:235–244. doi:10.1016/j.humpath.2018.06.029
18. Yang M, Li L, Wang J, et al. Heterogeneous nuclear ribonucleoproteins (hnRNPs) and human transformer-2-beta1 (hTra2-beta1)-regulated estrogen receptor-alpha improves prognosis of endometrial cancer. Eur J Gynaecol Oncol. 2014;35(6):701–707.
19. Hermes N, Kewitz S, Staege MS. Preferentially Expressed Antigen in Melanoma (PRAME) and the PRAME family of leucine-rich repeat proteins. Curr Cancer Drug Targets. 2016;16(5):400–414. doi:10.2174/1568009616666151222151818
20. Han DSC, Ni M, Chan RWY, et al. The biology of cell-free DNA fragmentation and the roles of DNASE1, DNASE1L3, and DFFB. Am J Hum Genet. 2020;106(2):202–214. doi:10.1016/j.ajhg.2020.01.008
21. Banno K, Kisu I, Yanokura M, et al. Hereditary endometrial cancer: lynch syndrome. Curr Obstet Gynecol Rep. 2013;2(1):11–18. doi:10.1007/s13669-012-0029-0
22. Meyer LA, Broaddus RR, Lu KH. Endometrial cancer and lynch syndrome: clinical and pathologic considerations. Cancer Control. 2009;16(1):14–22. doi:10.1177/107327480901600103
23. Møller P, Seppälä T, Bernstein I, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut. 2017;66(3):464–472. doi:10.1136/gutjnl-2015-309675
24. Wang Y, Wang Y, Li J, et al. Lynch syndrome related endometrial cancer: clinical significance beyond the endometrium. J Hematol Oncol. 2013;6(1):22. doi:10.1186/1756-8722-6-22
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.