Back to Journals » OncoTargets and Therapy » Volume 11
HMGB1/RAGE axis mediates the apoptosis, invasion, autophagy, and angiogenesis of the renal cell carcinoma
Authors Wu CZ, Zheng JJ, Bai YH, Xia P, Zhang HC, Guo Y
Received 4 March 2018
Accepted for publication 26 April 2018
Published 1 August 2018 Volume 2018:11 Pages 4501—4510
DOI https://doi.org/10.2147/OTT.S167197
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Cun-Zao Wu,1 Jian-Jian Zheng,2 Yong-Heng Bai,2 Peng Xia,1 Hai-Cong Zhang,3 Yong Guo1
1Department of Transplantation Center, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China; 2Department of Key Laboratory of Diagnosis and Treatment of Severe Hepato-Pancreatic Diseases of Zhejiang Province, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China; 3Department of Pathology, The Fifth Hospital of Shijiazhuang, Shijiazhuang, China
Background: High mobility group box 1 protein (HMGB1) is a sort of non-histone protein in chromatin, which plays an important role in tumor proliferation, invasion, and immune escape. HMGB1-RAGE (receptor for advanced glycation end products) interactions have been reported to be important in a number of cancers.
Methods: CCK8, flow cytometry and qRT-PCR were used to detected cell viability, apoptosis and gene expression, respectively.
Results: In the present study, we demonstrated that HMGB1/RAGE axis regulated the cell proliferation, apoptosis, and invasion of the renal cell carcinoma (RCC). Further, we discovered that HMGB1/RAGE axis increased the expression of autophagic proteins LC3 and Beclin-1 in RCC. Finally, we used a coculture model of human umbilical vein endothelial cells with RCC cell lines to find out that HMGB1 also increased the expression of VEGF and VEGFR2 in human umbilical vein endothelial cells. An in vivo study further confirmed that HMGB1 knockdown inhibited RCC tumor growth.
Conclusion: Our results illustrated that HMGB1/RAGE axis mediated RCC cell viability, apoptosis, invasion, autophagy, and angiogenesis, which provides a novel theoretical basis for using HMGB1 as the target in RCC.
Keywords: HMGB1, RCC, apoptosis, invasion, autophagy, angiogenesis
Corrigendum for this paper has been published
Introduction
Renal cell carcinoma (RCC) is one of the most common urological malignant tumors in the world. RCC constitutes about 3% of all human cancers1 and is the most common kidney tumor.2 A continuing rise in incidence has been reported with about 64,000 new estimated and 14,400 death cases in the United States in 2017.3 RCC is generally known to be poorly responsive to conventional radiotherapy and chemotherapy. Furthermore, RCC is usually diagnosed at advanced stages combined with local invasion and systematic metastasis. Therefore, a study on the molecular mechanism of RCC tumorigenesis, progression, and metastases formation is meaningful and imperative. Over the past few years, novel oncotargets for RCC have been identified,4 which may be helpful to develop new intervention strategies.
High mobility group box 1 (HMGB1) proteins are pervasive and abundant nuclear proteins with a great diversity of functions in the cell. The proteins were purified from nuclei for the first time in the 1970s.5 The HMGB1 gene belongs to the high mobility group protein family, whose protein product contains two 80-amino acid DNA-binding domains (A-box and B-box) and a negatively charged C-terminus.6 HMGB1 in the nucleus functions as a chromatin structural protein, while extracellular HMGB1 acts as a proinflammatory cytokine.7,8 HMGB1 is also well known by its multi-functionality, the disorder of which might cause pathologic conditions including Alzheimer’s disease,9 arthritis,10 and cancer.11
The overexpression of HMGB1 has been observed in a number of human tumor types, and it has been found to play an important role in cancer progression and prognosis. It associates highly with invasion and metastasis in several tumors, such as osteosarcoma,12 hepatocellular carcinoma,13 oral squamous cell carcinoma,14 prostate cancer,15 and gastrointestinal cancer.16 HMGB1 overexpression correlates highly with tumor development and participates in proliferation, invasion, and migration of cancer cells.17 HMGB1 also impairs the sensitivity of cancer cells to chemotherapeutic drugs in vitro.18 Receptor for advanced glycation end products (RAGE) is a cell-surface receptor of the immunoglobulin superfamily with multiple ligands that binds to HMGB1 and other ligands, including amyloids, AGEs, and S100 proteins.19 Moreover, the HMGB1–RAGE signaling was reported to be involved in several malignancies.20,21 As a consequence, the release of HMGB1 was considered as an inducement for acute antineoplastic inflammation, which was initiated against the tumor during chemotherapy.22 As a result, HMGB1 may serve as a biomarker of inflammation, as well as a prognostic marker for cancer progression.
In this study, we employed the RCC cell lines A498 and ACHN to investigate the role of HMGB1. We discovered that HMGB1 regulated the cell viability, apoptosis, and invasion of RCC. Furthermore, we investigated the signaling pathway activated by HMGB1 and identified that p62, LC3, Beclin1, and VEGF contributed to the regulation of HMGB1 in RCC autophagy.
Materials and methods
Cell culture and reagents
RCC cell lines, including Caki-1, A498, and ACHN, were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Human umbilical vein endothelial cells (HUVEC) was also provided by ATCC. The cells were cultured in culture media as recommended by the ATCC, supplemented with 10% fetal bovine serum. The cells were grown in a humidified atmosphere containing a 95% air, 5% CO2 mixture at 37°C. Anti-p62 (ab91526), anti-LC3I/II (ab128025), anti-Beclin1 (ab62557), anti-RAGE (ab3611), and anti-HMGB1 (ab79823) antibodies were purchased from Abcam (Cambridge, UK), and anti-VEGF (AF5131), anti-VEGFR2 (AF6281), and anti-β-actin (AF7018) antibodies were obtained from Affinity Biosciences (Cambridge, UK). Anti-RAGE neutralizing antibody (sRAGE) was provided by Abcam.
Gene silencing and overexpression in the A498 and ACHN cell lines
The cells were seeded into 6-well plates (2.5×105 cells/well) and were allowed to adhere for 24 h before transfection. Using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA), the cells were stably transfected with siRNAs directed against HMGB1 (HMGB1-siRNAs; GenePharma, Shanghai, China) or nontargeted control siRNA (GenePharma). Vector control and pcDNA3.1-HMGB1 plasmid were stably transfected into A498 and ACHN cells using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. After 6 h transfection, culture media containing the reagent mixture were removed and replaced with fresh complete medium, and then the cells were used for further experiments.
RNA extraction and real-time quantitative reverse-transcription PCR (qRT-PCR)
Trizol reagent was used to extract total RNA from the cell samples. cDNA was synthesized using reverse transcriptase (Thermo Fisher Scientific) following the manufacturer’s instructions. qRT-PCR was performed using SYBR Green qPCR kit (Thermo Fisher Scientific). The relative levels were calculated with the 2−ΔΔCT method. Sample Ct values were normalized to Ct values of β-actin RNA, all of which were calculated from triplicate reactions. The following primer pairs were used: HMGB1, 5′-AGTGCTCAGAGAGGTGGA-3′ and 5′-TTTGGGAGGGATATAGGT-3′; Beclin1, 5′-CTGCCGTTATACTGTTCT-3′ and 5′-TGTCTTCAATCTTGCCTT-3′; RAGE, 5′-GTGTCCTTCCCAACGGCTC-3′ and 5′-ATTGCCTGGCACCGGAAAA-3′; LC3, 5′-AACATGAGCGAGTTGGTCAAG-3′ and 5′-GCTCGTAGATGTCCGCGAT-3′; and β-actin, 5′-TGATCCACATCTGCTGGAAGGT-3′ and 5′-GACAGGATGCAGAAGGAGATTACT-3′.
CCK8 assay
The transfected cell suspension (3,000 cells/well, 100 μL) was dispensed in 96-well plates and incubated in an incubator for 24, 48, and 72 h (humidified atmosphere, 37°C, 5% CO2). Subsequently, the cells were then incubated with 10 μL of CCK8 (Dojindo Laboratories, Kumamoto, Japan) per well at 37°C for 4 h in the incubator. The absorbance at 460 nm (A460) was then examined using a scanning multiwell spectrophotometer (Thermo Scientific).
Flow cytometry Annexin V/FITC binding assay
Flow cytometry Annexin V/FITC Binding Assay (Dojindo Laboratories) was used to detect apoptosis. The transfected cell suspension (3,000 cells/well, 100 μL) was dispensed in 96-well plates and incubated in the incubator for 48 h. The cells were separated with 0.25% trypsin and centrifuged at 1,000 rpm for 5 min. Then, the cells were washed and resuspended with binding buffer in a microfuge tube. Five microliter FITC-conjugated Annexin V and 10 μL of PI were added to the tubes. The mixture was then incubated at dark for 10 min. The percent of early apoptotic, late apoptotic, and necrotic cells were determined using the flow cytometry (Beckman Gallios, Brea, CA, USA). The results were representative of three independent experiments with triplicate samples.
Transwell invasion assays
Evaluation of cell migration and invasion was examined using Transwell Permeable Supports (Corning Inc., Corning, NY, USA). Briefly, the transfected cells were allowed to grow to confluence. About 105 cells/well were resuspended in 100 μL serum-free medium and plated onto 8 μm Transwell filter inserts of 24-well plates coated with Matrigel (10 mg/L, BD Biosciences, San Jose, CA, USA) for 24 h in triplicate for invasion assays. The lower chambers contained 500 μL chemoattractant, which was the medium containing 10% fetal bovine serum. After the experiment, the cells in the upper chamber were removed with a cotton swab. The invasive cells at the bottom of the membrane were fixed with methanol and then stained with 0.1% crystal violet for 20 min. Finally, the number of invasive cells was counted.
Western blot analysis
About 5×106 cells were gathered after transfection for 48 h and lysed in RIPA buffer (Cell Signaling Technology, Danvers, MA, USA) in the presence of protease inhibitor (PMSF) and phosphatase inhibitor (Na-orthovanadate and NaF). The whole cell extracts were analyzed by SDS-PAGE and transferred to a nitrocellulose membrane using a transfer apparatus according to the manufacturer’s protocols (Bio-Rad). After incubation with 10% nonfat milk in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.1% Tween 20) for 1 h, the membrane was washed twice for 10 min with TBST and incubated with antibodies against p62 (1:1,000), LC3 (1:1,000), Beclin1 (1:2,000), RAGE (1:2,000), HMGB1 (1:1,000), VEGF (1:1,000), VEGFR2 (1:2,000), or β-actin (1:2,000, affinity biosciences) at 4°C overnight. Membranes were washed three times for 10 min and incubated with a 1:5,000 dilution of horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibodies for 1 h. Blots were washed with TBST 3 times for 10 min and developed with the ECL system (Millipore, Darmstadt, Germany) according to the manufacturer’s protocols. Results were normalized to the internal control β-actin.
Animal study
Seven to eight weeks old BALB/c-nu mice were provided SLAC (Shanghai, China) and housed in barrier facilities on a 12 h light/dark cycle. The mice were subcutaneously injected with A498 (5×106 cells/mouse). The tumor-bearing mice were randomly divided into 2 groups when the tumor volumes reached about 200 mm3. HMGB1 control or HMGB1 siRNA2 (twice a week) was intratumorally injected into the mice. The equation (length × width2)/2 was used to calculate the tumor volumes in mice weekly. All animals were sacrificed using CO2 after a 4-week treatment. The animal study protocol was fully approved by the Committee on the Ethics of Animal Experiments of The First Affiliated Hospital of Wenzhou Medical University. In addition, we strictly followed the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Statistical analysis
Statistical analysis was conducted using SPSS 19.0 (IBM Corporation, Armonk, NY, USA). Bar graphs were drawn using GraphPad Prism 7 (GraphPad Software, Inc., La Jolla, CA, USA). All the data are expressed as mean values ± SD. A paired Student’s t-test was used to assess differences between 2 groups. P<0.05 was used to indicate statistical significance. All experiments were performed in triplicate.
Results
HMGB1 regulated the cell viability of A498 and ACHN
Many studies have revealed that extracellular HMGB1 stimulated and induced responses in several cancer cell lines, related to inflammation, proliferation, and migration.10 In order to investigate the function of HMGB1 in RCC, siRNA and anti-RAGE neutralizing antibody were used to knockdown HMGB1 and RAGE expressions, respectively. Since the expressions of HMGB1 and RAGE were much higher in A498 and ACHN than in Caki-1, these 2 cell lines were selected for further experiments (Figure 1A). As shown in Figure 1B, HMGB1 siRNA-2 exerted the best effect in suppressing HMGB1 expression in A498 and ACHN cells. Thereby, the HMGB1 siRNA2 was chosen for the subsequent experiments. In addition, plasmid pcDNA3.1-HMGB1 was transfected into RCC cells to upregulate the expression of HMGB1 (Figure 1C). We next examined the effect of HMGB1 knockdown on RCC cells viability using CCK8 assay. Compared to control groups, HMGB1 siRNA2 significantly decreased cell viability, whereas the overexpression of HMGB1 promoted the cell viability (Figure 1D). Moreover, sRAGE suppressed cell viability compared to control groups, while HMGB1-siRNA attenuated the suppression (Figure 1D). All these results indicated that HMGB1 and RAGE were involved in cell proliferation in RCC cancer.
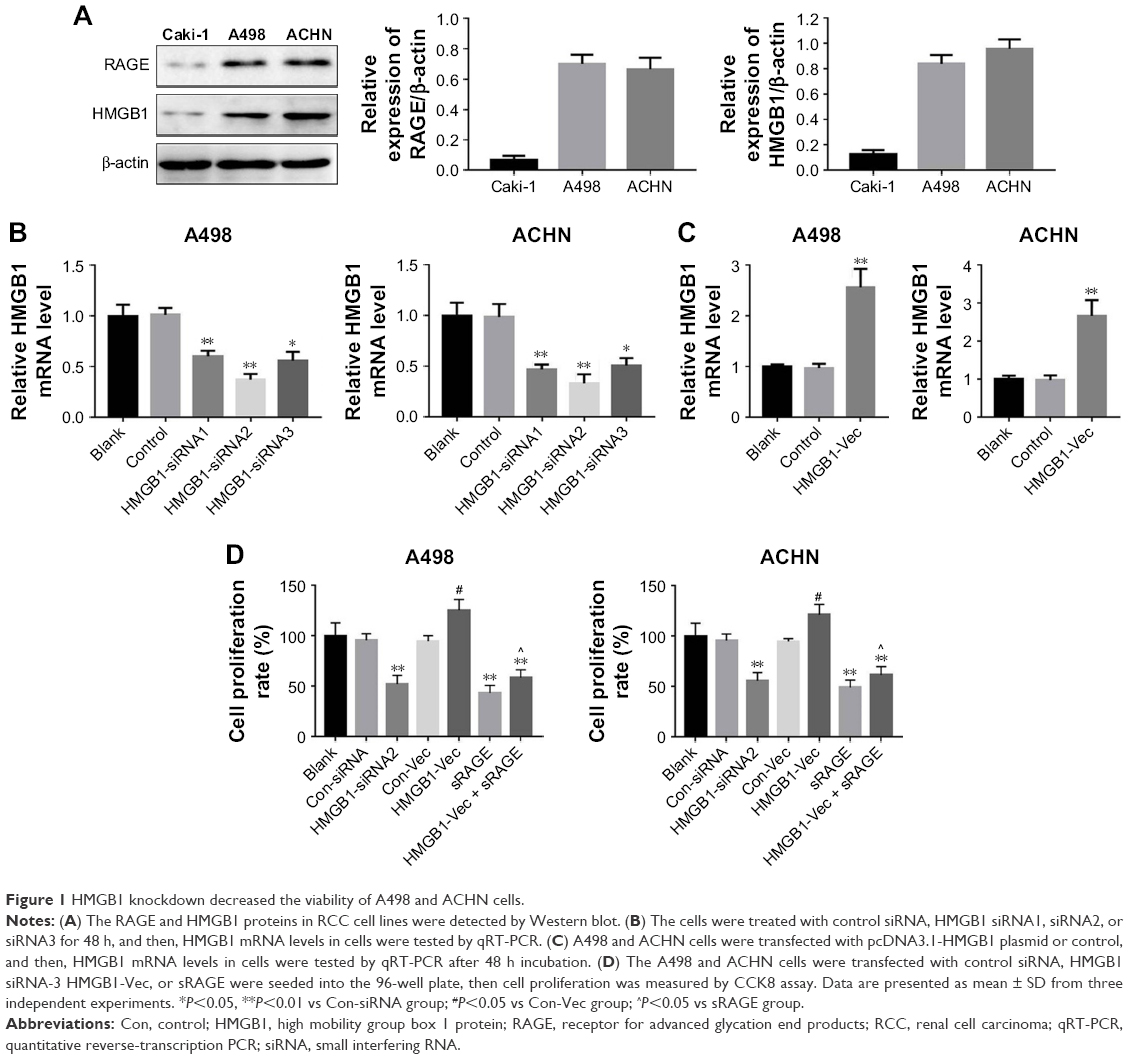 | Figure 1 HMGB1 knockdown decreased the viability of A498 and ACHN cells. |
HMGB1 knockdown induced the apoptosis of RCC cell lines
To further examine the antiproliferation effect of HMGB1 on RCC cells, the Annexin V-FITC/PI staining assay was performed. As shown in Figure 2A and B, HMGB1-siRNA or sRAGE significantly induced RCC cell apoptosis, which could be reversed by HMGB-1 overexpression. In addition, both Con-siRNA and HMGB1-Vec had very limited toxicity to A498 and ACHN cells. These results suggested that suppression of HMGB1/RAGE axis induced apoptosis of RCC cell lines.
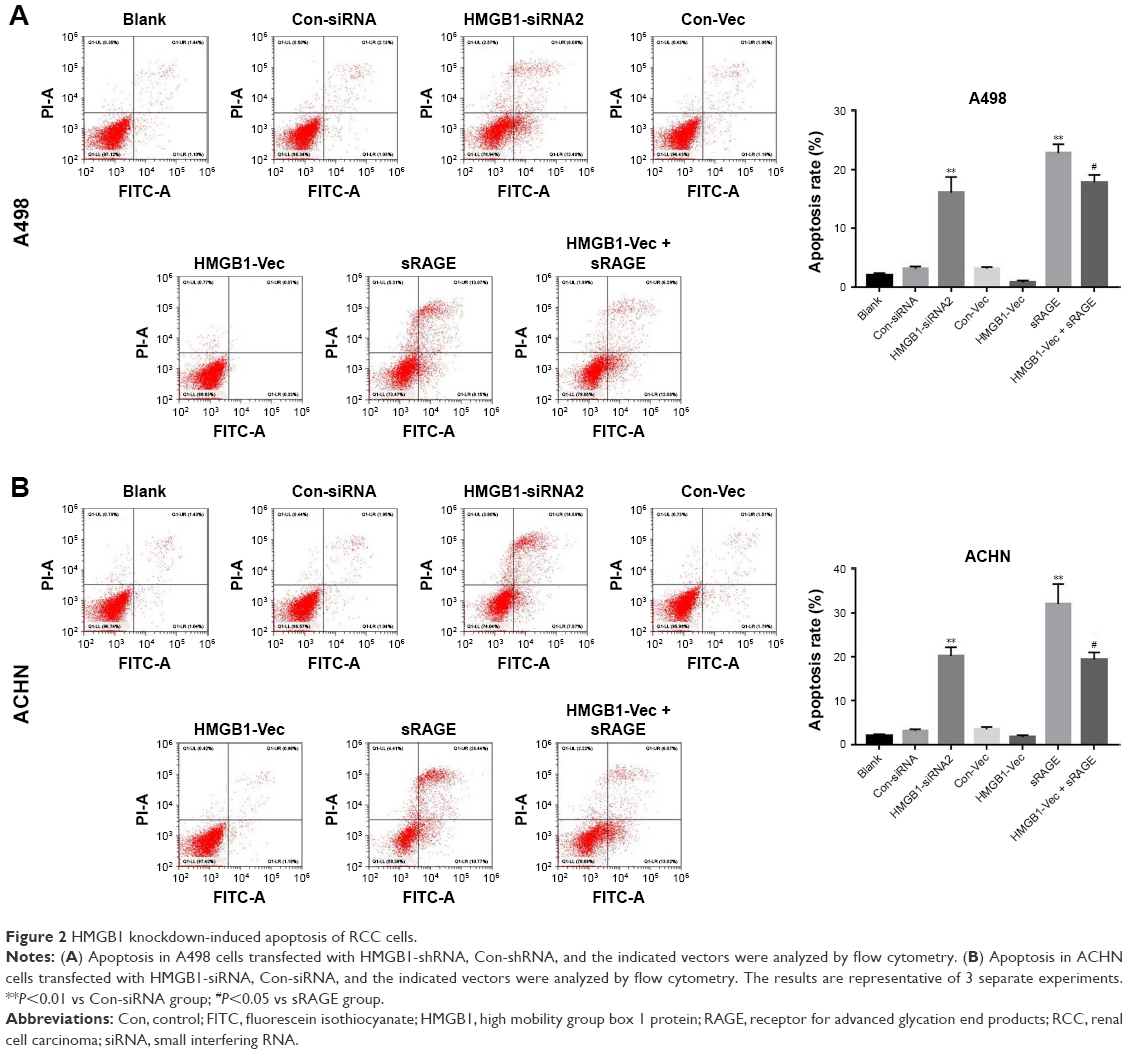 | Figure 2 HMGB1 knockdown-induced apoptosis of RCC cells. |
HMGB1 knockdown suppressed the invasion of RCC cell lines
Many studies have suggested that HMGB1 acts as an important regulator during the invasion of cancer cells.23 In order to determine whether HMGB1 plays a direct role during the motility of RCC, matrigel invasion assays were performed. The results indicated that transfection with HMGB1 siRNA or sRAGE significantly reduced the number of A498 cells that invaded through the Matrigel-coated membrane (Figure 3A and B). Nevertheless, overexpression of HMGB1 prompted the invasive ability of the A498 cells. As shown in Figure 3C and D, similar results were observed in the ACHN cells. These data indicated that inhibition of HMGB1/RAGE axis decreased the invasive ability of the RCC cells.
 | Figure 3 HMGB1 knockdown suppressed the invasion of RCC cells. |
HMGB1 regulated the expression of RAGE and autophagic proteins in human RCC cell lines
HMGB1 in the extracellular milieu could act as a paracrine/autocrine factor through interaction with its receptors, especially RAGE.10 Therefore, the expression of RAGE and autophagic proteins were also detected by Western blot analysis. As shown in Figure 4A and B, knockdown of the HMGB1 dramatically inhibited the expression of RAGE and autophagic proteins (LC3II and Beclin1). On the contrary, overexpression of HMGB1 upregulated the levels of RAGE, LC3II, and Beclin1 in RCC cells and downregulated the level of p62 in the cells (Figure 4A and B).
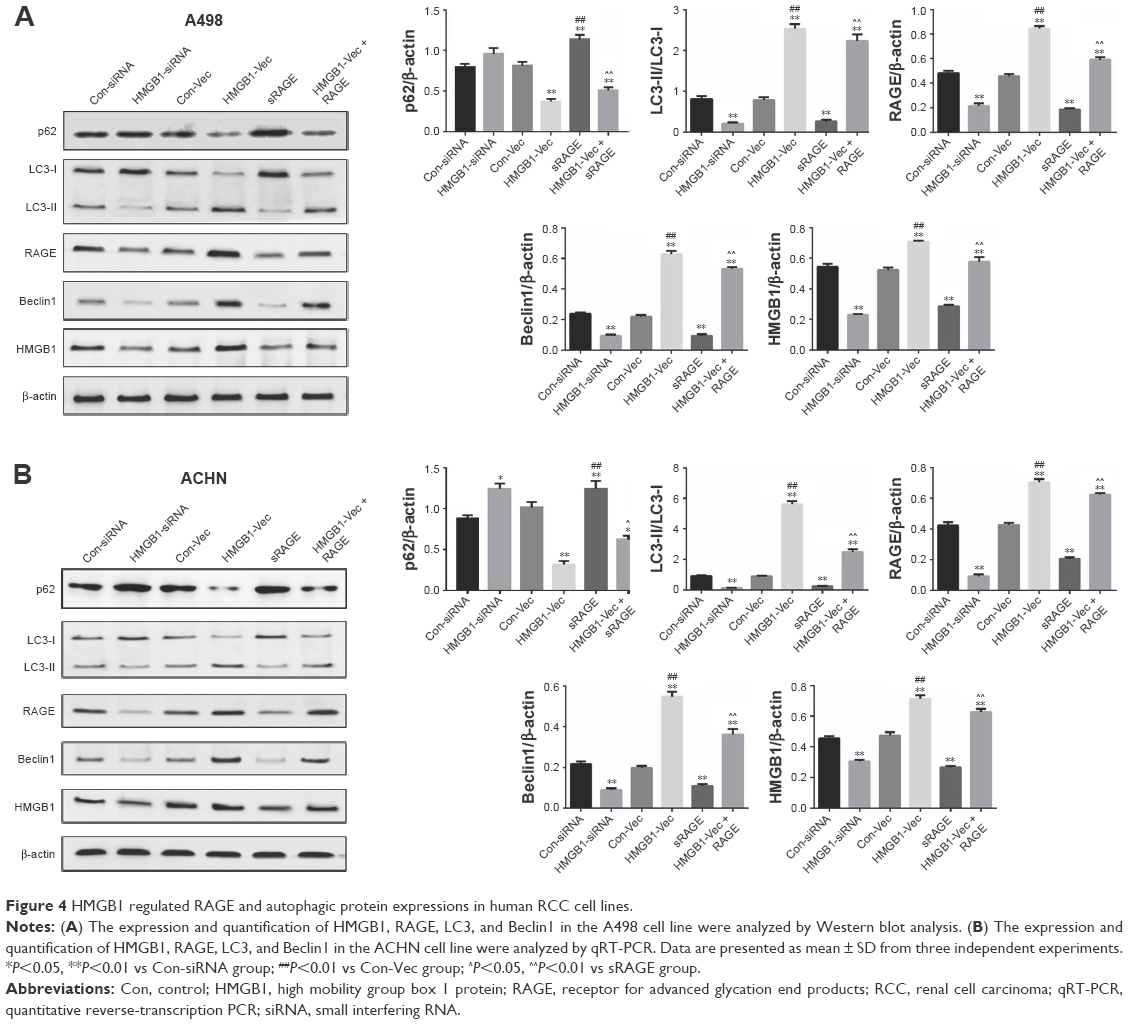 | Figure 4 HMGB1 regulated RAGE and autophagic protein expressions in human RCC cell lines. |
HMGB1 regulated the RCC-stimulated VEGF and VEGFR2 expression in HUVEC
The induction of angiogenesis can be mediated by a variety of molecules released by tumor cells. Vascular endothelial growth factor (VEGF) is generally confirmed to be one of the most important stimulators. The VEGF pathway is also identified as a key angiogenic signal. Exogenously added HMGB1 was found to play a positive role in promoting tumor angiogenesis, with the evidence of increased VEGF expression.24 The interaction between the cancer cells and endothelial cells has been implicated in tumor angiogenesis and metastasis.25 Therefore, a coculture model of HUVEC with RCC cell lines was established to study the effect of HMGB1 on the expression of VEGF and VEGFR2 in HUVEC. As indicated in Figure 5A and B, HMGB1 siRNA2 or sRAGE downregulated the mRNA expression of VEGF and VEGFR2 in HUVEC. In contrast, overexpression of HMGB1 in RCC cell lines upregulated the mRNA levels of VEGF and VEGFR2 in HUVEC. In addition, the protein expressions of VEGF and VEGFR2 in HUVEC were also detected by Western blot analysis. Consistent with the qRT-PCR results, the HMGB1 siRNA2 and sRAGE downregulated the proteins of VEGF and VEGFR2 in HUVEC, while the overexpression of HMGB1 upregulated the same (Figure 5C and D).
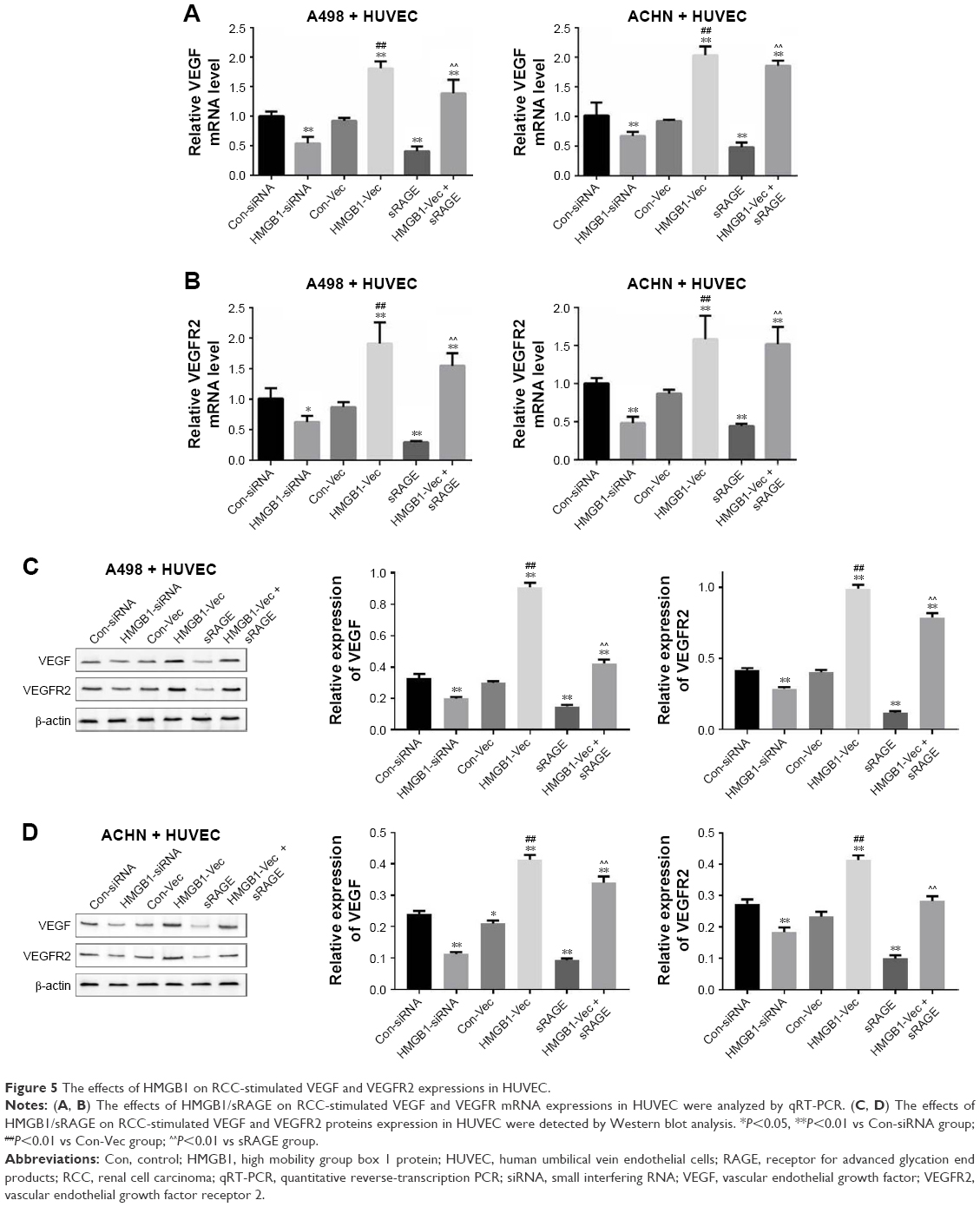 | Figure 5 The effects of HMGB1 on RCC-stimulated VEGF and VEGFR2 expressions in HUVEC. |
HMGB1 knockdown inhibited RCC tumor growth in vivo
A RCC A498 xenograft model was established in order to further evaluate the role of HMGB1 in vivo. After 4 weeks of treatment, HMGB1 knockdown significantly inhibited RCC tumor growth in vivo (Figure 6A). Meanwhile, the tumor weight in the HMGB1 knockdown group was 0.37 g, which was much less than that of the control (Figure 6B and C). During the process of in vivo study, no loss in body weight was detected in these 2 groups (Figure 6D). These results were consistent with in vitro data, which confirmed the antiapoptosis effect of HMGB1 in RCC.
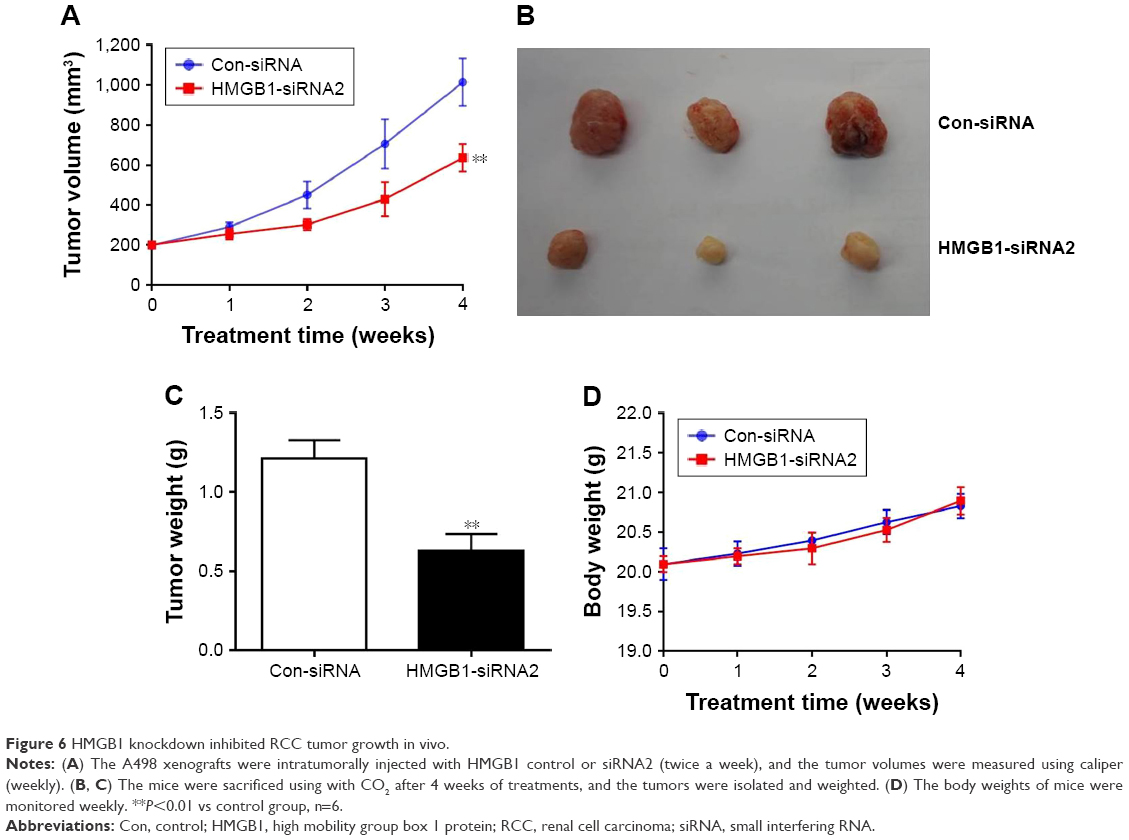 | Figure 6 HMGB1 knockdown inhibited RCC tumor growth in vivo. |
Discussion
RCC is the most common solid cancer in the adult kidney, which accounts for about 90% of kidney neoplasms and 3% of all adult malignancies.26 RCC acts as a challenge for the clinicians since one-third of the RCC patients are diagnosed with metastasis, while the remaining 20%–40% of patients will also develop metastasis as time goes by. Detecting new RCC biomarkers can provide future directions for interventional therapeutic targets of RCC and improve a patient’s prognosis.
HMGB1 is a kind of non-histone protein in chromatin, found abundantly in eukaryotic cell nuclei. Recently, some studies demonstrated that HMGB1 was highly expressed in a variety of solid tumors, including melanoma, breast cancer, colorectal cancer, cervical cancer, and bladder cancer.27,28 HMGB1 was also found to promote the proliferation and development of RCC.29 It is reported that the suppression of HMGB1 expression could attenuate autophagy and potently enhance apoptosis in bladder cancer cells.30 In line with the previous reports, the present results showed that the suppression of HMGB1/RAGE axis resulted in a declined RCC cell viability. The Annexin V-FITC assay also showed that the knockdown of HMGB1/RAGE axis induced the apoptosis of RCC cells.
Autophagy plays an important role during the major recycling and was an important way of cell component degradation.31 Multiple pathological conditions are triggered by the dysfunction of autophagy.31 HMGB1 was reported to play an important part in autophagy, DNA damage repair, and chemoresistance.32 Previous reports suggested that HMGB1 regulated chemotherapeutics-induced autophagy.33 In the current report, we found that the knockdown of HMGB1/RAGE axis regulated the autophagic proteins p62, LC3, and Beclin-1, which play important roles in the autophagy process.
The interaction between HMGB1 and RAGE was reported to promote the proliferation and invasion of tumor cells.10,27 In line with the previous reports, our results showed that the knockdown of HMGB1 could suppress the invasion of RCC cells. Invasion and angiogenesis are the two hallmarks of cancer.34 One of the most well-known angiogenesis inducers is VEGF. The results of our study are in accordance with the previous reports which show35 that the suppression of HMGB1 could downregulate the expression of VEGF and VEGFR2. Vijayaraghavan et al36 reported a novel and promising therapeutic strategy to inhibition of CDK4/6 and autophagy to treat breast. Similar to this novel strategy, inhibition of autophagy and angiogenesis by blocking of HMGB1/RAGE axis might be a promising way to treat patients with RCC. Meanwhile, DeVorkin et al37 found that inhibition of autophagy could increase the efficacy of sunitinib in clear cell ovarian carcinoma. Therefore, our findings may provide a future direction for combination of HMGB1 inhibition with other therapeutic treatments for RCC. However, the interaction between autophagy and angiogenesis mediated by HMGB1/RAGE axis in RCC is still unclear, and further studies need to be done to explore this.
In this study, we concluded that HMGB1 proteins act as key regulators in the progression and angiogenesis of RCC carcinoma and hence could serve as potential diagnostic and therapeutic targets.
Acknowledgment
Special thanks to the faculty of Key Laboratory of Diagnosis and Treatment of Severe Hepato-Pancreatic Diseases of Zhejiang Province, the First Affiliated Hospital of Wenzhou Medical University; thanks to the faculty of Department of Pathology, the Fifth Hospital of Shijiazhuang. This study was supported by Wenzhou Municipal Science and Technology Bureau (Y20160032, Y20160127), Zhejiang medical and health science and technology project (2017KY454).
Disclosure
The authors report no conflicts of interest in this work.
References
Ramana J. RCDB: renal cancer gene database. BMC Res Notes. 2012;5:246. | ||
Simard EP, Ward EM, Siegel R, Jemal A. Cancers with increasing incidence trends in the United States: 1999 through 2008. CA Cancer J Clin. 2012;62(2):118–128. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Zhu H, Mao JH, Wang Y, et al. Dual inhibition of BRD4 and PI3K-AKT by SF2523 suppresses human renal cell carcinoma cell growth. Oncotarget. 2017;8(58):98471–98481. | ||
van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis. 2008;11(1):91–99. | ||
Ferrari S, Finelli P, Rocchi M, Bianchi ME. The active gene that encodes human high mobility group 1 protein (HMG1) contains introns and maps to chromosome 13. Genomics. 1996;35(2):367–371. | ||
Hsieh MJ, Hsieh YH, Lin CW, Chen MK, Yang SF, Chiou HL. Transcriptional regulation of Mcl-1 plays an important role of cellular protective effector of vincristine-triggered autophagy in oral cancer cells. Expert Opin Ther Targets. 2015;19(4):455–470. | ||
Zhao L, An R, Yang Y, et al. Melatonin alleviates brain injury in mice subjected to cecal ligation and puncture via attenuating inflammation, apoptosis, and oxidative stress: the role of SIRT1 signaling. J Pineal Res. 2015;59(2):230–239. | ||
Takata K, Kitamura Y, Kakimura J, et al. Role of high mobility group protein-1 (HMGB1) in amyloid-β homeostasis. Biochem Biophys Res Commun. 2003;301(3):699–703. | ||
Ulloa L, Messmer D. High-mobility group box 1 (HMGB1) protein: friend and foe. Cytokine Growth Factor Rev. 2006;17(3):189–201. | ||
Tang L, Chai W, Ye F, et al. HMGB1 promotes differentiation syndrome by inducing hyperinflammation via MEK/ERK signaling in acute promyelocytic leukemia cells. Oncotarget. 2017;8(16):27314–27327. | ||
Liu K, Huang J, Ni J, et al. MALAT1 promotes osteosarcoma development by regulation of HMGB1 via miR-142-3p and miR-129-5p. Cell Cycle. 2017;16(6):578–587. | ||
Wu JH, Guo JP, Shi J, et al. CMA down-regulates p53 expression through degradation of HMGB1 protein to inhibit irradiation-triggered apoptosis in hepatocellular carcinoma. World J Gastroenterol. 2017;23(13):2308–2317. | ||
Lin CW, Chou YE, Yeh CM, Yang SF, Chuang CY, Liu YF. A functional variant at the miRNA binding site in HMGB1 gene is associated with risk of oral squamous cell carcinoma. Oncotarget. 2017;8(21):34630–34642. | ||
Zhang YX, Yuan YQ, Zhang XQ, Huang DL, Wei YY, Yang JG. HMGB1-mediated autophagy confers resistance to gemcitabine in hormone-independent prostate cancer cells. Oncol Lett. 2017;14(5):6285–6290. | ||
Song B, Song WG, Li ZJ, et al. Effect of HMGB1 silencing on cell proliferation, invasion and apoptosis of MGC-803 gastric cancer cells. Cell Biochem Funct. 2012;30(1):11–17. | ||
Huber R, Meier B, Otsuka A, et al. Tumor hypoxia promotes melanoma growth and metastasis via high mobility group box-1 and M2-like macrophages. Sci Rep. 2016;6:29914. | ||
Ma Y, Kang S, Wu X, Han B, Jin Z, Guo Z. Up-regulated HMGB1 in the pleural effusion of non-small cell lung cancer (NSCLC) patients reduces the chemosensitivity of NSCLC cells. Tumori. Epub 2017 Sep 1. | ||
Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. | ||
Kang R, Tang D, Schapiro NE, et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010;17(4):666–676. | ||
Leclerc E, Heizmann CW, Vetter SW. RAGE and S100 protein transcription levels are highly variable in human melanoma tumors and cells. Gen Physiol Biophys. 2009;28(Spec No Fucus):F65–F75. | ||
Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–1059. | ||
Taguchi A, Blood DC, del Toro G, et al. Blockade of RAGE-amphoterin signalling suppresses tumor growth and metastases. Nature. 2000;405(6784):354–360. | ||
Qu W, Wang Y, Wu Q, Liu J, Hao D. Emodin inhibits HMGB1-induced tumor angiogenesis in human osteosarcoma by regulating SIRT1. Int J Clin Exp Med. 2015;8(9):15054–15064. | ||
Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012;2(12):1091–1099. | ||
Ljungberg B, Campbell SC, Choi HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60(4):615–621. | ||
Ellerman JE, Brown CK, de Vera M, et al. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res. 2007;13(10):2836–2848. | ||
Lin L, Zhong K, Sun Z, Wu G, Ding G. Receptor for advanced glycation end products (RAGE) partially mediates HMGB1-ERKs activation in clear cell renal cell carcinoma. J Cancer Res Clin Oncol. 2012;138(1):11–22. | ||
Li J, Sun J, Rong R, et al. HMGB1 promotes myeloid-derived suppressor cells and renal cell carcinoma immune escape. Oncotarget. 2017;8(38):63290–63298. | ||
Yin H, Yang X, Gu W, et al. HMGB1-mediated autophagy attenuates gemcitabine-induced apoptosis in bladder cancer cells involving JNK and ERK activation. Oncotarget. 2017;8(42):71642–71656. | ||
Guan JL, Simon AK, Prescott M, et al. Autophagy in stem cells. Autophagy. 2013;9(6):830–849. | ||
Yang GL, Zhang LH, Bo JJ, et al. Increased expression of HMGB1 is associated with poor prognosis in human bladder cancer. J Surg Oncol. 2012;106(1):57–61. | ||
Pan B, Chen D, Huang J, et al. HMGB1-mediated autophagy promotes docetaxel resistance in human lung adenocarcinoma. Mol Cancer. 2014;13:165. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Feng Y, Ke J, Cao P, et al. HMGB1-induced angiogenesis in perforated disc cells of human temporomandibular joint. J Cell Mol Med. 2018;22(2):1283–1291. | ||
Vijayaraghavan S, Karakas C, Doostan I, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun. 2017;8:15916. | ||
DeVorkin L, Hattersley M, Kim P, et al. Autophagy inhibition enhances sunitinib efficacy in clear cell ovarian carcinoma. Mol Cancer Res. 2017;15(3):250–258. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.