Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
HMGB1/CXCL12-Mediated Immunity and Th17 Cells Might Underlie Highly Suspected Autoimmune Epilepsy in Elderly Individuals
Authors Han Y ![]() , Yang L
, Yang L ![]() , Liu X
, Liu X ![]() , Feng Y
, Feng Y ![]() , Pang Z
, Pang Z ![]() , Lin Y
, Lin Y ![]()
Received 17 December 2019
Accepted for publication 24 April 2020
Published 19 May 2020 Volume 2020:16 Pages 1285—1293
DOI https://doi.org/10.2147/NDT.S242766
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yu-Ping Ning
Yuxiang Han, Liling Yang, Xiaoyun Liu, Yabo Feng, Zaiying Pang, Youting Lin
Departments of Neurology, Shandong Provincial Hospital Affiliated to Shandong University, Ji’nan 250021, People’s Republic of China
Correspondence: Youting Lin Tel/Fax +86 531 68776354
Email [email protected]
Purpose: Late-onset epilepsy due to autoimmune dysfunction has been reported. However, definitive diagnosis requires positive antibody results. As a result, patients with negative antibody results, but presenting with classical manifestation of autoimmune epilepsy, may be managed as suspected cases. In this study, we aim to isolate and profile the concentration of cytokines/chemokines in the cerebrospinal fluid (CSF) and the serum to ascertain if they could act as alternative diagnostic biomarkers.
Patients and Methods: Twenty patients aged ≥ 50 years were considered in this study. Ten patients were diagnosed with suspected autoimmune epilepsy (sAE) based on clinic manifestation, electroencephalogram, magnetic resonance imaging, and with negative antibody results of the serum and the CSF. The equivalent control group exhibited neurological disorders due to non-inflammatory pathologies. Serum and CSF were analyzed for cytokines/chemokines concentration, including interleukin (IL)-6, IL-10, IL-17, chemokine (C-X-C motif) ligand (CXCL)12 and CXCL13, as well as high-mobility group box protein 1 (HMGB1) and B cell activation factor (BAFF)).
Results: The CSF levels of IL-6, IL-17, HMGB1, and CXCL12 were significantly higher in the sAE group than in the control group. There was no difference in the CSF levels of IL-10, CXCL13 and BAFF. The serum levels of HMGB1 and CXCL12 were elevated in the sAE group compared with the control group, and there was no statistical difference in the serum levels of IL-6, IL-10, IL-17, CXCL13, and BAFF between the two groups.
Conclusion: Our study shows that cytokines/chemokines may act as alternative biomarkers for diagnosis of sAE. The activation of both HMGB1/CXCL12-mediated immunity and T helper cells 17 (Th17) cells may be playing a central role in the pathogenesis of sAE. We suggest that cytokines/chemokines be treated as adjuvant biomarkers, instead of solely relying on antibody screening test. However, a larger cohort in a prospective approach is required to validate our findings.
Keywords: seizure, autoimmune, cytokine, chemokine, epilepsy
Introduction
Autoimmune etiology might be common for late-onset seizure,1 also called autoimmune epilepsy,2 which is resistant to anti-epileptic drugs (AEDs) but responds to early immunotherapy or tumor resection. A variety of new antibodies have been recognized in recent years as specific biomarkers to make an early diagnosis. However, some highly suspected autoimmune epilepsy (sAE) cases with unknown antibodies remain as obstacles for practical neurologists to diagnose merely based on clinical manifestations, let alone to make an early decision on treatment. This obstacle is particularly a problem for elderly patients who are prone to comorbidities such as diabetes or arteriosclerosis, because the therapeutic benefits relative to the side effects are difficult to evaluate before initiation of aggressive immune treatment. It would be of great importance to explore other potential biomarkers for sAE to initiate early and aggressive treatment, especially for elderly patients.
Cytokines and chemokines are key intercellular mediators of inflammation. Several studies have shown some inflammatory cytokine/chemokines to be potential biomarkers of autoimmune central nervous system (CNS) disorders.3 In response to inflammation, cytokines function as messengers among cells. Based on the cytokine profile, naïve T cells can be divided into distinct subsets, such as T helper (Th)1, Th2, Th17, T follicular helper, and T regulatory cells, which can promote distinct types of inflammatory responses in different immunological diseases.4 Chemokines comprise a superfamily of small proteins. Chemokines receptors together with chemokines are critical regulators in recruiting leukocytes and other cell types to sites of inflammation into the CNS.5 Thus, cytokine/chemokines might reflect the underlying immunopathology, and they have been widely studied in CNS inflammatory processes. For example, B cell markers, such as chemokine (C-X-C motif) ligand (CXCL)13 and B cell activation factor (BAFF), have been shown to be elevated in antibody-associated CNS disorders, such as anti- N-methyl-D-aspartate receptor (NMDAR) encephalitis and neuromyelitis optica (NMO).6,7 Moreover, interleukin (IL)-4, IL-5, IL-6, IL-17, chemoattractant cytokine ligand (CCL) 17, and CXCL8, which are Th2 and Th17 cell cytokines, are elevated in inflammatory CNS disease, such as acute disseminated encephalomyelitis (ADEM) and NMO. A large number of studies have shown increased levels of cytokines in the blood and cerebrospinal fluid analysis (CSF) from patients with multiple sclerosis (MS), such as tumor necrosis factor alpha (TNF-α), IL-17, CXCL8, IL-23, and CXCL10.8 Our previous research suggested that CXCL13 might be a potential biomarker for active neuroinflammation in anti- leucine-rich glioma-inactivated protein 1 (LGI1) encephalitis.9 With respect to anti-NMDAR encephalitis, in addition to the elevated level of CXCL13, a wider range of cytokine/chemokines associated with Th cells are upregulated, such as IL-6, IL-17, IL-10 and high-mobility group box protein 1 (HMGB1),7,10-12 which are known as B cell enhancers. Thus, we postulated that these cytokine/chemokines, which are indicators of immune activation, are likely to serve as biomarkers in terms of diagnosis and therapy for sAE.
We therefore conducted an initial study to screen a series of cytokine/chemokines in the serum and CSF of patients with sAE in an attempt to identify potential biomarkers.
Patients and Methods
Ten patients with late-onset epilepsy who were diagnosed with sAE and 10 with noninflammatory CNS diseases treated at Shandong Provincial Hospital between December 2017 and April 2019 were included in the study. The study was approved by the Ethics Committee of Shandong Provincial Hospital. Written informed consents were obtained from all patients, and this study was conducted in accordance with the Declaration of Helsinki.
In the current study, late-onset epilepsy was defined as those that first seizure occurred at the age over 50 years old. To determine the underlying etiology, all patients had to complete an electroencephalography (EEG, international 10–20-system of electrode placement), brain magnetic resonance Imaging (MRI), CSF analysis, and necessary neoplasm screening. Both serum and CSF were tested routinely for antibodies (Abs) against the NMDAR, LGI1, contactin-associated proteinlike 2 (CASPR2), c-aminobutyric acid B receptor (GABABR), a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), and glycine receptor (GlyR). The other serum analysis included Abs against glutamic acid decarboxylase (GAD), Hu, Yo, Ri, Ma2, CV2, and amphiphysin, as well as antinuclear antibody, anti-double stranded DNA, anti-neutrophil cytoplasmic antibodies, anti-ribonucleoprotein, and anti-Smith antibodies. Patients who did not complete these tests were not included. We included patients based on the criteria as follows:2,13-15 (1) late-onset seizures with an unknown etiology; (2) an autoimmune etiology suspected on the basis of clinical presentation (multiple seizure types, faciobrachial dystonic seizures, AED resistance, history of recent or past neoplasia, cognition disorder, and psychiatric behavior abnormality), inflammatory CSF (elevated CSF protein >45 mg/dl and/or lymphocytic pleocytosis >5 cells/dl), MRI characteristics suggesting inflammation (usually T2/fluid-attenuated inversion recovery [FLAIR] hyperintense signal alterations prominently in the mesial temporal lobes; (3) negative neural autoantibodies in both serum and CSF; (4) reasonable exclusion of alternative causes; and (5) immunotherapy as introduced, and long-term follow-up was based on local protocol, thus once monthly for the first 6 months, and thereafter once every two months. Follow-up was done either physically or by telephone. Report of reduction in seizure frequency by 50% and above as compared to prior to treatment, therapy was considered responsive, and otherwise non-responsive. The clinical data were independently reviewed by two neurologists, and ambiguous points were discussed to ultimately achieve agreement with the final diagnosis. Ten patients who fulfilled the criteria were ultimately included in this study.
The control group consisted of 10 samples of contemporaneous patients with non-inflammatory neurological disorders, including cerebral vascular disease (n=4), Parkinson’s Syndrome (n=2), insomnia (n=2), hereditary ataxia (n=1), and frontotemporal dementia (n=1).
The serum of 1 patient with sAE was not available, and 9 serum and 10 CSF samples were obtained immediately after the patients were hospitalized prior to immunotherapy. Samples were stored at −80°C until the assays were performed. The same procedure was performed for the control patients. The cytokine/chemokines concentrations, including IL-6, IL-10, IL-17, CXCL12, CXCL13, BAFF and HMGB1, were measured by enzyme-linked immunosorbent assays (ELISAs) using human ELISA kits (Abcam, Cambridge, UK). According to the manufacturer’s instructions, samples were diluted and measured. The results were calculated using standard curves. Data are expressed as the median (inter-quartile range) and statistically analyzed using Prism (version 8.0). The Mann–Whitney test was used to compare the levels of cytokines/chemokines between groups. P < 0.05 was considered significantly different.
Results
The ratio of male to female was 7/3 in both groups. The average age was 59.5 (range 51~71) in sAE group and 56.9 (range 50~71) in the control, and there was no difference in the distribution of age and gender between patients with sAE and the controls. The detailed clinical features of the 10 patients with sAE are listed in Table 1.
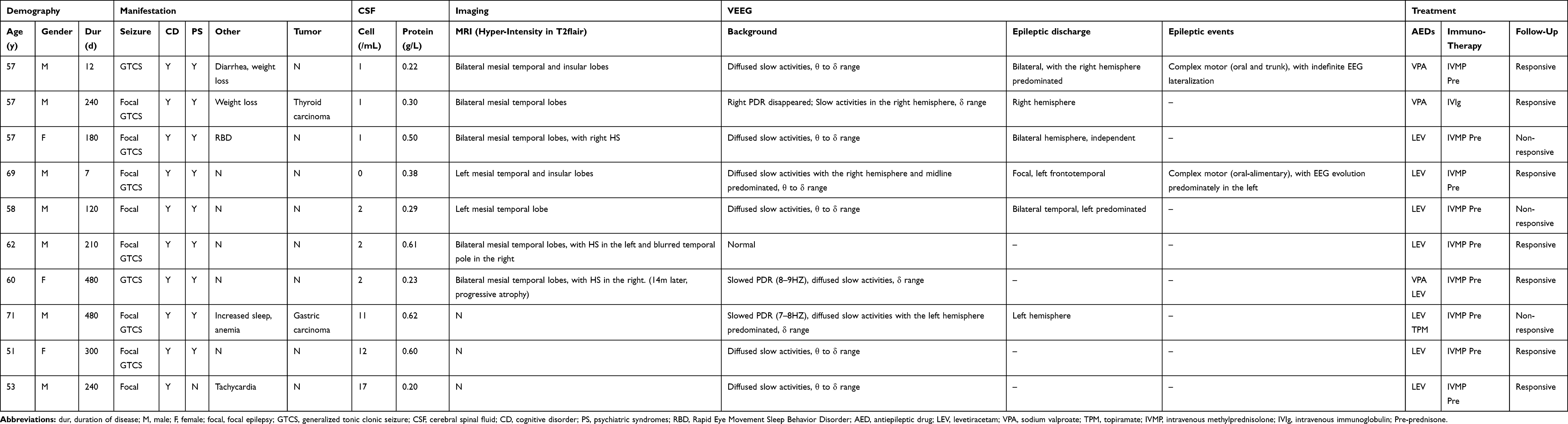 |
Table 1 Clinical Data of Patient Group |
The CSF levels of IL-6, IL-17, HMGB1, and CXCL12 were significantly higher in patients with sAE than in those in the control group, while the BAFF, IL-10 and CXCL13 levels were not significantly different from the control group (Table 2 and Figure 1).
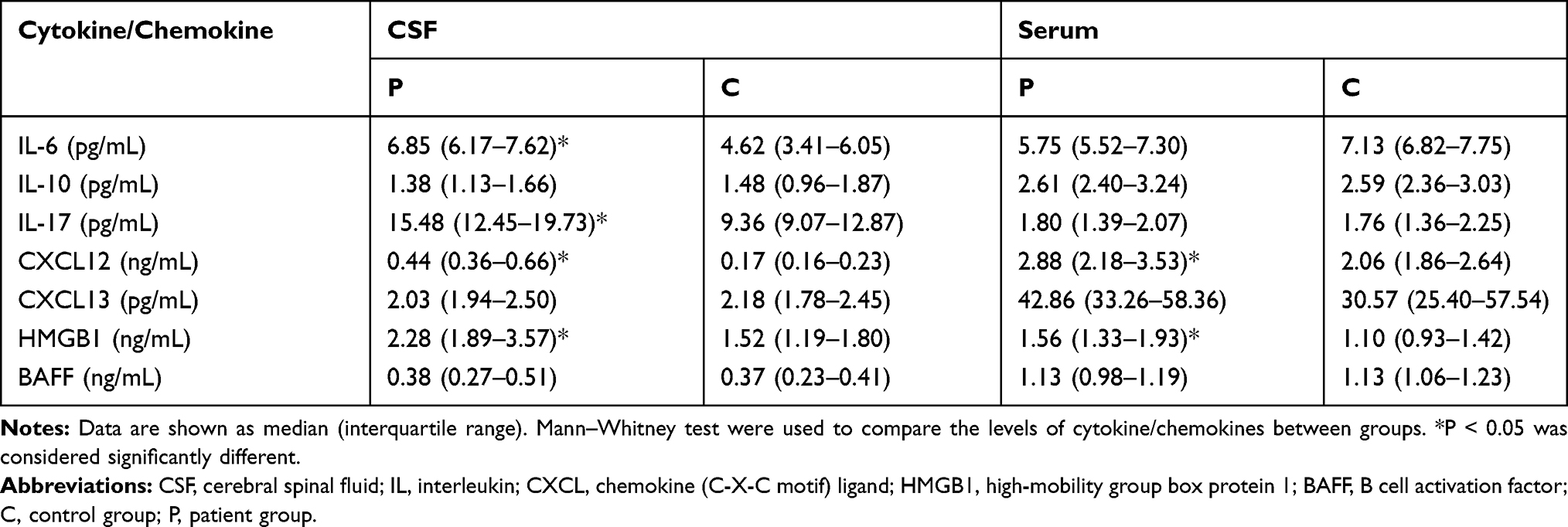 |
Table 2 Levels of Cytokines and Chemokines in the CSF and the Serum |
 |
Figure 1 CSF concentrations of cytokines/chemokines: IL-6 (A), IL-10 (B), IL-17 (C), CXCL12 (D), CXCL13 (E), HMGB1 (F) and BAFF (G). Higher levels of IL-6 (A), IL-17 (C), CXCL12 (D) and HMGB1 (F) in the CSF are found in late-onset sAE patients. Note: *P < 0.05 was considered significantly different.Abbreviations: CSF, cerebral spinal fluid; IL, interleukin; CXCL12, stromal cell-derived factor-1; CXCL13, C-X-C motif chemokine 13; HMGB1, high-mobility group box protein 1; BAFF, B cell activation factor; C, control group; P, patient group. |
The serum HMGB1 and CXCL12 levels were significantly elevated in patients with sAE compared with the control group. There were no significant differences in the levels of IL-6, IL-17, BAFF, IL-10, and CXCL 13 between the sAE and control groups (Table 2 and Figure 2).
 |
Figure 2 Blood concentrations of cytokines/chemokines: IL-6 (A), IL-10 (B), IL-17 (C), CXCL12 (D), CXCL13 (E), HMGB1 (F) and BAFF (G). Higher levels of CXCL12 (D) and HMGB1 (F) in the serum are found in late-onset sAE patients. Note: *P < 0.05 was considered significantly different.Abbreviations: IL, interleukin; CXCL12, stromal cell-derived factor-1; CXCL13, C-X-C motif chemokine 13; HMGB1, high-mobility group box protein 1; BAFF, B cell activation factor; C, control group; P, patient group. |
Discussion
In this study, we demonstrated elevated levels of HMGB1, IL-6, IL-17, and CXCL12 in the CSF as well as of HMGB1 and CXCL12 in the serum of patients with sAE. There was no difference in the other serum/CSF cytokines or chemokines between the two groups, including IL-10, CXCL13, and BAFF. We proposed that the inflammatory cytokines/chemokines could serve as potential biomarkers for active neuroinflammation in sAE, and might indicate aggressive immunotherapy.
It has been proposed that the profiles of cytokine/chemokines in the CSF/serum reflect the distinct mechanism underlying autoimmune CNS diseases.8,11 We speculate that Th17 cells are recruited in sAE on the basis of the elevation of IL-6/IL-17 in the CSF. IL-6 induces the development of Th17 cells from naive T cells together with transforming growth factor-β (TGF-β), while IL-6 inhibits TGF-β-induced T regulatory cell differentiation.16,17 An overproduction of IL-6 leads to autoimmune diseases, such as MS and rheumatoid arthritis (RA), in which Th17 cells are considered to be the primary cause of pathology and a therapeutic target.18–20 Thus, the upregulation of CSF IL-6/IL-17 may suggest that Th17 cells could be having an important role in the pathogenesis in sAE.
CXCL12, CXCL13, and BAFF are markers of B cell activation in neuroinflammatory CNS disorders;8,21 however, the mechanisms and consequences of elevated CXCL12 could be complicated due to multiple modulatory mechanisms, such as changes in expression, bioavailability, and post-translational modifications, as well as changes in its receptors CXC chemokine receptor 4 (CXCR4), atypical chemokine receptor 3 (ACKR3), and glycosaminoglycans (GAGs).22 With respect to CNS disorders, little is known about CXCL12. The CXCL12/CXCR4 signaling pathway has been reported to suppress inflammation and promote the repair of myelin/neurons in MS models;23–25 however, recent studies have shown that increased levels of CXCL12 together with other cytokines/chemokines are detected in the CSF of MS patients with higher levels of grey matter damage at the time of diagnosis.26 A specific Th cell population co-expressing CXCR4 offers both a diagnostic marker and therapeutic target in MS,27 demonstrating the pro-inflammatory roles. In this study we detected an elevation of CXCL12 with no change in the other B cell markers (CXCL13 and BAFF), which is quite different from previous reports about antibody-mediated autoimmune encephalitis in which CXCL13 was shown to be a potential biomarker of active CNS inflammation,28 and the major determinant for B cell recruitment to the CSF.29,30 Interestingly, another study showed that elevated CXCL12 might be associated with leucocyte extravasation and the maintenance of chronic neuroinflammation, while CXCL13 was more likely to be an indicator of the active process but not CXCL12.31 Thus, although the upregulation of CXCL12 might participate in neuroinflammation but not be sufficient as a proof of B cell activation in sAE. Details need to be further explored to elucidate the exact function of CXCL12 and its receptors.
HMGB1 is a nonhistone DNA-binding nuclear protein that acts as a damage- associated molecular pattern (DAMP). As a nuclear molecule, HMGB1 plays a key role in modulating DNA architecture and transcriptional regulation, while extracellular HMGB1 induces several responses in chronic inflammation, including the release of proinflammatory cytokines, cell proliferation, and cell infiltration.32,33 In particular, the function of HMGB1 in recruiting inflammatory cells requires the cooperation of CXCL12 and its receptor CXCR4, and HMGB1/CXCL12 promotes sustaining inflammation.33–35 The inhibition of HMGB1/CXCL12/CXCR4 could suppresses neuroinflammation,36,37 indicating its role in the pathogenesis of central nervous system (CNS) inflammatory diseases. Thus, the simultaneous elevation of HMGB1 and CXCL12 in the serum and CSF of the sAE group has several implications. As members of the proinflammatory signaling pathway, the simultaneous elevation of HMGB1 and CXCL12 could be an indicator of the underlying inflammatory mechanism. Combined with the elevated IL6/17 in the CSF, it is plausible to speculate that the HMGB1/CXCL12-related autoimmune mechanism might lead to leukocyte extravasation through the blood-brain barrier (BBB) and the upregulation of cytokines/chemokines within the CNS in sAE. Furthermore, they might serve as biomarkers for neuroinflammation and even potential therapeutic targets to be developed. In addition, the function of HMGB1/CXCL12 in mediating sustained inflammation might have a clinical significance in warning of the possibility of a chronic course no matter how long the disease duration was judged by the clinical presentation, and this was coincident with the clinical observation that the insidious onset of the neuropsychiatric comorbidity was a common feature for sAE.
As a retrospective and preliminary study, there were several pitfalls that need further exploration. The relationship of the cytokines/chemokines and prognosis was not included because of the small sample size and the heterogeneity of etiology, for instance, with or without performing oncological surgery. The patients included in this study underwent routine Ab detection based on clinical need, but were not tested for all known Abs. Thus, Ab-mediated immune epilepsy could not be excluded, even though the biomarkers of B cells were not significant. Nevertheless, negative results should be modestly considered because of the limited cytokines/chemokines analyzed in this study.
Conclusion
In a nutshell, our study shows that cytokines/chemokines may act as alternative biomarkers for diagnosis of sAE. The activation of both HMGB1/CXCL12-mediated immunity and T helper cells 17 (Th17) cells may be playing a central role in the pathogenesis of sAE. However, a larger cohort in a prospective approach is required to validate our findings.
Acknowledgments
The authors thank Dr. Geoffrey Joseph Changwe for his advice in English language copy editing.
Disclosure
The authors report no conflicts of interest in this work.
References
1. von Podewils F, Suesse M, Geithner J, et al. Prevalence and outcome of late-onset seizures due to autoimmune etiology: a prospective observational population-based cohort study. Epilepsia. 2017;58(9):1542–1550. doi:10.1111/epi.13834
2. Dubey D, Singh J, Britton JW, et al. Predictive models in the diagnosis and treatment of autoimmune epilepsy. Epilepsia. 2017;58(7):1181–1189. doi:10.1111/epi.13797
3. Weissert R. Adaptive immunity is the key to the understanding of autoimmune and paraneoplastic inflammatory central nervous system disorders. Front Immunol. 2017;8:336. doi:10.3389/fimmu.2017.00336
4. Akdis M, Burgler S, Crameri R, et al. Interleukins, from 1 to 37, and interferon-gamma: receptors, functions, and roles in diseases. J Allergy Clin Immunol. 2011;127(3):701–770. doi:10.1016/j.jaci.2010.11.050
5. Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36(5):705–716. doi:10.1016/j.immuni.2012.05.008
6. Okada K, Matsushita T, Kira J, Tsuji S. B-cell activating factor of the TNF family is upregulated in neuromyelitis optica. Neurology. 2010;74(2):177–178. doi:10.1212/WNL.0b013e3181c919ee
7. Liba Z, Kayserova J, Elisak M, et al. Anti-N-methyl-D-aspartate receptor encephalitis: the clinical course in light of the chemokine and cytokine levels in cerebrospinal fluid. J Neuroinflammation. 2016;13(1):55. doi:10.1186/s12974-016-0507-9
8. Kothur K, Wienholt L, Brilot F, Dale RC. CSF cytokines/chemokines as biomarkers in neuroinflammatory CNS disorders: a systematic review. Cytokine. 2016;77:227–237. doi:10.1016/j.cyto.2015.10.001
9. Lin YT, Yang X, Lv JW, Liu XW, Wang SJ. CXCL13 is A biomarker of Anti-Leucine-Rich Glioma-inactivated protein 1 encephalitis patients. Neuropsychiatr Dis Treat. 2019;15:2909–2915. doi:10.2147/NDT.S222258
10. Byun JI, Lee ST, Moon J, et al. Distinct intrathecal interleukin-17/interleukin-6 activation in anti-N-methyl-d-aspartate receptor encephalitis. J Neuroimmunol. 2016;297:141–147. doi:10.1016/j.jneuroim.2016.05.023
11. Kothur K, Wienholt L, Mohammad SS, et al. Utility of CSF cytokine/chemokines as markers of active intrathecal inflammation: comparison of demyelinating, anti-NMDAR and enteroviral encephalitis. PLoS One. 2016;11(8):e0161656. doi:10.1371/journal.pone.0161656
12. Ai P, Zhang X, Xie Z, et al. The HMGB1 is increased in CSF of patients with an Anti-NMDAR encephalitis. Acta Neurol Scand. 2018;137(2):277–282. doi:10.1111/ane.12850
13. Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391–404. doi:10.1016/S1474-4422(15)00401-9
14. Toledano M, Britton JW, McKeon A, et al. Utility of an immunotherapy trial in evaluating patients with presumed autoimmune epilepsy. Neurology. 2014;82(18):1578–1586. doi:10.1212/WNL.0000000000000383
15. Lv RJ, Ren HT, Guan HZ, Cui T, Shao XQ. Seizure semiology: an important clinical clue to the diagnosis of autoimmune epilepsy. Ann Clin Transl Neurol. 2018;5(2):208–215. doi:10.1002/acn3.520
16. Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi:10.1038/nature04753
17. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–189. doi:10.1016/j.immuni.2006.01.001
18. Serada S, Fujimoto M, Mihara M, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105(26):9041–9046. doi:10.1073/pnas.0802218105
19. Nishimoto N, Yoshizaki K, Miyasaka N, et al. Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 2004;50(6):1761–1769. doi:10.1002/art.20303
20. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. 2010;40(7):1830–1835. doi:10.1002/eji.201040391
21. Lepennetier G, Hracsko Z, Unger M, et al. Cytokine and immune cell profiling in the cerebrospinal fluid of patients with neuro-inflammatory diseases. J Neuroinflammation. 2019;16(1):219. doi:10.1186/s12974-019-1601-6
22. Janssens R, Struyf S, Proost P. Pathological roles of the homeostatic chemokine CXCL12. Cytokine Growth Factor Rev. 2018;44:51–68. doi:10.1016/j.cytogfr.2018.10.004
23. Zilkha-Falb R, Kaushansky N, Kawakami N, Ben-Nun A. Post-CNS-inflammation expression of CXCL12 promotes the endogenous myelin/neuronal repair capacity following spontaneous recovery from multiple sclerosis-like disease. J Neuroinflammation. 2016;13:7. doi:10.1186/s12974-015-0468-4
24. Meiron M, Zohar Y, Anunu R, Wildbaum G, Karin N. CXCL12 (SDF-1alpha) suppresses ongoing experimental autoimmune encephalomyelitis by selecting antigen-specific regulatory T cells. J Exp Med. 2008;205(11):2643–2655. doi:10.1084/jem.20080730
25. Miljkovic D, Stanojevic Z, Momcilovic M, Odoardi F, Flugel A, Mostarica-Stojkovic M. CXCL12 expression within the CNS contributes to the resistance against experimental autoimmune encephalomyelitis in albino oxford rats. Immunobiology. 2011;216(9):979–987. doi:10.1016/j.imbio.2011.03.013
26. Magliozzi R, Howell OW, Nicholas R, et al. Inflammatory intrathecal profiles and cortical damage in multiple sclerosis. Ann Neurol. 2018;83(4):739–755. doi:10.1002/ana.25197
27. Galli E, Hartmann FJ, Schreiner B, et al. GM-CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat Med. 2019;25(8):1290–1300. doi:10.1038/s41591-019-0521-4
28. Leypoldt F, Hoftberger R, Titulaer MJ, et al. Investigations on CXCL13 in anti-N-methyl-D-aspartate receptor encephalitis: a potential biomarker of treatment response. JAMA Neurol. 2015;72(2):180–186. doi:10.1001/jamaneurol.2014.2956
29. Rainey-Barger EK, Rumble JM, Lalor SJ, Esen N, Segal BM, Irani DN. The lymphoid chemokine, CXCL13, is dispensable for the initial recruitment of B cells to the acutely inflamed central nervous system. Brain Behav Immun. 2011;25(5):922–931. doi:10.1016/j.bbi.2010.10.002
30. Kowarik MC, Cepok S, Sellner J, et al. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J Neuroinflammation. 2012;9:93. doi:10.1186/1742-2094-9-93
31. Krumbholz M, Theil D, Cepok S, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain. 2006;129(Pt 1):200–211. doi:10.1093/brain/awh680
32. Gorgulho CM, Romagnoli GG, Bharthi R, Lotze MT. Johnny on the spot-chronic inflammation is driven by HMGB1. Front Immunol. 2019;10:1561. doi:10.3389/fimmu.2019.01561
33. Cecchinato V, D’Agostino G, Raeli L, et al. Redox-mediated mechanisms fuel monocyte responses to CXCL12/HMGB1 in active Rheumatoid Arthritis. Front Immunol. 2018;9:2118. doi:10.3389/fimmu.2018.02118
34. Schiraldi M, Raucci A, Munoz LM, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. 2012;209(3):551–563. doi:10.1084/jem.20111739
35. De Leo F, Quilici G, Tirone M, et al. Diflunisal targets the HMGB1/CXCL12 heterocomplex and blocks immune cell recruitment. EMBO Rep. 2019;e47788.
36. Hei Y, Chen R, Yi X, Long Q, Gao D, Liu W. HMGB1 neutralization attenuates hippocampal neuronal death and cognitive impairment in rats with chronic cerebral hypoperfusion via suppressing inflammatory responses and oxidative stress. Neuroscience. 2018;383:150–159. doi:10.1016/j.neuroscience.2018.05.010
37. Das S, Mishra KP, Chanda S, Ganju L, Singh SB. CXCR7: a key neuroprotective molecule against alarmin HMGB1 mediated CNS pathophysiology and subsequent memory impairment. Brain Behav Immun. 2019;82:319–337. doi:10.1016/j.bbi.2019.09.003
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.