Back to Journals » OncoTargets and Therapy » Volume 13
Highly Efficient Targeting of EGFR-Expressing Tumor Cells with UniCAR T Cells via Target Modules Based on Cetuximab®
Authors Jureczek J, Feldmann A, Bergmann R ![]() , Arndt C, Berndt N, Koristka S, Loureiro LR
, Arndt C, Berndt N, Koristka S, Loureiro LR ![]() , Mitwasi N, Hoffmann A, Kegler A, Bartsch T, Bachmann M
, Mitwasi N, Hoffmann A, Kegler A, Bartsch T, Bachmann M ![]()
Received 8 January 2020
Accepted for publication 19 April 2020
Published 12 June 2020 Volume 2020:13 Pages 5515—5527
DOI https://doi.org/10.2147/OTT.S245169
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Justyna Jureczek,1– 3 Anja Feldmann,3 Ralf Bergmann,3,4 Claudia Arndt,3 Nicole Berndt,3 Stefanie Koristka,3 Liliana Rodrigues Loureiro,3,5 Nicola Mitwasi,3 Anja Hoffmann,3 Alexandra Kegler,1– 3 Tabea Bartsch,3 Michael Bachmann1– 3,5,6
1German Cancer Consortium (DKTK), Dresden, Germany; 2German Cancer Research Center (DKFZ), Heidelberg, Germany; 3Department of Radioimmunology, Institute of Radiopharmaceutical Cancer Research, Helmholtz-Zentrum Dresden-Rossendorf (HZDR), Dresden, Germany; 4Department of Biophysics and Radiation Biology, Semmelweis University, Budapest, Hungary; 5National Center for Tumor Diseases (NCT), German Cancer Research Center (DKFZ), Heidelberg; Faculty of Medicine and University Hospital Carl Gustav Carus, Technische Universität Dresden, Dresden; Helmholtz-Zentrum Dresden - Rossendorf (HZDR), Dresden, Germany; 6Tumor Immunology, University Cancer Center (UCC), University Hospital Carl Gustav Carus Dresden, Technische Universität Dresden, Dresden, Germany
Correspondence: Michael Bachmann
Helmholtz-Zentrum Dresden-Rossendorf e.V. Managing Director, Institute of Radiopharmaceutical Cancer Research, Bautzner Landstraße 400, Dresden 01328, Germany
Tel +49 351 260 3170
Fax +49 351 260 3232
Email [email protected]
Introduction: Since epithelial growth factor receptor (EGFR) overexpression is linked to a variety of malignancies, it is an attractive target for immune therapy including chimeric antigen receptor (CAR)-engineered T cells. Unfortunately, CAR T cell therapy harbors the risk of severe, even life-threatening side effects. Adaptor CAR T cell platforms such as the previously described UniCAR system might be able to overcome these problems. In contrast to conventional CARs, UniCAR T cells are per se inert. Their redirection towards target cells occurs only in the presence of a tumor-specific target molecule (TM). TMs are bifunctional molecules being able to recognize a tumor-associated antigen and to cross-link the CAR T cell via a peptide epitope recognized by the UniCAR domain.
Materials and Methods: Here, we compare αEGFR TMs: a nanobody (nb)-based αEGFR TM derived from the camelid αEGFR antibody 7C12 with a murine and humanized single-chain fragment variable (scFv) based on the clinically used antibody Cetuximab®.
Results: In principle, both the nb- and scFv-based TM formats are able to redirect UniCAR T cells to eliminate EGFR-expressing tumor cells in an antigen-specific and TM-dependent manner. However, the scFv-based αEGFR TM was significantly superior to the nb-based TM especially with respect to lysis of tumor cells.
Discussion: Improved efficiency of the scFv-based TM allowed the redirection of UniCAR T cells towards tumor cells expressing high as well as low EGFR levels in comparison to nb-based αEGFR TMs.
Keywords: EGFR, UniCAR, CAR T cells, adaptor CARs, solid tumors, immunotherapy
Introduction
EGFR overexpression or mutation is common in various types of tumors, including several human epithelial malignancies like lung, colon, kidney, and head and neck carcinomas as well as pancreatic tumors.1 High EGFR expression is correlated with poor prognosis and treatment resistance.2,3 Therefore, EGFR and the EGFR signaling pathway represent attractive targets for tumor therapy. In principle, two types of EGFR-targeting strategies have been established clinically based on either anti-EGFR monoclonal antibodies (mAbs) or small-molecule EGFR inhibitors.4,5 Nevertheless, both therapeutic approaches struggle with some limitations. For instance treatment resistance frequently occurs in patients most likely caused by mutations within the EGFR itself or other kinases involved in the EGFR signaling pathway, as well as other compensation mechanisms that may overcome the EGFR inhibition.6,7 In order to circumvent some of these problems, retargeting of genetically modified chimeric antigen receptor (CAR)-expressing T cells to EGFR-positive cells came into focus of immunotherapies especially since the approval of CAR T cell therapies directed against the CD19 antigen for the clinical use in hematological malignancies (Kymriah™ (Tisagenlecleucel-T; CTL019, CART-19) and Yescarta™ (Axicabtagene ciloleucel; KTE-C19)).8,9 Encouraged by the success of anti-CD19-directed CAR T cells in certain hematological malignancies, notable efforts are being made for the application of CAR T cells also for solid tumor treatment. Up to date, around 200 clinical trials are ongoing with CAR T cell-based products.10 However, clinical efficacy of CAR T cells against solid tumors is highly challenging due to some physical and biochemical hurdles. For example, infiltration of CAR T cells into the tumor is limited and their function is impaired due to the immunosuppressive microenvironment.11,12 Furthermore, CAR T cells can also cause severe to life-threatening side effects like on-target/off-tumor toxicities in patients, in case the targeted tumor-associated antigen (TAA) is not limited to malignant but also expressed on healthy cells.13,14 In particular, EGFR is such a common target that is overexpressed on many tumor tissues but also expressed on normal cells.
As conventional CAR T cell reactivity cannot be controlled in patients there is an urgent need to develop safety strategies to minimize CAR T cell adverse reactions. Encouraged by the task to develop a “control mode” for CAR T cells, we introduced a modular CAR system termed UniCAR in 2014.15 The UniCAR strategy splits the signaling and antigen-binding domains of conventional CARs into two separate components: (I) the UniCAR T cell and (II) the target module (TM) (Figure 1). The extracellular domain of the UniCAR is not directed against a TAA on the tumor cell but against a short peptide motif of 10 amino acids (E5B9 tag) derived from the human nuclear autoantigen La/SS-B.16 For recognition of the target antigen, the UniCAR T cell needs its ON switch-TM, carrying the specificity for a TAA and the E5B9 tag for UniCAR T cell recognition. Consequently, T cells engineered to express UniCARs remain inactive after infusion and exert their cytotoxic potential only in the presence of specific TMs cross-linking them towards tumor cells.15,17 An additional advantage of the switchable and controllable UniCAR system is its high flexibility with respect to the tumor and target entity as well as the TM format,18,19 because it can be easily redirected to a broad variety of tumor targets, simply by exchanging the TM. Furthermore, different TM structures can be used in the UniCAR system ranging from small peptide molecules (PET tracers), nanobodies (nbs), single-chain fragments variable (scFvs) to size- and half-life-extended IgG backbones.
 |
Figure 1 Switchable UniCAR system. The UniCAR system is composed of two separate units: (I) T cells genetically engineered with UniCARs comprising a humanized αE5B9 scFv as extracellular domain and (II) a target module (TM) carrying an antigen-binding domain (eg a single-chain fragment variable (scFv)) that is directed against a specific tumor-associated antigen (TAA) and coupled to the E5B9 peptide epitope (E5B9 tag). UniCAR T cells per se are unable to bind to target cells (OFF switch). The cross-linkage of UniCAR T cells with their targets is mediated by a specific TM resulting in UniCAR T cell activation and tumor cell killing (ON switch). Here, we present novel TMs directed against EGFR. |
Recently, we described a novel TM directed against EGFR based on the αEGFR nb 7C12.20 Although the nb-based TM could be expressed by both prokaryotic and eukaryotic expression systems, the prokaryotic expression reduced its specific activity most likely due to hydrophobic aggregation caused by the lack of post-translational modifications such as glycosylation.21,22 Though well functional as TM a camelid-derived protein may be more immunogenic than a humanized scFv. As the chimeric mAb Cetuximab® (IMC C-225) is already clinically approved, we decided to construct additional αEGFR TMs that are based on the variable heavy and light chains of Cetuximab®.23 In order to minimize immunogenicity, we humanized the murine αEGFR scFv sequence of Cetuximab® by CDR grafting as described previously.24 Then, we compared the functionality of the murine and humanized Cetuximab®-derived scFv TMs with the previously designed nb-based αEGFR TM.
Here, we present a novel humanized scFv-based αEGFR TM constructed from the sequence of the clinically used mAb Cetuximab®. Most importantly, the novel scFv-based αEGFR TM shows superior efficiency to the previously published nb-based αEGFR TM especially with respect to killing of tumor cells expressing low levels of EGFR.
Materials and Methods
PBMCs and Cell Lines
Target cancer cell lines A431, MDA-MB-435S and Ab-producing murine 3T3, were purchased from American Type Culture Collection and maintained according to supplier recommendations, as already described.20 Panc-89 and L363 cells were cultured in RPMI 1640 medium supplemented with 10% FCS, 1% non-essential amino acids, 1 mM sodium pyruvate, 2 mM N-acetyl-L-alanyl-L-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin (Biochrom, Berlin). A431 and Panc-89 cells were transduced with firefly luciferase, resulting in A431 luc or Panc-89 luc cells, according to procedures described previously.20 Human T cells used in this study were isolated from peripheral blood mononuclear cells (PBMCs) from buffy coats of healthy donors and cultured following the protocols described previously.25 All procedures using human materials were performed in accordance with local regulations, guidelines and approval from the local ethics committee of the Medical Faculty Carl Gustav Carus Dresden of the TU Dresden (EK27022006). All healthy blood donors provided written informed consent.
Lentiviral Transduction of T Cells
UniCAR T cells were obtained via lentiviral transduction of human T cells activated using CD3/CD28 magnetic beads (Dynabeads™ Human T-Activator CD3/CD28, Invitrogen) as described previously.26 For the experiments, three different constructs were used: (I) the signaling UniCAR (UniCAR CD28/ζ), (II) the UniCAR lacking an intracellular signaling domain (UniCAR stop) and the mock construct expressing only the reporter EGFP (vector control). Transduced T cells were sorted on a FACSAria III (BD Biosciences, Heidelberg, Germany) for EGFP expression (purity >95%) and cultured in RPMI 1640 complete medium supplemented with 200 U/mL IL-2 (Proleukin® S, Novartis Pharmaceuticals, Horsham, UK), 5 ng/mL IL-7 and 5 ng/mL IL-15 (ImmunoTools, Friesoythe, Germany).
Generation, Expression, Purification and Characterization of the Novel EGFR-Specific Target Modules
Recently, we described a nb-based αEGFR TM.20 As mentioned above its TAA-binding domain is derived from the camelid single-domain Ab (sdAb) αEGFR HCAb Clone 7C12. In order to directly compare the sdAb-based TM with TMs based on the scFv of the αEGFR mAb Cetuximab®, the murine or the humanized sequences of the variable heavy (VH) and light chain (VL) of Cetuximab®27 were fused via glycine-serine linkers. Then, the UniCAR E5B9 tag and an oligo-His-tag were added to the C-terminus of these murine and humanized αEGFR scFvs. The resulting recombinant αEGFR TM genes were transduced into murine 3T3 cells and permanent cell lines were established secreting the respective TM into the cell culture supernatant. TMs were purified from cell culture supernatants using Ni-NTA affinity chromatography and eluted as previously reported.28 Elution fractions were finally dialyzed against 1xPBS (Biochrom GmbH, Berlin, Germany) for 24 h and subsequently characterized by SDS-PAGE and immunoblotting.
Binding Assays
EGFR expression of target cell lines and specific binding of obtained αEGFR TMs were analyzed using flow cytometry. Determination of EGFR density on the target cells was performed via staining with mouse α-human EGFR IgG1 mAb (clone AY13; BioLegend, Fell, Germany) and goat-α-mouse IgG-Pacific Blue™ (Thermo Fisher Scientific, Germany). EGFR quantification was performed using the QIFIKIT® (Agilent Technologies, Böblingen, Germany) following the manufacturer’s instructions. Specific binding of TMs to the target cells was detected via UniCAR E5B9 tag. Tumor cells (2x105) were incubated for 1 h at 4°C with 25 ng/µL of the respective TM, washed with PBS and subsequently stained for 30 min at 4°C with mouse αE5B9 mAb (5 µg/mL). Detection was assessed using goat-α-mouse IgG-Pacific Blue™ (Thermo Fisher Scientific). As a negative control, cells were incubated only with the detection Abs. In order to estimate the equilibrium dissociation constant value (Kd) of the respective TM, target cells were incubated with increasing TM concentrations ranging between 0.1 ng/µL and 100 ng/µL. Cells were analyzed using MACSQuant® Analyzer and the MACSQuantify® software (Miltenyi Biotec GmbH, Bergisch-Gladbach, Germany). Based on the relative median of fluorescence intensity of the stained cells binding curves were prepared and corresponding Kd values were calculated using the GraphPad Prism 8.0 software (GraphPad Software Inc., La Jolla, CA, USA).
In vitro Tumor Cell Lysis
The cytotoxic potential of redirected UniCAR T cells towards EGFR-positive tumor cells via TM was assessed using standard chromium release assays. The 51Cr-labelled target cells (A431 or MDA-MB 435S) were incubated with UniCAR CD28/ζ T cells at an effector to target cell (E:T) ratio of 5:1 with or without the respective TM. Supernatants were collected after 24 h and released 51Cr was measured in a beta counter (PerkinElmer LAS GmbH). Specific tumor cell lysis was calculated as previously described.29 Besides chromium release assays, tumor cell killing was also analyzed by using a bioluminescence-based killing assay (luciferase assay) as described previously.28
Cytokine Secretion
Cytokine release was measured from cell-free supernatants of 24 h co-cultures by ELISA using the OptEIA™ Human IFN-γ, Human TNF, Human GM-CSF and Human IL-2 ELISA Kits (BD Biosciences) as described previously30 or MACSPlex Cytokine 12 Kit (Miltenyi Biotec GmbH) according to the manufacturer’s instructions.
Tumor Xenograft Model
Tumor xenograft model in five-week-old female Rj:NMRI-Foxn1nu/nu mice (Janvier Labs, St. Berthevin, France) was established to investigate in vivo functionality of αEGFR TM. Mice were divided into three groups each containing five animals. All groups were subcutaneously injected into the right flank with 1x106 A431 luc target cells. In the control groups, tumor cells were injected alone (group 1) or together with 1x106 UniCAR CD28/ζ T cells in the presence of an irrespective TM (group 2). Mixtures of tumor cells, UniCAR CD28/ζ T cells and αEGFR TM were injected into animals of the treated group (group 3). Tumor cell killing in anesthetized mice was investigated by bioluminescence imaging as described previously.31 Data evaluation was performed by using the analysis software Bruker MI and Multispectral (Bruker, Karlsruhe, Germany). The bioluminescence activity of region of interest over the tumor area was analyzed and background signal was subtracted. Acquired values (photons/s/mm2) were normalized to that obtained on the day of injection (day 0).30 All animal studies were approved by the Landesdirektion Dresden (24–9165.40-4. 24.9168.21-4/2004-1) and performed at the Helmholtz-Zentrum Dresden-Rossendorf (HZDR) in accordance with the guidelines of German Regulations for Animal Welfare.
Statistical Analysis
Statistical analysis of all data was completed using GraphPad Prism 8.0 software (GraphPad Software Inc., La Jolla, CA, USA). Flow cytometry data was analyzed using FlowJo v.10.
Results
Construction, Purification and Biochemical Characterization of Novel αEGFR TMs
UniCAR T cells can be redirected to EGFR-expressing tumor cells by Ab-based TMs as schematically shown in Figure 1. In a previous study, we used nb-based TMs derived from the VHH domain of the camelid Ab clone 7C12 fused to the UniCAR E5B9 tag.20 As mentioned above, the novel αEGFR TMs were constructed from scFvs derived from Cetuximab®. One TM is based on the original murine sequence of the variable heavy and light chains of Cetuximab® (Mu scFv αEGFR TM).27 The other one represents the corresponding fully humanized scFv TM (Hu scFv αEGFR TM) which was obtained by CDR grafting to minimize its immunogenicity.27 Finally, we compared side-by-side their biochemical properties and functionality with the previously published nb-based αEGFR TM. The structural differences between the nb-based and novel scFv-based TMs are schematically shown in Figure 2A and B.
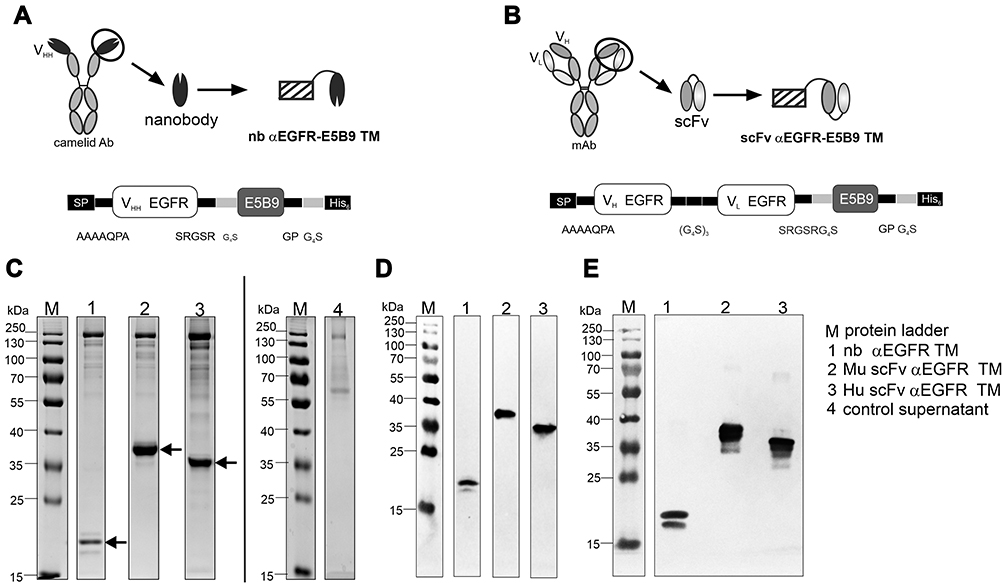 |
Figure 2 Construction and biochemical characterization of αEGFR TMs. The pre-existing nanobody (nb) αEGFR target module (TM) consists of one single camelid antibody-derived (clone 7C12) domain to enable EGFR binding and the E5B9 tag for UniCAR recognition (A). Novel murine (Mu) and humanized (Hu) αEGFR TMs consist of the single-chain fragment variable (scFv) of the αEGFR mAb Cetuximab and the E5B9 tag (B). The recombinant TMs were further equipped with 6xHis residues (His6) for protein purification and detection (A, B). Amino acids of linker elements are indicated (A, B). Recombinant TMs were expressed by 3T3 cells after transduction. Eluted and dialyzed fractions were separated via SDS-PAGE and stained with Coomassie brilliant blue G-250 (C) or detected after immunoblotting via αHis mAb (D) or αE5B9 mAb (E) on a nitrocellulose membrane. Arrows indicate the recombinant TMs. Supernatant of non-transduced 3T3 wild type cells was analyzed as a negative control (control supernatant) in the Coomassie-stained gel (C). (VH, variable heavy chain domain; VL, variable light chain domain). |
All recombinant fusion proteins contain an N-terminal signaling peptide (SP) for secretion, the C-terminal E5B9 tag for UniCAR recognition and a His tag for purification and detection. All TMs were expressed in murine 3T3 cells and isolated from the cell culture supernatants using Ni-NTA affinity chromatography. The size, concentration and purity of TMs were analyzed by SDS-PAGE and immunoblotting. Gels were stained with Coomassie brilliant blue G-250. As shown in Figure 2C, D and E, all TMs were expressed at concentrations between 1 and 5 µg/mL. Purified TMs migrated with an apparent molecular mass of 30–35 kDa for the scFv TMs and 20 kDa for the nb TM. These data are in agreement with their theoretical values of 30.4 kDa for the Mu scFv, 29.5 kDa for the Hu scFv and 15 kDa for the nb-based TM. Immunoblot analysis confirmed that all constructs were expressed as full-size proteins and could be detected via their C-terminal His tag (Fig. 2D) and UniCAR E5B9 tag (Fig. 2E). In addition to the dominant TM bands, some high molecular weight impurities were observed in the Coomassie-stained SDS gel (Figure 2C). Most probably, these proteins were not aggregates of the recombinant TMs, since they did not react neither with the αHis Ab nor with the αE5B9 Ab. Similar impurities were co-purified in previous studies for other TMs.20,28 Moreover, it was shown that these impurities do neither interfere nor influence functional retargeting assays.20,28
Antigen-Specific Binding of αEGFR TMs
In order to evaluate the binding characteristics of the αEGFR TMs, flow cytometry binding assays were performed using A431 tumor cells. Firstly, the expression of EGFR was confirmed on A431 cells by using a commercially available αEGFR mAb (Figure 3A, black histogram). Afterwards, binding of the TMs was detected via their UniCAR E5B9 tag. As shown in Figure 3A (grey histograms), all of the TMs were able to bind to A431 target cells. Increasing concentrations of each of the TMs were used to estimate the Kd value for TM binding (Figure 3B). Estimated Kd for nb αEGFR TM was 77 nM, 45 nM for Mu scFv αEGFR TM and 40 nM for Hu scFv αEGFR TM. All TMs demonstrated a dose-dependent binding and similar binding affinities in the nanomolar range. Furthermore, these binding data confirm the accessibility of the UniCAR E5B9 tag of the TMs for UniCAR recognition after their binding to tumor cells what is essential for the functionality of the TMs within the UniCAR system. As shown in Supplementary Figure S1A, the nb-based and scFv-based TM did not bind to EGFR-negative tumor cells confirming antigen-specificity of the αEGFR TMs.
 |
Figure 3 Binding capability of αEGFR TMs to A431 cancer cells. In order to analyze the binding capability of the indicated TMs to EGFR-expressing A431 tumor cells, flow cytometry binding assays were performed. A431 cells were incubated with αEGFR mAb and detected via goat-α-mouse IgG-Pacific BlueTM to confirm EGFR expression on A431 cells (A). Furthermore, A431 cells were incubated with 25 ng/µL of respective target modules (TMs) and specific TM binding was detected via the E5B9 tag using the αE5B9 mAb in combination with the detection Ab mentioned above (A). Percentage (%) and median fluorescence intensity (MFI) of positively stained cells were shown (A). Target cells incubated with the detection Abs in the absence of any TM served as a negative control. Equilibrium dissociation constant (Kd) was evaluated using increasing concentrations of respective TMs (B). The means of relative MFI with SEM are shown for three independent experiments (B). |
Eradication of EGFR-Positive Tumor Cells by Redirected UniCAR T Cells
In order to demonstrate functionality of the novel scFv-based αEGFR TMs with respect to redirection of UniCAR T cells to kill tumor cells, chromium release assays were performed (see Materials and Methods). As shown in Figure 4A, all αEGFR TMs were able to successfully redirect UniCAR T cells leading to an efficient lysis of A431 tumor cells. In contrast, in the absence of any TM no tumor cell lysis was measured proving that UniCAR T cell activity can be controlled by TM availability. In addition, we observed that only T cells armed with the signaling UniCAR CD28/ζ construct possessed cytotoxic activity in the presence of the respective TM. T cells equipped with the negative control UniCAR stop construct lacking intracellular signaling domains or the EGFP vector control alone did not cause lysis of the target cells. At the used TM concentration maximal tumor cell lysis of 40 to 50 % induced by redirected UniCAR T cells was comparable for all TMs. Subsequently, we used titrated TM concentrations in order to estimate the range of working concentrations and further characterize the TM efficiencies. As anticipated, the therapeutic efficacy strictly correlated with the concentration of the respective TM (Figure 4B). Interestingly, the novel scFv-based αEGFR TMs worked clearly more efficient than the nb αEGFR TM. The plateau of specific lysis for nb αEGFR TM was reached at around 5 nM concentration, whereas for both scFvs only around 0.1 nM were needed. Furthermore, the comparison of the half-maximal effective concentration (EC50) values confirmed that the efficiency of the novel scFv-based TMs was superior to the nb-based TM. In detail, the EC50 values were estimated as 1.8 nM for nb αEGFR TM, 9 pM for Mu scFv αEGFR TM and 7 pM for Hu scFv αEGFR TM. As the murine and humanized scFv-based TMs performed equally well, the humanization procedure obviously had not influenced TM functionality. Moreover, we have demonstrated that specific tumor cell lysis increased with increasing effector to target cell (E:T) ratios, whereas killing even occurred at low E:T ratio of 2:1 (Figure 4C). In order to further confirm specificity of the αEGFR TMs, we have used EGFR-negative tumor cells (L363) in combination with UniCAR T cells and αEGFR TMs in killing assays. As shown in the Supplementary Figure S1B, αEGFR TMs did not mediate tumor cell killing of EGFR-negative cells confirming that UniCAR T cells were redirected by the αEGFR TMs in an antigen-specific manner.
In vivo Activity of Redirected UniCAR T Cells and αEGFR TMs
Next, a mouse tumor xenograft model was used to prove the ability of tumor eradication by TM-redirected UniCAR T cells in vivo. Since the in vivo functionality of the nb αEGFR TM was already demonstrated,20 and both the Mu scFv and Hu αEGFR TM performed equally well in vitro, we limited the mouse experiment to the Mu scFv αEGFR TM. A pool of fifteen female Rj:NMRI-Foxn1nu/nu mice were divided into three cohorts, each consisting of five animals. Mice of the control groups were injected with firefly luciferase-expressing EGFR-positive A431 tumor cells alone (group 1) or as a mixture with UniCAR CD28/ζ T cells and an irrelevant TM (group 2). Mice of the treatment group were co-injected with A431 luc cells, UniCAR CD28/ζ T cells and the Mu scFv αEGFR TM (group 3). Optical imaging of the bioluminescent signal was measured on the day of injection (day 0) and in the subsequent 3 days (day 1, day 2, day 3). As depicted in Figure 4D, luciferase activity could be continuously detected in the control groups, whereas the bioluminescent signal significantly decreased in the treated mice. Thus, tumor cell eradication was only observed in the treated group (A431 luc + UniCAR CD28/ζ + Mu scFv αEGFR) confirming the ability of Mu scFv αEGFR TM to effectively eliminate tumor cells in vivo by antigen-specific activation of UniCAR CD28/ζ T cells.
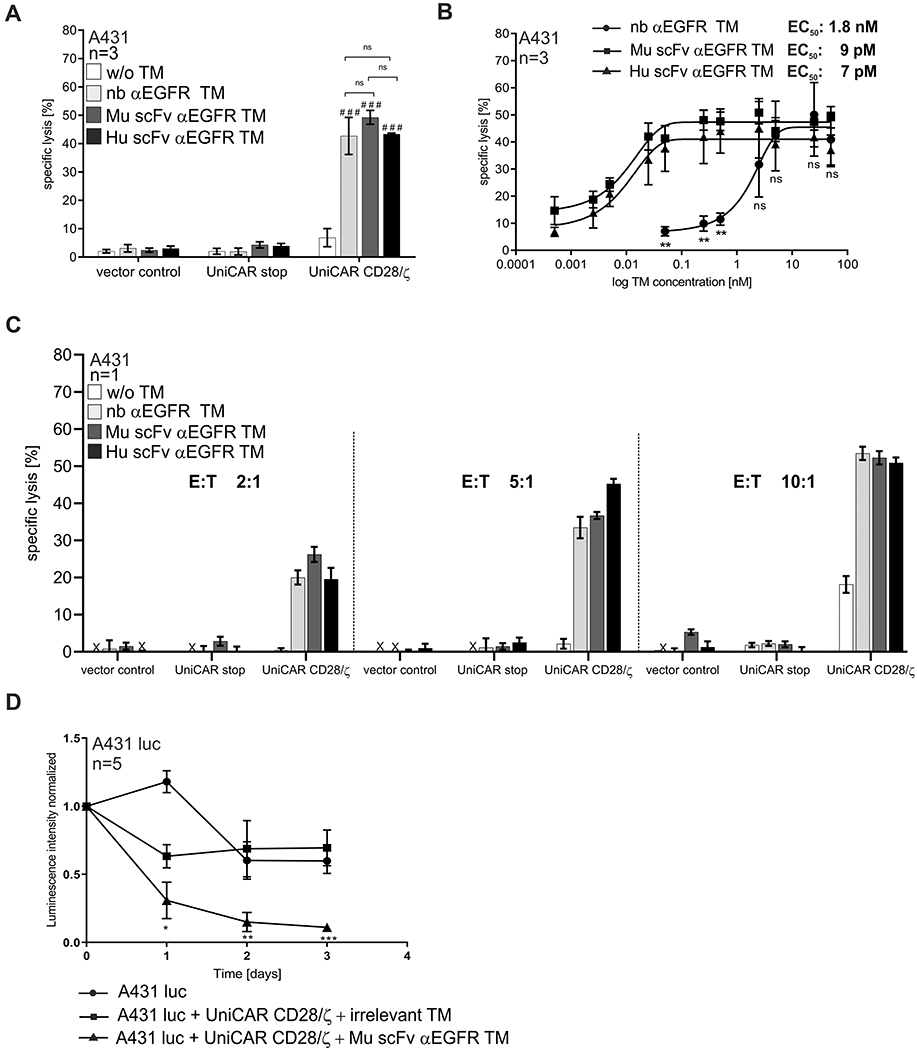 |
Figure 4 Killing of A431 tumor cells by UniCAR T cells redirected by αEGFR TMs in vitro and in vivo. Chromium release cytotoxicity assays were performed to compare the functionality of the nb- and scFv-based αEGFR target modules (TMs) with respect to the redirection of UniCAR T cells towards EGFR-expressing A431 tumor cells. Therefore, 51Cr-loaded A431 cells were incubated for 24 h in the presence or absence of 50 nM of the respective TM together with UniCAR CD28/ζ T cells at an effector to target cell (E:T) ratio of 5:1 (A, B). Mock T cells expressing only EGFP (vector control) or UniCAR stop T cells lacking the intracellular signaling domain (UniCAR stop) served as controls. Bars represent the mean specific lysis with SEM of three individual donors (A). Using UniCAR CD28/ζ T cells under the same conditions with increasing concentrations of respective TMs, half-maximal effective concentration (EC50) values were estimated. Mean specific lysis with SEM of three individual donors for each concentration is shown (B). Additionally, indicated T cells were incubated with A431 tumor cells at different E:T ratios in the presence or absence of indicated TMs (C). Bars represent the mean specific lysis with SEM of triplicates for one representative donor (C). In vivo functionality of αEGFR TM was analyzed in a xenograft mouse model (D). Immunodeficient Rj:NMRI-Foxn1nu/nu mice were injected into the left flank with 1x106 A431 luciferase-transduced tumor cells (A431 luc) alone or as a mixture with 1x106 UniCAR CD28/ζ T cells in the presence of an irrelevant target module (TM) or the Mu scFv αEGFR TM resulting in three experimental groups with five animals per group. Tumor cell killing in anesthetized mice was investigated by bioluminescence imaging at indicated time points. Quantitative signal analysis was performed using Bruker Molecular Imaging software (Bruker, BioSpin Corporation) and presented as mean with SEM of each experimental group at indicated time points (D). Statistical significance was evaluated by two-way ANOVA (A) or one-way ANOVA (B, D) with Bonferroni’s multiple comparison test (###p < 0.001, ns (p ≥ 0.05) not significant with respect to control w/o TM or *p < 0.05, **p < 0.01, ***p < 0.001, ns (p ≥ 0.05) not significant between indicated TMs (A, B) or in comparison to the control group with irrelevant TM (D). |
Cytokine Production by UniCAR T Cells in Combination with αEGFR TMs
In order to investigate whether redirected UniCAR T cells release pro-inflammatory cytokines, we performed ELISA assays using supernatants of 24 h co-cultures of UniCAR CD28/ζ T cells with or without EGFR-positive A431 target cells (E:T ratio 5:1) in the absence or presence of respective TMs. Secretion of the cytokines GM-CSF, IFN-γ, TNF-α and IL-2 was verified for three individual donors. As clearly shown in Figure 5A, redirected UniCAR T cells were triggered to secrete the pro-inflammatory cytokines GM-CSF, IFN-γ, TNF-α and IL-2 in the presence of one of the αEGFR TMs. Total cytokine amounts were comparable between the nb- or scFv-based αEGFR TMs. However, as already observed in our previous studies, the absolute cytokine amounts differed between individual donors. Furthermore, we confirmed that cytokine secretion strictly depends on the cross-linkage of UniCAR T cells with EGFR-positive tumor cells via appropriate TMs, since cytokine amounts did not increase in the control groups without any TM or in the absence of A431 tumor cells. Subsequently, we further analyzed an extended list of cytokines using the human MACSPlex Cytokine 12 Kit (Miltenyi Biotec GmbH) for T cell redirection via the Mu scFv αEGFR TM. In doing so, we confirmed that the pro-inflammatory cytokines GM-CSF, IFN-γ, TNF-α and IL-2 were predominantly secreted, whereas IL-4, IL-5, IL-6, IL-9, IL-10, IL-12, IL-17A, and IFN-α were not induced or only secreted in marginal concentrations (Figure 5B). In conclusion, UniCAR T cells are able to produce pro-inflammatory cytokines in an antigen-specific and TM-dependent manner.
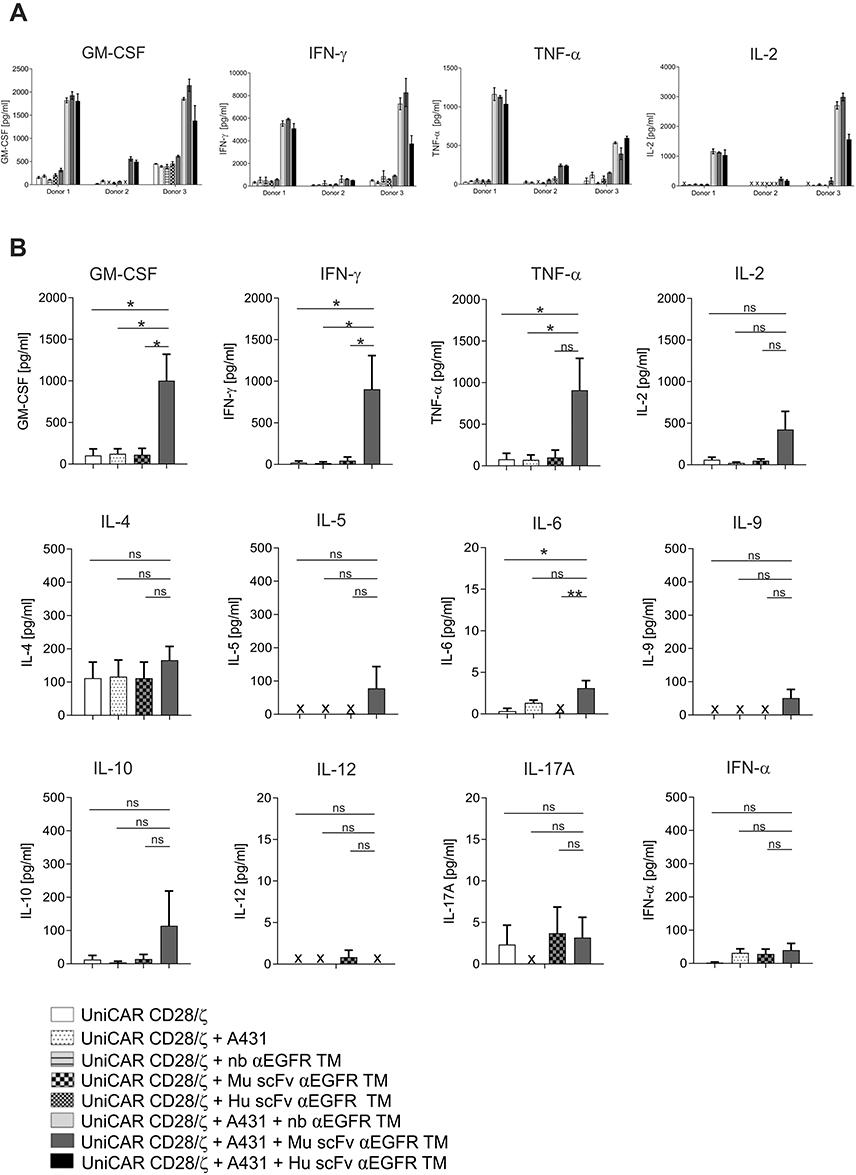 |
Figure 5 Cytokine secretion by EGFR-redirected UniCAR T cells. Cytokine concentration was measured by ELISA in cell-free supernatants of co-cultures of UniCAR CD28/ζ T cells with A431 target cells at an effector to target cell ratio of 5:1 and respective αEGFR target modules (TMs) for 24 h (A). Data represent the mean and SD of triplicates for three independent donors (A). Furthermore, 12 different cytokines were measured in cell-free supernatants of UniCAR T cells incubated with or without A431 tumor cells and the Mu scFv αEGFR TM under the same conditions using a bead-based flow cytometry MACSPlex Cytokine Kit (B). Mean and SEM for three individual donors are shown. Statistical analysis was performed by one-way ANOVA with Dunnett’s multiple comparison test (*p < 0.05, **p < 0.01, ns (not significant)) (B). (x, not detectable). |
Elimination of EGFRlow-Expressing Tumor Cells by UniCAR T Cells Redirected via the Novel scFv-Based αEGFR TM
Revealing significant differences in the efficiency of nb-based versus scFv-based αEGFR TMs tested using EGFRhigh-expressing A431 cells (Figure 4B), we asked if superior efficiency facilitates the targeting of tumor cells presenting a low EGFR density level. For this purpose, the EGFRlow-expressing MDA-MB-435S cell line was selected based on QIFIKIT® analysis (Figure 6A). In contrast to EGFRhigh-expressing A431 tumor cells (Figure 3A), binding of nb- as well as scFv-based αEGFR TMs to EGFRlow-expressing MDA-MB-435S cells was barely detectable (Figure 6B). Interestingly, the murine and humanized scFv-based TM induced a significantly higher specific lysis of EGFRlow-expressing MDA-MB-435S tumor cells than the nb-based TM in comparison to the background lysis without any TM (Figure 6C and D). This finding is in accordance with the superior efficiency of the scFv-based αEGFR TM already observed for EGFRhigh-expressing A431 tumor cells (Figure 4B).
 |
Figure 6 Killing of EGFRlow-expressing tumor cells by UniCAR T cells redirected by αEGFR TMs. The estimated number of EGFR molecules per A431 or MDA-MB-435S tumor cell was evaluated using the QIFIKIT® (A). MDA-MB-435S tumor cells were stained with αEGFR mAb and detected via goat-α-mouse IgG-Pacific BlueTM to determine EGFR surface expression by flow cytometry (B). Binding of indicated αEGFR TMs to the MDA-MB-435S target cell line was analyzed by staining and flow cytometry (B). Therefore, cells were incubated with αEGFR TM, the αE5B9 mAb and goat-α-mouse IgG-Pacific BlueTM (B). As a negative control, cells were stained with the detection Ab alone (white graphs) (B). Percentage (%) and median fluorescence intensity (MFI) of positively stained cells were shown (B). In a standard chromium release cytotoxicity assay, EGFRlow-expressing MDA-MB-435S tumor cells were incubated with T cells genetically modified to express EGFP (vector control), non-signaling UniCAR stop or signaling UniCAR CD28/ζ in the presence or absence of indicated αEGFR TMs at an effector to target cell ratio of 5:1 (C). Mean specific lysis and SEM are shown for two individual donors in the presence of Hu scFv αEGFR TM and three individual donors in the presence of nb or Mu scFv αEGFR TM (C). In addition, MDA-MB-435S tumor cells were co-cultured together with UniCAR CD28/ζ T cells and increasing concentrations of the respective αEGFR TMs under the same conditions (D). Mean specific lysis and SEM are shown. Statistical analysis was performed by two-way ANOVA with Tukey’s multiple comparison test (C) or paired t-test for each specific concentration (D). (#p < 0.05, ##p < 0.01, ###p < 0.001 with respect to control w/o TM or *p < 0.05, **p < 0.01 between indicated TMs. (ns, not significant)). |
Discussion
Currently, various immunotherapeutic strategies for the elimination of cancer cells are under investigation with the aim of increasing safety and tumor specificity without damaging healthy tissues. EGFR is one of these examples as it is upregulated on many tumor cells but low levels of EGFR are also detectable on healthy cells. As a frontrunner in immunotherapy, the chimeric mouse-human mAb Cetuximab® was the first EGFR-targeting mAb clinically approved for the treatment of metastatic colorectal cancer and later also for metastatic non-small cell lung cancer as well as head and neck carcinomas.32,33 Clinical efficiency of Cetuximab® paved the way for the development of further therapeutics targeting EGFR, including mAbs (eg panitumumab, nimotuzumab), tyrosine kinase inhibitors (TKIs) (eg Gefitinib, Erlotinib, Lapatinib) and lately αEGFR CAR T cells.5,34,35
The use of CAR T cells has already been shown to be highly effective in the treatment of B cell lineage malignancies.36 However, side effects and tumor escape mechanisms still remain big challenges reducing the therapeutic success of this promising tool in the fight against cancer.37 As living drugs, conventional CAR T cells are hard to steer and their uncontrollable proliferation could unpredictably increase side effects of the CAR T cell therapy including cytokine release syndrome, on-target/off-tumor toxicity or neurotoxicity. If not treated properly, these adverse reactions could be even lethal.38,39 In order to manage these problems, novel strategies are urgently needed. Especially for targeting of TAAs like EGFR, switchable adaptor CAR systems that allow an on-/off-switch as well as a fine-tuning of CAR T cell activity may be suitable to steer CAR T cell activity and thereby may minimize on-target/off-tumor toxicities.40–42
One adaptor CAR platform with favorable pharmacokinetics of TMs is the previously described UniCAR system. It combines high anti-tumor efficiency with low immunogenicity, high flexibility, good controllability and increased safety properties in one platform.15,17,18,26,43 Cross-linkage of UniCAR T cells with antigen-presenting tumor cells via TMs results in UniCAR T cell activation, expansion, cytokine release and tumor cell killing in an antigen-specific and TM-dependent manner.18,20,26,44,45 The UniCAR system allows a flexible targeting of diverse tumor targets expressed on hematological or solid malignancies (e.g. CD19, CD123, CD33, PSCA, PSMA, GD2, STn, and others)18,28,46,47 by different TMs varying in their structure, specificity and binding valence. In general, we learnt that TMs based on different structures, like nb, scFv, IgG, as well as small peptide molecules (PET tracers), can be successfully used for redirection of UniCAR T cells.18 Consequently, a huge playground exists to establish TMs with different design, size, structures and valences.
The herein presented scFv-based αEGFR TM, both the murine (Mu scFv αEGFR TM) and the humanized (Hu scFv αEGFR TM) one, were efficiently produced by permanent 3T3 cells with sufficient yield and purity. Besides the dominant recombinant TM fraction, some high molecular weight impurities were observed after SDS-PAGE and Coomassie-staining in the purified protein samples (Figure 2C). As these co-isolated proteins do neither react with the anti-E5B9 mAb nor with Abs directed to the His tag they are most probably unrelated to the TMs and do not represent high molecular weight aggregates of the TMs. Bearing in mind that the Ni-NTA affinity chromatography is a one-step purification for the enrichment of His-tagged recombinant proteins, this technique commonly leads to an additional co-isolation of non-His-tagged protein impurities as also seen in previous studies.20,28 Their presence, however, is not critical at least not in preclinical studies. According to our data, all tested αEGFR TMs were able to bind to EGFR (Figure 3A). Moreover, the affinities of all tested TMs were comparable (Figure 3B). Most importantly, all αEGFR TMs were successful in the redirection of UniCAR T cells to kill EGFR-expressing tumor cells in vitro and in vivo (Figure 4). Fortunately, even humanization did not impair the functionality of the scFv-based TM. Even more of interest, the scFv-based TMs possessed significant superior efficiency compared to the nb-based TM. The EC50 value of the scFv TMs was around 200-260-fold lower than that of the nb-based TM. In addition to EGFRhigh-expressing tumor cells (Figure 4), we also tested tumor cells with an intermediate (Figure S2) and low EGFR expression (Figure 6). As shown in Figures 4 and 6 and Supplementary Figure S2, UniCAR T cells can be effectively redirected by the scFv-based TMs towards tumor cells expressing different EGFR density levels. Remarkably, the improved efficacy of scFv-based TMs allowed us to target EGFRlow tumor cells which were not attacked by nb-based TMs.20,31 However, one should keep in mind that targeting EGFRlow tumor cells increases also the risk of damaging healthy tissues where EGFR is expressed at low levels. However, this risk should be manageable by titration of the TM. Beside cytotoxic potential, UniCAR T cells mainly secrete the pro-inflammatory cytokines IFN-γ, GM-CSF, TNF-α, and IL-2 (Figure 5) promoting immunoreactions, persistence and proliferation of immune cells as well as tumor cell killing. This can be helpful to overcome the immunosuppressive microenvironment in solid tumors and to increase the anti-tumor effect. Similar to IL-4, IL-5, IL-9, IL-10, IL-12, IL-17A, and IFN-α, IL-6, a major player in the cytokine release syndrome, was not secreted in relevant amounts (Figure 5B). As shown previously for other UniCAR T cell/TM interactions,44,45 redirection of UniCAR T cells with the here presented novel αEGFR TMs also resulted in their expansion (data not shown). Noteworthy, UniCAR T cell activity is strictly dependent on the presence of the respective TM and target antigen confirming controllability and specificity of the UniCAR system.
Conclusion
We established novel scFv-based αEGFR TMs showing superior efficiency in the redirection of UniCAR T cells towards tumor cells expressing high as well as low EGFR levels in comparison to nb-based αEGFR TMs.
Acknowledgments
We thank Julia Lagler for the technical support with the in vitro experiments.
Author Contributions
J.J., A.F, R.B., N.B., M.B., planned, performed and analyzed the experiments. J.J. and A.F. arranged the figures of the manuscript. J.J., A.F, C.A., R.B., N.B., S.K., L.R.L, N.M., A.H., A.K., T.B discussed the data and provided critical suggestions. J.J., M.B. and A.F. wrote the manuscript. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
M.B. has invented the UniCAR system and holds patents related to the UniCAR system. He is co-founder and shareholder of the company GEMoaB Monoclonals GmbH, which owns the IP, related to the UniCAR system. M.B. declares no non-financial competing interests. All the other authors declare no financial and non-financial competing interests.
References
1. Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12:3–20. doi:10.1002/1878-0261.12155
2. Roskoski R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi:10.1016/J.PHRS.2013.11.002
3. Xu MJ, Johnson DE, Grandis JR. EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev. 2017;36(3):463–473. doi:10.1007/s10555-017-9687-8
4. Dassonville O, Bozec A, Fischel JL, Milano G. EGFR targeting therapies: monoclonal antibodies versus tyrosine kinase inhibitors. Similarities and differences. Crit Rev Oncol Hematol. 2007;62:53–61. doi:10.1016/j.critrevonc.2006.12.008
5. Rawla P, Barsouk A, Hadjinicolaou AV, Barsouk A. Immunotherapies and targeted therapies in the treatment of metastatic colorectal cancer. Med Sci. 2019;7(8):83. doi:10.3390/medsci7080083
6. Castillo L, Etienne-Grimaldi MC, Fischel JL, Formento P, Magné N, Milano G. Pharmacological background of EGFR targeting. Ann Oncol. 2004;15(7):1007–1012. doi:10.1093/annonc/mdh257
7. Dienstmann R, De Dosso S, Felip E, Tabernero J. Drug development to overcome resistance to EGFR inhibitors in lung and colorectal cancer. Mol Oncol. 2011;6(1):15–26. doi:10.1016/j.molonc.2011.11.009
8. O’Leary MC, Lu X, Huang Y, et al. FDA approval summary: tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin Cancer Res. 2019;25(4):1142–1146. doi:10.1158/1078-0432.CCR-18-2035
9. Bouchkouj N, Kasamon YL, de Claro RA, et al. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin Cancer Res. 2019;25(6):1702–1708. doi:10.1158/1078-0432.CCR-18-2743
10. Strohl WR, Naso M. Bispecific T-cell redirection versus Chimeric Antigen Receptor (CAR)-T cells as approaches to kill cancer cells. Antibodies. 2019;8(3):41. doi:10.3390/antib8030041
11. Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. 2019;10:
12. Zhang E, Gu J, Xu H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol Cancer. 2018;17(1):1–12. doi:10.1186/s12943-018-0759-3
13. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management. Blood Rev. 2019;34:45–55. doi:10.1016/J.BLRE.2018.11.002
14. Gauthier J, Turtle CJ. Insights into cytokine release syndrome and neurotoxicity after CD19-specific CAR-T cell therapy. Curr Res Transl Med. 2018;66(2):50–52. doi:10.1016/j.retram.2018.03.003
15. Koristka S, Cartellieri M, Feldmann A, et al. Flexible antigen specific redirection of human regulatory T cells via a novel universal chimeric antigen receptor system. Blood. 2014;124(21):3494. doi:10.1182/blood.V124.21.3494.3494
16. Koristka S, Cartellieri M, Arndt C, et al. Retargeting of regulatory T cells to surface-inducible autoantigen La/SS-B. J Autoimmun. 2013;42:105–116. doi:10.1016/j.jaut.2013.01.002
17. Cartellieri M, Feldmann A, Koristka S, et al. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016;6(8):e458–488. DOI:10.1038/bcj.2016.61
18. Bachmann M. The UniCAR system: a modular CAR T cell approach to improve the safety of CAR T cells. Immunol Lett. 2019;211:13–22. doi:10.1016/j.imlet.2019.05.003
19. Feldmann A, Arndt C, Koristka S, Berndt N, Bergmann R, Bachmann MP. Conventional CARs versus modular CARs. Cancer Immunol Immunother. 2019;68(10):1713–1719. doi:10.1007/s00262-019-02399-5
20. Albert S, Arndt C, Feldmann A, et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology. 2017;6(4):1–17. doi:10.1080/2162402X.2017.1287246
21. Li H, D’Anjou M. Pharmacological significance of glycosylation in therapeutic proteins. Curr Opin Biotechnol. 2009;20(6):678–684. doi:10.1016/j.copbio.2009.10.009
22. Vendel MC, Favis M, Snyder WB, et al. Secretion from bacterial versus mammalian cells yields a recombinant scFv with variable folding properties. Arch Biochem Biophys. 2012;526(2):188–193. doi:10.1016/j.abb.2011.12.018
23. Cohen MH, Chen H, Shord S, et al. Approval summary: cetuximab in combination with cisplatin or carboplatin and 5-fluorouracil for the first-line treatment of patients with recurrent locoregional or metastatic squamous cell head and neck cancer. Oncologist. 2013;18(4):460–466. doi:10.1634/theoncologist.2012-0458
24. Stamova S, Koristka S, Keil J, et al. Cancer immunotherapy by retargeting of immune effector cells via recombinant bispecific antibody constructs. Antibodies. 2012;1(2):172–198. doi:10.3390/antib1020172
25. Jureczek J, Bergmann R, Berndt N, et al. An oligo-His-tag of a targeting module does not influence its biodistribution and the retargeting capabilities of UniCAR T cells. Sci Rep. 2019;9(1):1–15. doi:10.1038/s41598-019-47044-4
26. Feldmann A, Arndt C, Bergmann R, et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget. 2017;8(19):31368–31385. doi:10.18632/oncotarget.15572
27. Akbari B, Farajnia S, Zarghami N, et al. Design, expression and evaluation of a novel humanized single chain antibody against epidermal growth factor receptor (EGFR). Protein Expr Purif. 2016;127:8–15. doi:10.1016/j.pep.2016.06.001
28. Mitwasi N, Feldmann A, Bergmann R, et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget. 2017;8(65):108584–108603. doi:10.18632/oncotarget.21017
29. Feldmann A, Stamova S, Bippes CC, et al. Retargeting of T cells to prostate stem cell antigen expressing tumor cells: comparison of different antibody formats. Prostate. 2011;71(9):998–1011. doi:10.1002/pros.21315
30. Koristka S, Kegler A, Bergmann R, et al. Engrafting human regulatory T cells with a flexible modular chimeric antigen receptor technology. J Autoimmun. 2018;90:116–131. doi:10.1016/j.jaut.2018.02.006
31. Albert S, Arndt C, Koristka S, Berndt N. From mono- to bivalent: improving theranostic properties of target modules for redirection of UniCAR T cells against EGFR- expressing tumor cells in vitro and in vivo. Oncotarget. 2018;9(39):25597–25616.
32. Wong SF. Cetuximab: an epidermal growth factor receptor monoclonal antibody for the treatment of colorectal cancer. Clin Ther. 2005;27(6):684–694. doi:10.1016/j.clinthera.2005.06.003
33. Moreira J, Tobias A, O’Brien MP, Agulnik M. Targeted therapy in head and neck cancer: an update on current clinical developments in epidermal growth factor receptor-targeted therapy and immunotherapies. Drugs. 2017;77(8):843–857. doi:10.1007/s40265-017-0734-0
34. Capdevila J, Elez E, Macarulla T, Ramos FJ, Ruiz-Echarri M, Tabernero J. Anti-epidermal growth factor receptor monoclonal antibodies in cancer treatment. Cancer Treat Rev. 2009;35(4):354–363. doi:10.1016/j.ctrv.2009.02.001
35. Tamada K, Geng D, Sakoda Y, et al. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. 2012;18(23):6436–6445. doi:10.1158/1078-0432.CCR-12-1449
36. Guedan S, Ruella M, June CH. Emerging cellular therapies for cancer. Annu Rev Immunol. 2019;37(1):145–171. doi:10.1146/annurev-immunol-042718-041407
37. Srivastava S, Riddell SR. Chimeric antigen receptor T cell therapy: challenges to bench-to-bedside efficacy. J Immunol. 2018;200(2):459–468. doi:10.4049/jimmunol.1701155
38. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi:10.1038/MT.2010.24
39. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011. doi:10.1038/mto.2016.11
40. Urbanska K, Lynn RC, Stashwick C, Thakur A, Lum LG, Powell DJ. Targeted cancer immunotherapy via combination of designer bispecific antibody and novel gene-engineered T cells. J Transl Med. 2014;12(1):347. doi:10.1186/s12967-014-0347-2
41. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173(6):1426–1438. doi:10.1016/j.cell.2018.03.038
42. Ma JSY, Kim JY, Kazane SA, et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci. 2016;113(4):E450–E458. doi:10.1073/pnas.1524193113
43. Arndt C, Bachmann M, Bergmann R, Berndt N, Feldmann A, Koristka S. Theranostic CAR T cell targeting: a brief review. J Label Compd Radiopharm. 2019;62(8):533–540. doi:10.1002/jlcr.3727
44. Kegler A, Koristka S, Bergmann R, et al. T cells engrafted with a UniCAR 28/z outperform UniCAR BB/z-transduced T cells in the face of regulatory T cell-mediated immunosuppression. Oncoimmunology. 2019;8:9. doi:10.1080/2162402X.2019.1621676
45. Fasslrinner F, Arndt C, Koristka S, et al. Midostaurin abrogates CD33-directed UniCAR and CD33-CD3 bispecific antibody therapy in acute myeloid leukaemia. Br J Haematol. 2019:1–6. doi:10.1111/bjh.15975.
46. Bachmann D, Aliperta R, Bergmann R, et al. Retargeting of UniCAR T cells with an in vivo synthesized target module directed against CD19 positive tumor cells. Oncotarget. 2018;9(7):7487–7500. doi:10.18632/oncotarget.23556
47. Loureiro LR, Feldmann A, Bergmann R, et al. Development of a novel target module redirecting UniCAR T cells to Sialyl Tn-expressing tumor cells. Blood Cancer J. 2018;8(9):4–9. doi:10.1038/s41408-018-0113-4
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.