Back to Journals » The Application of Clinical Genetics » Volume 12
High prevalence of genetic determined familial hypercholesterolemia in premature coronary artery disease
Authors Pirazzi C, Håkansson L, Gustafsson C, Omerovic E ![]() , Wiklund O, Mancina RM
, Wiklund O, Mancina RM ![]()
Received 26 January 2019
Accepted for publication 14 March 2019
Published 24 May 2019 Volume 2019:12 Pages 71—78
DOI https://doi.org/10.2147/TACG.S202942
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Carlo Pirazzi,1 Lina Håkansson,1 Carola Gustafsson,1 Elmir Omerovic,1,2 Olov Wiklund,2 Rosellina Margherita Mancina2
1Department of Cardiology, Sahlgrenska University Hospital, Gothenburg, Sweden; 2Department of Molecular and Clinical Medicine, Institute of Medicine, Sahlgrenska Academy, Wallenberg Laboratory, University of Gothenburg, Gothenburg, Sweden
Background: Premature coronary artery disease (CAD) is a major cause of mortality and morbidity. Increased low-density lipoprotein-cholesterol (LDL-C) level is a major risk factor for CAD and thus the main target for its prevention. Familial Hypercholesterolemia (FH) is a genetic inherited disorder characterized by high LDL-C, and subsequent premature CAD development. Early drug treatment with lipid-lowering medications in FH prevents cardiovascular disease onset.
The FH prevalence in the Northern European general population is 0.3%, and it is estimated that it explains 20% of premature CAD cases in individuals with familial clustering. Despite the wide number of papers showing the prevalence of clinical FH in cardiovascular disease, the prevalence of genetic FH in individuals with premature CAD is not yet well known. Here, we examined the prevalence of genetically determined FH in individuals with premature CAD.
Patients and methods: 66 patients who underwent coronary angiography with suspected premature acute coronary syndrome (age <50 years for men and <55 years for women) underwent genetic screening to identify FH-causing mutations. All patients underwent physical and clinical examinations. Information about family and personal history, drug therapy and habits were also collected.
Results: We found FH-causative mutations in 3/66 (4.5%) screened individuals with premature CAD. When considering individuals with confirmed CAD after coronary angiography, the FH mutation prevalence was 6.1% (3/49). After excluding individuals with classical risk factors for CAD other than hypercholesterolemia, the FH mutation prevalence raised to 15.8% (3/19).
Conclusion: In conclusion, we found that individuals with premature CAD have a more than 15-fold increased prevalence of FH mutations compared to the general population.
Keywords: young, myocardial infarction, CAD, acute coronary event, FH
Introduction
Premature coronary artery disease (CAD) is a major cause of mortality and morbidity. In 2002, a Danish study showed that the incidence of CAD among people between 30 and 54 of age was 1% for men and 0.4% for women per year.1 Increased serum low-density lipoprotein-cholesterol (LDL-C) level is a major risk factor for CAD and thus the main target for its prevention.1 However, an acute exposure to high levels of serum LDL-C is not sufficient to determine the CAD onset. Indeed, prolonged exposure time to elevated levels of LDL-C is the main risk factor for acute coronary syndrome.2
Familial Hypercholesterolemia (FH) is an inherited disorder of lipid metabolism characterized by high levels of LDL-C, and subsequent early CAD development. In northern Europe, the prevalence of FH in the general population is thought to be approximately 1/200.3
The most common causes of FH are mutations in LDL-receptor (LDLR), Apolipoprotein B (APOB) and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes,3–5 but a genetic diagnosis is reached only in 40–70% of cases.6 The frequency of FH-causing mutations at Northern European general population level was estimated to be 0.3% while the frequency of clinical FH is estimated to be 0.5%.3,7 This discrepancy suggests that approximately 40% of individuals with clinical diagnosis of FH do not have causative mutations in the three FH genes. For these individuals, it has been suggested that a polygenic form of the disease, rather than a monogenic, is the cause for the raised LDL-C levels. The polygenic form of FH can be defined based on a weighted LDL-C-raising gene score (SNP score) calculated by the presence of common LDL-C-raising alleles, each affecting LDL-C levels by only a small extent, identified by the Global Lipid Genetics Consortium.8
Early drug treatment with lipid-lowering medications prevents the onset of cardiovascular disease.9,10 Therefore, the early identification of FH patients is crucial to start an effective prevention strategy.
It is estimated that FH explains 20% of premature CAD cases in individuals with familial clustering.11,12 Many studies describe the prevalence of clinical FH in individuals with premature CAD.13–16 The clinical diagnosis is a very useful tool for general population screening. However, it shows limitations in identifying FH patients with a causing mutation. Genetic diagnosis is the gold standard to identify individuals with FH, and it allows the family screening also in individuals with borderline/moderately high serum LDL-C levels. With this study, we examined the prevalence of genetically determined FH in 66 individuals with premature CAD (<50 years of age for men and <55 years of age for women).
Patients and methods
Selection criteria
A total of 218 patients who underwent coronary angiography with suspected premature acute coronary syndrome (age <50 years for men and <55 years for women) were identified at Sahlgrenska University Hospital between 2008 and 2013. To all of them, it was offered to test for FH by DNA analysis and it was explained that identification of an FH-causing mutation in the patient may help early identification and treatment of family members with FH to prevent a myocardial infarction. Although repeated letters and telephone calls, there was a poor compliance to the study. Specifically, 66 patients out of 218 (30%) underwent genetic screening to identify FH causing mutations. Of these, 49 had a confirmed diagnosis of premature CAD after coronary angiography. The study conforms to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the regional ethics committee of Gothenburg (Dnr 486–13). All subjects gave their written informed consent to participate in the study.
Data collection
All patients were given a questionnaire to complete and underwent a physical and clinical examination at the admittance. Specifically, we collected information about family and personal history, drug therapy and habits. Blood samples were collected and serum lipid levels (including total cholesterol, LDL-C, HDL-C and triglycerides) were determined by routine techniques at the admittance, at the Laboratory for Clinical Chemistry, Sahlgrenska University Hospital, Gothenburg. LDL-C levels before treatment were available in 91% individuals (60 out of 66). LDL-C levels for those patients in whom the pretreatment values were not available (n=6) were quantified based on the estimated effect of the different statins.17 The diagnosis of cardiovascular disease was established based on coronary angiography finding. The Dutch Lipid Clinic Network score was calculated at the admittance as previously described.3
Sequencing
DNA was isolated from whole blood and targeted sequenced using SEQPRO LIPO RS (Progenika Biopharma, Derio, Spain,
SNP score calculation for polygenic hypercholesterolemia assessment
For patients negative for FH-causing mutations, the LDL-raising SNP score was calculated using weighted sums for six selected SNPs.8 Genotyping for the 6 SNPs was performed in duplicate by TaqMan 5ʹ-nuclease assays (Life Technologies, Carlsbad, CA) with a concordance between duplicates and a success rate of 100%. All genotypes were all in Hardy-Weinberg equilibrium.
Potential polygenic FH was defined has a SNP score of ≥0.83 representing the top quintile score.
Statistical analysis
Continuous variables are shown as means ± standard deviations and categorical variables as numbers and proportions. p-values were calculated by Mann–Whitney non-parametric test for independent samples for continuous variables, or by Fisher exact test for categorical variables. Statistical analyses were performed using the IBM Statistical Package for Social Sciences (version 20.0; IBM SPSS Inc., Chicago, IL, USA). Two-sided p-values <0.05 were considered statistically significant.
Results
Characteristics of the study cohort
The study cohort included 66 individuals with diagnosis of premature acute coronary event (<50 years of age for men and <55 years of age for women) who underwent a coronary angiography between 2008 and 2013 in one of the three coronary units at Sahlgrenska University Hospital in Gothenburg. Clinical characteristics of the study cohort are shown in Table S1. Briefly, mean age of the study cohort was 43±6 years, BMI 27±5 kg/m2 and untreated LDL-C 3.71±1.45 mmol/L. Twelve (18%) individuals had diabetes, 40 (61%) had hypertension and 11 (17%) were smokers. Forty-one (62%) were finally diagnosed with myocardial infarction (STEMI or NSTEMI), 8 (12%) with unstable angina, stable angina or atherosclerosis, 9 (14%) with spasm angina and 8 (12%) with other diagnosis (non-atherosclerotic) including 2 systemic lupus erythematosus (SLE) vasculitis, 2 patent foramen ovale (PFO) with paradoxical embolizations, 1 anatomical variant, 1 Ehlers-Danlos syndrome, 1 drug-induced cardiotoxicity and 1 troponin T (TNT) elevation without clear cause. According to DLCN criteria, 9 (15%) individuals in the overall cohort had at least a probable FH diagnosis (Table S1). Of these, 2 were confirmed by genetic screening.
Prevalence of FH in patients with premature acute coronary syndrome
We found a causative mutation in 3 individuals out of 66 screened (prevalence 4.5%). Compared to an estimated frequency of genetic FH of 0.3% in the general Northern European population, this would account for a 15-fold enrichment.7 When considering individuals with a confirmed CAD after coronary angiography (myocardial infarction, unstable and stable angina and atherosclerosis), the FH mutation prevalence was 6.1% (3 out of 49). After excluding individuals with classical risk factors for CAD other than hypercholesterolemia (namely diabetes, hypertension and smoking), the FH mutation prevalence raised to 15.8% (Figure 1).
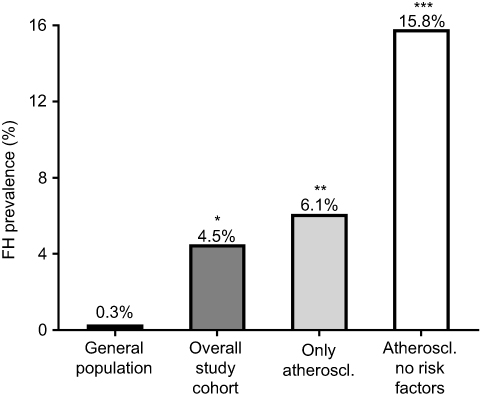 | Figure 1 Prevalence of genetically determined familial hypercholesterolemia (FH). The expected prevalence of FH in the general population is 0.3%.7 In our study cohort of premature cardiovascular disease, FH-causative mutations have been detected in 4.5% of the individuals accounting for a 15-fold enrichment compared to general population. When considering only individuals with a confirmed atherosclerotic disease, the prevalence increased to 6.1%. When individuals with other risk factors for cardiovascular disease (namely diabetes, smoking and hypertension) were further excluded from the analysis, the prevalence of genetically determined FH was 15.8% accounting for approximately a 53-fold enrichment compared to the general population. P-value calculated by Fisher exact test and referred to the comparison with the general population. * p=0.015, **p=0.007, ***p=0.0005. |
The genetic analysis in the 3 FH individuals revealed 2 mutations in the LDLR gene (1 pathogenic stop codon and 1 splicing site classified as variant of uncertain significance) and 1 mutation in the PCSK9 gene (pathogenic in-frame insertion resulting in a Leucine duplication and described as possible gain-of-function variant) (Table 1).18,19
 | Table 1 Identified FH-causing mutations in the study cohort |
Clinical characteristics of the study cohort stratified based on genetic diagnosis of FH are shown in Table 2. Briefly, mean age was 34±16 years for FH individuals and 43±6 years for non-FH. There were no differences between FH and non-FH in blood sugar or blood pressure. None of the FH individuals was smoker, whilst 17% of the non-FH group had smoking habit. Untreated LDL-cholesterol levels in individuals with FH mutation were 2-fold higher than in those without mutation (7.17±2.67 vs 3.55±1.17 mmol/L, respectively, P<0.005; Figure 2). When considering only individuals with angiography confirmed CAD, the LDL-C level difference between FH and non-FH individuals remained significant, with no differences in blood pressure and blood sugar levels between the groups. The mean LDL cholesterol level under ongoing statin treatment was 3.7 mmol/L for the FH group, accounting for a 45% reduction. Individuals with CAD and no FH mutation experienced an LDL-cholesterol level reduction of 0.8 mmol/L (from 3.6 to 2.8 mmol/L) under ongoing statin treatment.
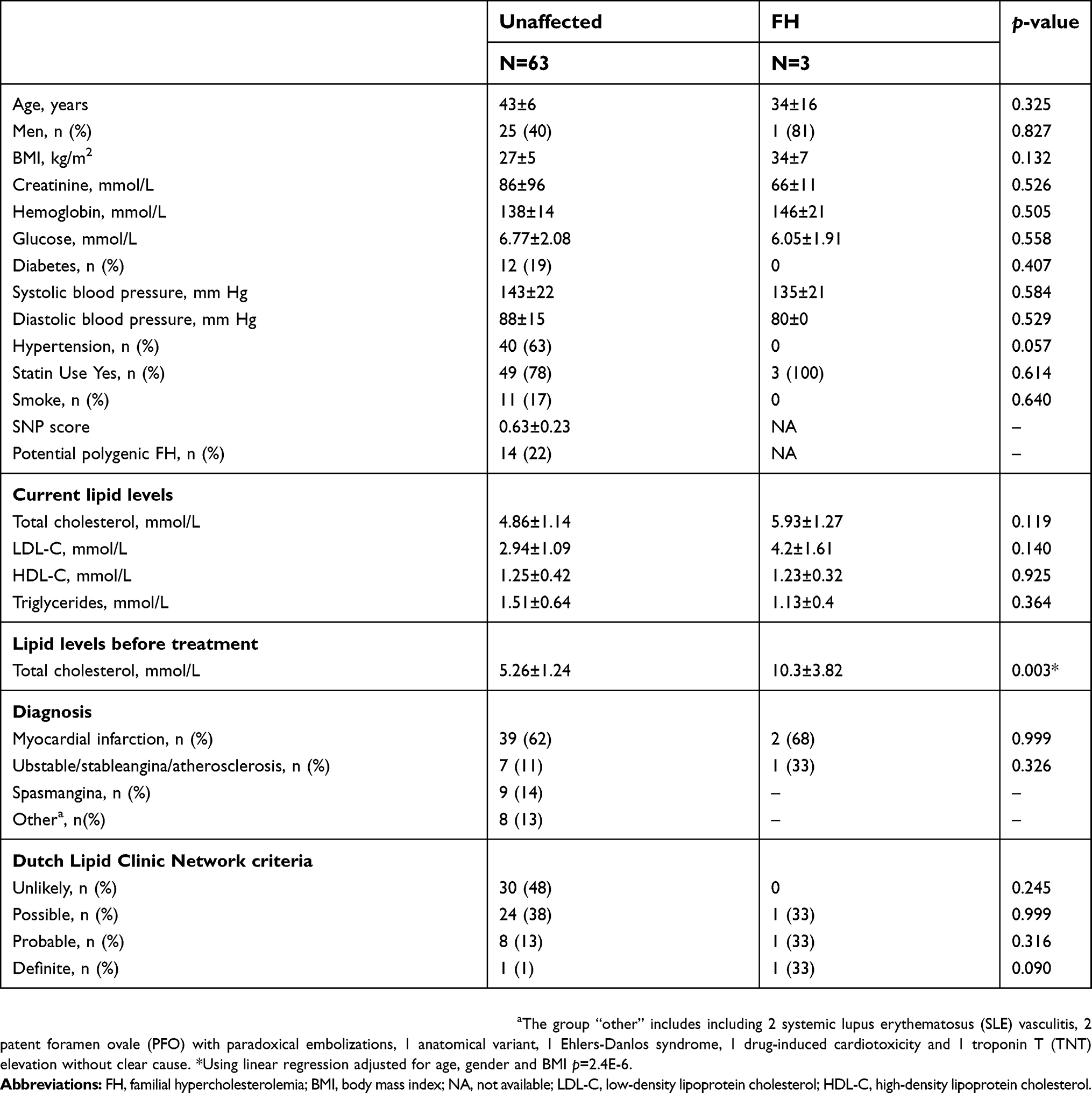 | Table 2 Clinical and biochemical characteristics of the study cohort stratified based on genetic diagnosis of FH |
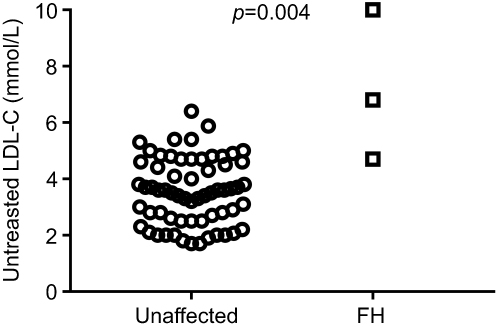 | Figure 2 Untreated LDL-cholesterol levels in individuals without or with FH mutation. Patient with genetically determined FH had higher LDL-C before statin treatment. p-value calculated by Mann–Whitney non-parametric test for independent samples. Using linear regression adjusted for age, gender and BMI p=0.004. |
Two of the 3 individuals with genetic FH diagnosis underwent PCI by stent delivery, one for STEMI and one for NSTEMI. These 2 individuals (both carriers of mutation on the LDLR) were 16 and 42 years old, respectively, with a family history of myocardial infarction. The third one had no new stenosis to be treated at the coronary angiography performed at inclusion, but had previously undergone PCI in 2003. This individual (carrier of the mutation on the PCSK9) was 42 years old and did not have family history of myocardial infarction.
All FH-negative individuals underwent genotyping for 6 LDL-raising SNPs and the weighted SNP score for polygenic hypercholesterolemia was calculated. The mean SNP score was 0.63±0.22. There was a tendency to positive correlation between SNP score and LDL-C, however not significant (Spearman r=0.173, p=0.180). Potential polygenic FH was defined as having a SNP score of ≥0.83 (corresponding to the top quintile score). Based on this definition, 14 patients out of 63 (22.3%) were considered as potential polygenic FH (Table 2).
Discussion and conclusions
In the present study, we describe the prevalence of FH mutations in 66 young individuals with diagnosis of acute coronary event who underwent coronary angiography. We show that the prevalence of FH mutation in this cohort is 4.5%. This number is approximately 15 times higher than the expected mutation rate in the general population. We also show that, when considering only individuals with a confirmed atherosclerotic disease, the prevalence rises to 6.1%, and it further increases to 15.8% when excluding those with other cardiovascular risk factors. These data are consistent with a recently published work that showed an enrichment of FH mutation in individuals with acute coronary syndrome, LDL-C>4.14 mmol/L at admission and under age 65.20 Compared to this study, our cohort is smaller, younger and was not selected based on any LDL-C threshold or ACS diagnosis. Our results are also consistent with the enrichment of mutations on LDLR described in individuals with premature myocardial infarction from the National Heart, Lung and Blood Institute’s Exome Sequencing Project from the USA.21 In this paper, authors performed whole exome next-generation sequencing on DNA from individuals with and without premature MI. They found that carriers for FH mutations on LDLR had a prevalence of ~2% in their MI cohort. They also have shown that carriers of rare non-synonymous mutations on LDLR had a 4.2-fold increased risk for MI while carriers of null alleles had a more than 13-fold increased risk for MI.
Previously, a study performed on young individuals with myocardial infarction or stable angina pectoris from the Finnish region of North Karelia had shown a 9% prevalence of 2 FH mutations very common in that area due to founder effect.22 The authors tested only for the 2 specific mutations by PCR assay. At the time the study was conducted, criteria for myocardial infarction diagnosis were slightly different from now due to unavailability of high sensitive troponin test.
More recently, in a larger (n=231) cohort of young myocardial infarction individuals from UK, an FH prevalence of 1.3% was described after testing for 48 common mutations in the LDLR gene and NGS of LDLR only in those with LDL-C >7 (mmol/L).23 The lower FH prevalence in this cohort might be explained by the fact that NGS sequencing was not performed on all the individuals and mutations in PCSK9 and APOB genes were not tested either.
Furthermore, in a recent series of 255 German subjects with myocardial infarction before 60 years of age, a prevalence of FH mutations of 5% was found by NGS of LDLR, APOB, PCSK9 and STAP1 genes.24
We hypothesize that the higher prevalence of FH mutation we find in CAD subjects in our cohort compared to the previous ones is due to our slightly different approach of selection, where we combine younger patients with a broad NGS sequencing of the 3 most important FH genes.
Despite the methodological differences between the aforementioned studies and our current work, all the findings are consistent with the hypothesis that FH prevalence in young individuals with CAD is much higher than in general population. This aspect has a two-fold importance. First, it identifies a population which would benefit from a broader genetic screening strategy. Second, it would allow implementing a very effective cascade screening strategy to strongly reduce cardiovascular risk in a high-risk population.
In the present study, we identified 2 mutations described as pathogenic (one stop gain in the LDLR gene and one in-frame insertion in the PCSK9 gene),19,25 and 1 splicing site in the LDLR gene classified as variant of uncertain significance (Table 1). The LDLR mutation of uncertain significance has been previously described to segregate with the phenotype in cascade screening, suggesting a pathogenic role.6 More specifically, the variant has been detected in 2/3 tested relatives of our proband: the 48-year-old father had LDL-C of 7.0 mmol/L; the 17-year-old brother, despite his young age, had LDL-C of 5.1 mmol/L.6 This finding strongly supports the pathogenicity of this variant.
Analyses of the clinical characteristics of the study cohort stratified by genetic FH diagnosis showed that those with FH mutation had as expected higher LDL-C levels both before and under statin treatment compared to those without. Treatment induced a 45% reduction in LDL-C for the FH group, while the non-FH group had only a 23% reduction. It is well established that FH subjects are usually treated more intensively with lipid-lowering medication. Interestingly, neither of the two groups was on target for secondary prevention. We did not find any other differences in risk factors among the two groups, most likely because of the small sample size. However, a tendency to lower blood sugar, triglycerides, diabetes prevalence, smoking and hypertension was present in those with FH.
Comparing the genetic test results with clinically determined FH diagnosis by DLCN criteria, we observed that 2 out of 3 genetically confirmed FH were classified as at least probable FH, while the third one as possible. On the other hand, one subject with definitive clinical diagnosis by DLCN had no genetic mutation. Our interpretation of these results is that the DLCN criteria is a very useful tool for general population screening, but a more effective strategy for genetic screening needs to be implemented to overcome the limits of clinical diagnosis. At the same time, further effort should be put into discovering new FH causative genes to reduce possible false negatives. The genetic diagnosis reduces the number of false negatives, allows screening of relatives and allows the identification of at-risk people early in life and before symptoms development. The early identification and treatment of FH individuals decreases the risk of CVD and results in life expectancy and quality of life for these at very-high-risk individuals compared to the general population. Furthermore, the genetic diagnosis gives a clear cut LDL-C target for treatment (2.5 mmol/L according to the ESC guidelines). These are major advantages of using FH genetic and why we support the utilization of the genetic screening together with the measurement of high LDL-C in clinical practice.
Using the LDL-raising SNP score, we identified 22% of the non-FH subjects as potential polygenic hypercholesterolemia. To our knowledge, there is not a SNP score cut-off that can be used to make a formal diagnosis of polygenic FH. In the present study, we used the SNP score top quintile as cut-off to identify potential polygenic FH subjects which does not correspond to a formal diagnosis. More studies need to be done to further characterize this subgroup of high-risk subjects and define which targets need to be achieved to optimize their treatment.
Finally, the analysis of diagnosis after coronary angiography showed a quite high prevalence of non-atherosclerotic diseases leading to the procedure. A high rate of spasm angina was detected, which is consistent with the young age and the high rate of female gender of our casuistic. Furthermore, we found 2 subjects with embolism from PFO and 2 with coronary artery engagement of SLE. Again, the high prevalence of non-atherosclerotic disease is in line with the young age of the cohort.
Major weaknesses of the current study are: the low number of participants and of carriers of FH mutations, and the absence of a control group which does not allow to make estimation of the risk in terms of Odds Ratio; the low attendance (30%), and the fact that an enrichment of FH cases in the studied group cannot be excluded. The attenders and non-attenders were of similar age and BMI. They had a similar lipid profile and similar diabetes prevalence. In a “worst case” scenario, the three FH patients were the only ones in the original group of 218. In that case, the FH frequency would be 1.4%, which still is 5 times higher than the expected mutation rate in the general population. This frequency would be even higher if only patients with atherosclerotic disease or subjects without other risk factors are analyzed.
In conclusion, we found a high prevalence of FH mutations in young subjects who underwent coronary angiography. Our results support the importance to implement an FH genetic screening program for patients at high risk for CAD and for their families, even independently of untreated LDL-C values. We also found that our cohort was generally undertreated in terms of LDL-C lowering targets.
Acknowledgments
This work was supported by the Wilhelm and Martina Lundgren Science Fund (RMM), and by project grant from Amgen and Sanofi-Aventis.
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
OW has received honoraria for educational lectures from AMGEN and Sanofi. The authors report no other conflicts of interest in this work.
References
1. Schnohr P, Jensen JS, Scharling H, Nordestgaard BG. Coronary heart disease risk factors ranked by importance for the individual and community. A 21 year follow-up of 12,000 men and women from The Copenhagen City Heart Study. Eur Heart J. 2002;23(8):620–626. doi:10.1053/euhj.2001.2842
2. Pletcher MJ, Bibbins-Domingo K, Liu K, et al. Nonoptimal lipids commonly present in young adults and coronary calcium later in life: the CARDIA (Coronary Artery Risk Development in Young Adults) study. Ann Intern Med. 2010;153(3):137–146. doi:10.7326/0003-4819-153-1-201007060-00004
3. Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478–3490. doi:10.1093/eurheartj/eht273
4. De Castro-Orós I, Pocoví M, Civeira F. The genetic basis of familial hypercholesterolemia: inheritance, linkage, and mutations. Appl Clin Genet. 2010;3:53–64.
5. Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. 2004;160(5):407–420. doi:10.1093/aje/kwh212
6. Maglio C, Mancina RM, Motta BM, et al. Genetic diagnosis of familial hypercholesterolaemia by targeted next-generation sequencing. J Intern Med. 2014;276(4):396–403. doi:10.1111/joim.12263
7. Wald DS, Bestwick JP. Preventive percutaneous coronary intervention and aspiration thrombectomy-updates in the management of ST-elevation myocardial infarction. J Thorac Dis. 2016;8(8):1908–1912. doi:10.21037/jtd.2016.07.73
8. Futema M, Shah S, Cooper JA, et al. Refinement of variant selection for the LDL cholesterol genetic risk score in the diagnosis of the polygenic form of clinical familial hypercholesterolemia and replication in samples from 6 countries. Clin Chem. 2015;61(1):231–238. doi:10.1373/clinchem.2015.238717
9. Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008;337:a2423. doi:10.1136/bmj.a2423
10. Marks D, Thorogood M, Neil HA, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis. 2003;168(1):1–14.
11. Goldstein JL, Schrott HG, Hazzard WR, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest. 1973;52(7):1544–1568. doi:10.1172/JCI107295
12. Neefjes LA, Ten Kate GJ, Alexia R, et al. Accelerated subclinical coronary atherosclerosis in patients with familial hypercholesterolemia. Atherosclerosis. 2011;219(2):721–727. doi:10.1016/j.atherosclerosis.2011.09.052
13. Li S, Zhang Y, Zhu CG, et al. Identification of familial hypercholesterolemia in patients with myocardial infarction: a Chinese cohort study. J Clin Lipidol. 2016;10(6):1344–1352. doi:10.1016/j.jacl.2016.05.006
14. Faggiano P, Pirillo A, Griffo R, et al. Prevalence and management of familial hypercholesterolemia in patients with coronary artery disease: the heredity survey. Int J Cardiol. 2018;252:193–198. doi:10.1016/j.ijcard.2017.10.105
15. Ohmura H, Fukushima Y, Mizuno A, et al. Estimated prevalence of heterozygous familial hypercholesterolemia in patients with acute coronary syndrome. Int Heart J. 2017;58(1):88–94. doi:10.1536/ihj.16-188
16. Rerup SA, Bang LE, Mogensen UM, et al. The prevalence and prognostic importance of possible familial hypercholesterolemia in patients with myocardial infarction. Am Heart J. 2016;181:35–42. doi:10.1016/j.ahj.2016.08.001
17. Law MR, Wald NJ, Rudnicka AR. Quantifying effect of statins on low density lipoprotein cholesterol, ischaemic heart disease, and stroke: systematic review and meta-analysis. BMJ. 2003;326(7404):1423. doi:10.1136/bmj.326.7402.1329-b
18. Dron JS, Hegele RA. Complexity of mechanisms among human proprotein convertase subtilisin-kexin type 9 variants. Curr Opin Lipidol. 2017;28(2):161–169. doi:10.1097/MOL.0000000000000386
19. Abifadel M, Bernier L, Dubuc G, et al. A PCSK9 variant and familial combined hyperlipidaemia. J Med Genet. 2008;45(12):780–786. doi:10.1136/jmg.2008.059980
20. Amor-Salamanca A, Castillo S, Gonzalez-Vioque E, et al. Genetically confirmed familial hypercholesterolemia in patients with acute coronary syndrome. J Am Coll Cardiol. 2017;70(14):1732–1740. doi:10.1016/j.jacc.2017.04.053
21. Do R, Stitziel NO, Won HH, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518(7537):102–106. doi:10.1038/nature13917
22. Koivisto UM, Hämäläinen L, Taskinen MR, Kettunen K, Kontula K. Prevalence of familial hypercholesterolemia among young north Karelian patients with coronary heart disease: a study based on diagnosis by polymerase chain reaction. J Lipid Res. 1993;34(2):269–277.
23. Wald DS, Bangash FA, Bestwick JP. Prevalence of DNA-confirmed familial hypercholesterolaemia in young patients with myocardial infarction. Eur J Intern Med. 2015;26(2):127–130. doi:10.1016/j.ejim.2015.01.014
24. Brænne I, Kleinecke M, Reiz B, et al. Systematic analysis of variants related to familial hypercholesterolemia in families with premature myocardial infarction. Eur J Hum Genet. 2016;24(2):191–197. doi:10.1038/ejhg.2015.100
25. Holla Ø, Kulseth MA, Berge KE, Leren TP, Ranheim T. Nonsense-mediated decay of human LDL receptor mRNA. Scand J Clin Lab Invest. 2009;69(3):409–417. doi:10.1080/00365510802707163
Supplementary material
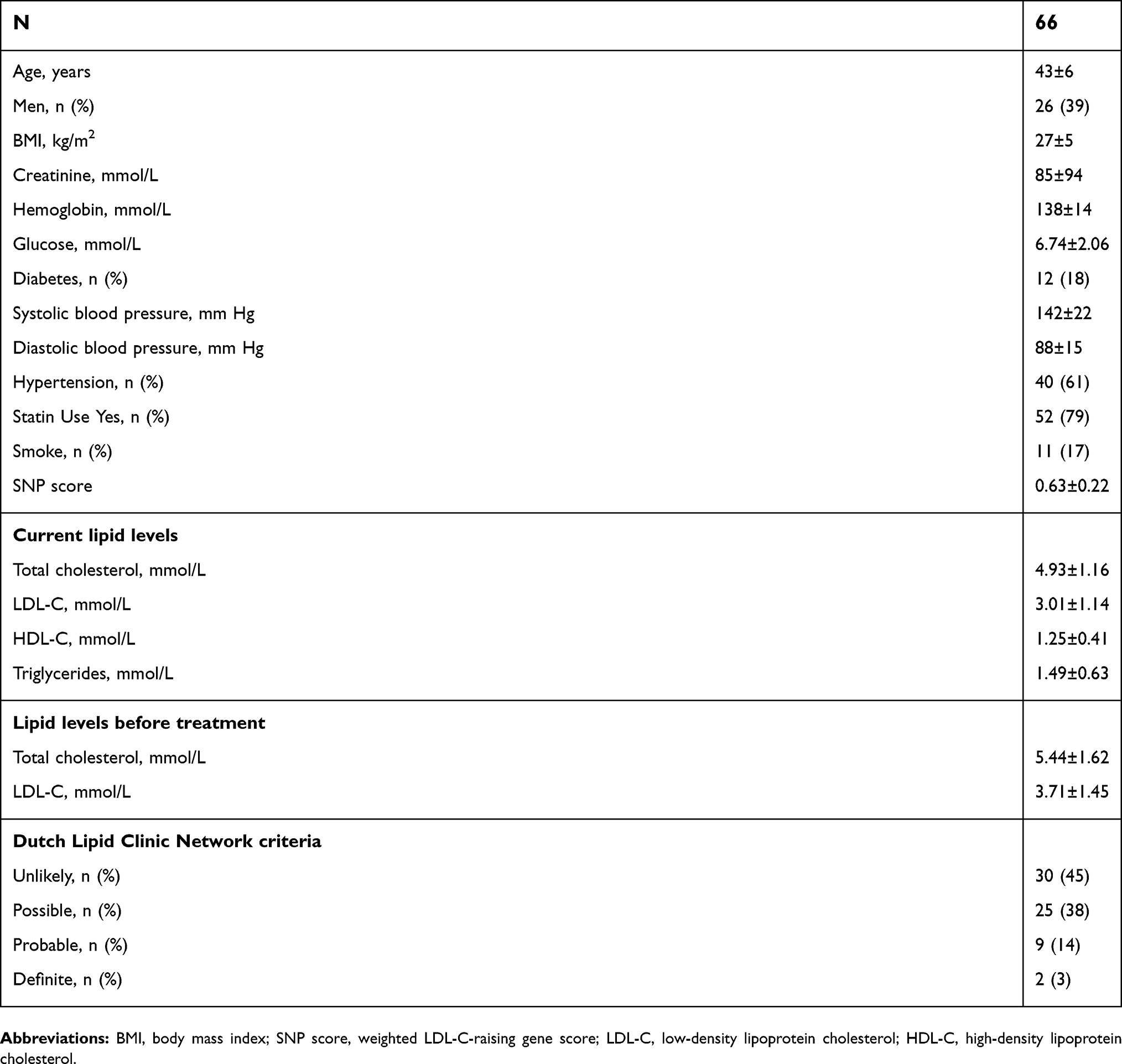 | Table S1 Clinical and biochemical characteristics of the study cohort |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.