")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 9
High glucose levels promote the proliferation of breast cancer cells through GTPases
Authors Hou Y, Zhou M, Xie J, Chao P, Feng Q, Wu J
Received 26 February 2017
Accepted for publication 9 May 2017
Published 13 June 2017 Volume 2017:9 Pages 429—436
DOI https://doi.org/10.2147/BCTT.S135665
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Yilin Hou,1 Man Zhou,1 Jing Xie,1 Ping Chao,1 Qiyu Feng,2 Jun Wu1
1Department of Endocrinology, The Third Hospital of Wuhan (Tongren Hospital of Wuhan University), Wuhan, People’s Republic of China; 2Eastern Hepatobiliary Surgery Institute, National Center for Liver Cancer, Shanghai, People’s Republic of China
Abstract: Hyperglycemia or diabetes mellitus (DM), which is characterized by high blood glucose levels, has been linked to an increased risk of cancer for years. However, the underlying molecular mechanisms of the pathophysiological link are not yet fully understood. In this study, we demonstrate that high glucose levels promote the proliferation of breast cancer cells by stimulating epidermal growth factor receptor (EGFR) activation and the Rho family GTPase Rac1 and Cdc42 mediate the corresponding signaling induced by high glucose levels. We further show that Cdc42 promotes EGFR phosphorylation by blocking EGFR degradation, which may be mediated by the Cbl proteins, whereas the Rac1-mediated EGFR phosphorylation is independent of EGFR degradation. Our findings elucidate a part of the underlying molecular mechanism of the link between high glucose levels and tumorigenesis in breast cancer and may provide new insights on the therapeutic strategy for cancer patients with diabetes or hyperglycemia.
Keyword: hyperglycemia, breast cancer, GTPases, Rac1, Cdc42, EGFR
Introduction
It has been known for years that diabetes increases the risk of developing many types of cancers and mortality of cancer patients.1–5 Recently, this area gains more attention due to both the rising global diabetes epidemic and current findings, which suggest that some antidiabetic agents appear to lower cancer risk and have potential roles in cancer therapy.5–7 Diabetes can be divided into two major subtypes, types 1 and 2. Type 2 diabetes is linked with increased risk of breast, colon and pancreatic cancer.3–5 Although the detailed pathophysiological link of diabetes and cancer has not yet been elucidated, studies to date have demonstrated that hyperglycemia is one of the key factors in the mechanisms postulated for the increased cancer risk in diabetes.8–10 In fact, it has been reported that high glucose levels promote both the proliferation and migration of cancer cells.11–14
Epidermal growth factor receptor (EGFR) activates mitogenic signaling pathways, which are critical in cell proliferation. Overexpression or hyperactivity of EGFR is a major characteristic of many human malignancies including breast cancer. Thus, EGFR is normally considered as a major oncogenic factor and an attractive therapeutic target.15–18 Due to the critical role in various cellular functions, EGF receptor signaling is tightly regulated by the careful balancing between receptor activation and signal downregulation, with the latter including receptor endocytosis, degradation and recycling. Following the binding of ligands such as EGF, the receptor dimerizes, autophosphorylates and then initiates the activation of various signaling pathways leading to cell proliferation. However, EGFR degradation is a means of signal downregulation and critical for the homeostasis of EGF/EGFR signaling pathways.15,16 Ubiquitination targets EGFR for degradation and the Cbl proteins catalyze this reaction.19,20 Moreover, the Rho family GTPase Cdc42, which serves as a molecular switch in cells,21,22 negatively regulates this Cbl-catalyzed EGFR ubiquitination.6,23–25 Then, the aberrant status of these proteins may disrupt the balance between EGFR recycling to the cell surface and degradation, resulting in receptor accumulation, excessive mitogenic signaling and eventually over-promoting cell proliferation.
In this report, we demonstrate that high glucose promotes the proliferation of breast cancer cells such as MDAMB231, SKBR3 and MCF-7 cells and EGFR activation mediates this high glucose-induced cell growth. Our data further indicate that high glucose levels stimulate EGFR activation through GTPase Rac1 and Cdc42. Cdc42 promotes EGFR phosphorylation by blocking EGFR degradation mediated by the Cbl proteins, whereas the Rac1-mediated EGFR phosphorylation is independent of EGFR degradation. The combination of these regulatory effects leads to the overgrowth of breast cancer cells under high glucose conditions.
Methods
Materials and reagents
Compounds AG1478 and PD153035 were purchased from Calbiochem (San Diego, CA, USA). Anti-EGFR, anti-phospho-EGFR, anti-c-Cbl and anti-phospho-Cbl antibodies were obtained from Millipore (Billerica, MA, USA). Anti-Cdc42 and anti-Rac1 antibodies were purchased from Abcam (Cambridge, UK). Anti-ubiquitin antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Constructs and cell culture
The small interfering RNAs (siRNAs) targeting Cdc42 or Rac1, and the control siRNA, were from Invitrogen (Carlsbad, CA, USA) and introduced into cells using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). HA-tagged Cdc42(F28L), Cdc42(Q61L) and Rac1(G12V) constructs are kindly gifts from Dr. Qiyu Feng, Cornell University. They were introduced into cells using Lipofectamine 2000.
Human breast cancer cell lines MDAMB231, SKRB3 and MCF-7 were purchased from American Type Culture Collection (ATCC) and maintained in DMEM at different glucose levels at 37°C in a humidified atmosphere containing 5% CO2. These cell lines were used between passage 8 and 12 to ensure cell line authentication.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis
The cell culture samples were washed with PBS and lysed in radioimmunoprecipitation assay (RIPA) buffer. A total of 20 μg protein was resolved on SDS-PAGE and transferred to polyvinylidene difluoride membranes (PerkinElmer Life Science, Waltham, MA, USA). After being blocked in 5% bovine serum albumin in tris-buffered saline containing 0.2%. Tween for half an hour, the membranes were incubated with a primary antibody overnight at 4°C, followed by incubation with a horseradish peroxidase-conjugated secondary antibody. The membranes were visualized using the enhanced chemiluminescence (ECL) system (Amersham Biosciences, Piscataway, NJ, USA). The Western blot analysis was quantified by ImageJ (NIH image) and the statistical analysis was performed using GraphPad Prism.
Cell growth assay
Cells were seeded (1104 cells per well) and cultured in 12-well plates under conditions required by experimental design (e.g., with or without serum, high glucose or low glucose). At the indicated time points, the cultured cells (three wells per time point) were washed with PBS, trypsinized, suspended and counted using a hemocytometer. Each cell growth assay was repeated independently at least three times. Data are presented as mean ± SD (error bars). Statistical analysis was performed using GraphPad Prism.
Cell-cycle progression assay
To determine BrdU incorporation, MDAMB231 cells were seeded in sterile chamber-well slides. AG1478 or PD153035 (0.25 μM) was added (24 hours after plating the cells) to the medium along with BrdU (50 μM). After 16 hours, immunofluorescence staining was performed using anti-BrdU monoclonal antibody and Hoechst.26 For each experiment, the percentage of BrdU positive cells was determined for >500 cells from multiple visual fields.
Cdc42/Rac1 activity assay
The activation of Cdc42 and Rac1 was conducted using a pull-down assay kit according to manufacturer instructions (NewEast Biosciences, Malvern, PA, USA).
Statistical analysis
All statistical analyses were performed using GraphPad Prism. Data are presented as mean ± SD (error bars) from at least three replicates. Student’s t-test was used to compare two groups. P values ≤0.05 is considered to have significance.
Results
High glucose promotes the proliferation of breast cancer cells
In order to determine the effect of glucose levels on the proliferation of breast cancer cells, we examined the growth rates of MDAMB231, SKBR3 and MCF-7 cells at two different glucose concentrations: the glucose level in high glucose medium (25 mM glucose, HG) is similar to the level of glucose commonly used in current cancer cell culture and the glucose level in the low glucose medium (5 mM glucose, LG) is close to normal physiological conditions of 4–8 mM.7–9 We found that the high glucose level (25 mM glucose) promotes the proliferation of MDAMB231, SKBR3 and MCF-7 cells in these cell growth assays (Figure 1A–C). This is consistent with previous reports about MCF-7 cell or other cancer cell lines.13,14,27
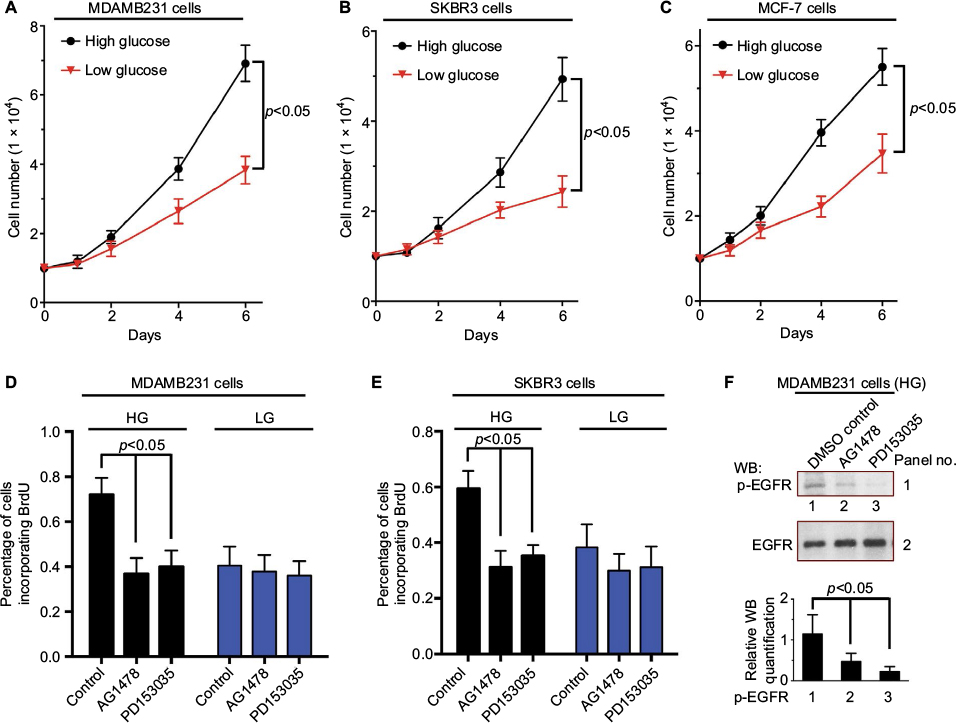 | Figure 1 High glucose promotes the proliferation of breast cancer cells. Notes: MDAMB231 (A), SKBR3 (B) or MCF-7 (C) cells (1×104) were seeded in 12-well plates and cultured in DMEM supplemented with 2% FBS under low glucose (5 mM) or high glucose (25 mM) condition. The number of cells was counted at the indicated times. (D, E) BrdU incorporation assays were performed on MDAMB231 (D) or SKBR3 (E) cells treated with either DMSO (control), AG1478 (0.25 μM) or PD153035 (0.25 μM) under low glucose (5 mM) or high glucose (25 mM) condition. The percentage of cells incorporating BrdU after 12 hours was shown. (F) MDAMB231 cells treated with either DMSO (control), AG1478 (0.25 μM) or PD153035 (0.25 μM) under high glucose (25 mM) condition (same as the treatment in D). The effects of AG1478 and PD153035 on EGFR phosphorylation were shown. Abbreviations: DMSO, dimethyl sulfoxide; EGFR, epidermal growth factor receptor; FBS, fetal bovine serum; HG, high glucose; LG, low glucose; WB, western blot. |
The proliferation of MDAMB231 cells and SKBR3 cells was further investigated by BrdU incorporation assay. As shown in Figure 1D and E, the high glucose level (25 mM glucose) stimulated DNA synthesis (BrdU incorporation) in both MDAMB231 and SKBR3 cells. This result confirms that the elevated growth rate in these cell growth assays (Figure 1A–C) is the result of promoting cell proliferation rather than inhibiting the apoptosis of breast cancer cells. The addition of AG1478 or PD153035, which are specific inhibitors of EGFR tyrosine kinase activity,28 inhibited the EGFR phosphorylation and DNA synthesis stimulated by high glucose (25 mM glucose) (Figure 1D–F). These results indicate that EGF/EGFR signaling may mediate this high glucose-induced breast cancer cell proliferation.
High glucose induces EGFR activation in MDAMB231 cells
High glucose is among the various factors that are able to directly or indirectly activate EGFRs.13,29 As shown in Figure 2, MDAMB231 cells were continually cultured in low glucose (5 mM glucose) medium or exposed to high glucose (25 mM glucose) for 6 or 24 hours together with or without serum. Then, we assessed EGFR activation by the level of EGFR phosphorylation. Under serum starvation condition, high glucose stimulated EGFR phosphorylation (Figure 2, panel 1, lane 2, compared to lane 1). The presence of serum also increased the level of EGFR phosphorylation (Figure 2, panel 1, lane 5, compared to lane 1). Together with serum, high glucose further stimulated EGFR phosphorylation (Figure 2, panel 1, lanes 3 and 4, compared to lanes 5 and 6). Then, we examined the ubiquitination state of EGFR, which is critical for receptor degradation. Serum stimulation, rather than high glucose (25 mM glucose), caused an increase in EGFR ubiquitination (Figure 2, panel 3, lanes 5 and 6, compared to lanes 1 and 2). In fact, high glucose inhibited the serum-induced EGFR ubiquitination (Figure 2, panel 3, lanes 3 and 4, compared to lanes 5 and 6).
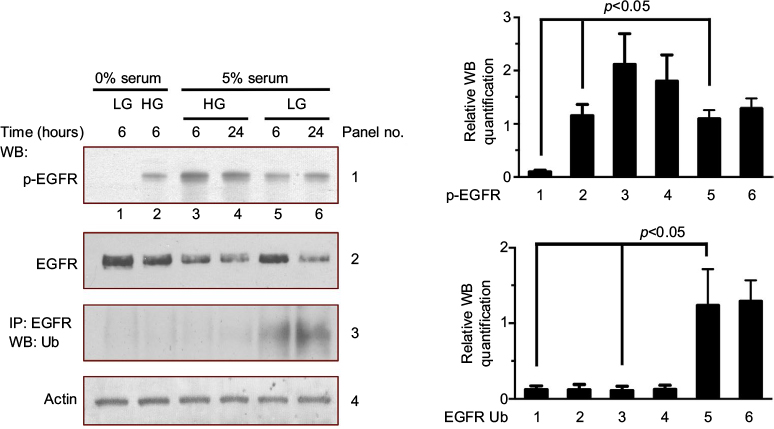 | Figure 2 High glucose induces EGFR activation in MDAMB231 cells. Notes: Serum-deprived MDAMB231 cells were cultured in low glucose condition (5 mM) for 12 hours and then were kept in this condition (control, lane 1) or exposed to high glucose (25 mM) with (5% FBS) or without serum. After 6 or 24 hours, these cells were harvested for the measurement of EGFR phosphorylation, EGFR levels and the ubiquitination of EGFR. The Western blot analysis of EGFR phosphorylation and EGFR ubiquitination were quantified and presented by the plots at the bottom. Abbreviations: EGFR, epidermal growth factor receptor; FBS, fetal bovine serum; HG, high glucose; LG, low glucose; WB, western blot. |
The data in Figure 2 suggest that both high glucose and serum can stimulate EGFR phosphorylation (Figure 2, panel 1, lane 2 vs lane 1 and lane 5 vs lane 1). Serum stimulation enables ubiquitination and degradation of EGFR. High glucose stimulates EGFR phosphorylation and blocks EGFR degradation.
High glucose influences c-cbl phosphorylation and GTPase Rac1 activity
EGFR degradation is catalyzed via a cascade of ubiquitination enzymes, especially, the E3 ubiquitin ligase activity of the Cbl adaptor proteins.19,20 We examined the c-cbl phosphorylation levels under different conditions. Both high glucose (25 mM glucose) and serum elevated c-cbl phosphorylation (Figure 3A, panel 1, lanes 2 and 3, compared to lane 1). However, when high glucose was combined with serum, high glucose inhibited the serum-induced c-cbl phosphorylation (Figure 3A, panel 1, lane 4, compared to lanes 2 and 3).
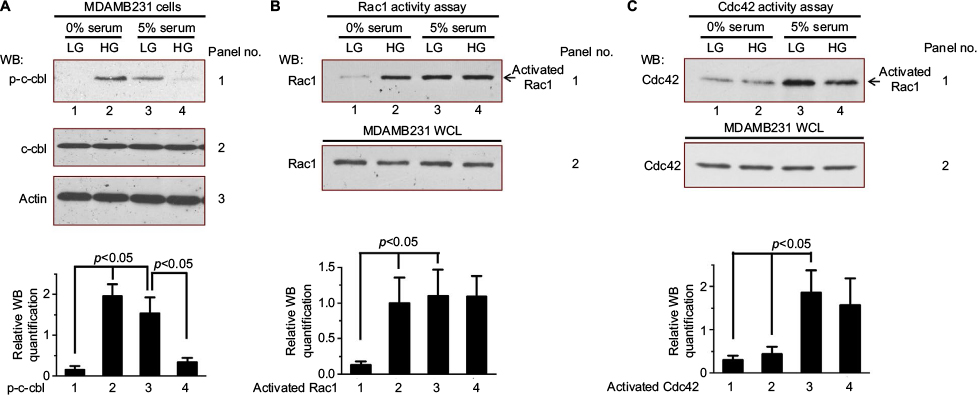 | Figure 3 High glucose influences c-cbl phosphorylation and GTPase Rac1 activation. Notes: (A) Serum-deprived MDAMB231 cells were cultured in low glucose condition (5 mM) for 12 hours and then were kept in this condition (control, lane 1) or exposed to high glucose (25 mM) with (5% FBS) or without serum. After 6 hours, these cells were harvested for measurement of c-cbl phosphorylation and total c-cbl levels. The Western blot analysis of c-cbl phosphorylation was quantified and presented by the plot at the bottom. (B, C) Serum-deprived MDAMB231 cells were treated same as (A). Then, these cells were harvested and the activities of Rac1 (B) and Cdc42 (C) were measured by pull-down activity assay. The Western blot analysis of activated Rac1 (B) and activated Cdc42 (C) were quantified and presented by the plots at the bottom. Abbreviations: FBS, fetal bovine serum; HG, high glucose; LG, low glucose; WB, western blot; WCL, whole cell lysates. |
Rho family GTPases, such as Rac1 and Cdc42, are molecular switches in mitogenic signaling cascades.21,22 It has been reported that Cdc42 regulates EGFR degradation through Cbl.6,23–25 Therefore, we examined whether high glucose (25 mM glucose) activates Rac1 and Cdc42. As shown in Figure 3B, both high glucose and serum stimulation increased the level of activated Rac1 (Figure 3B, panel 1, lane 2–4, compared to lane 1). Serum stimulation activated Cdc42 (Figure 3C, panel 1, lanes 3 and 4, compared to lane 1), whereas high glucose did not stimulate the activity of Cdc42 (Figure 3C, panel 1, lane 2, compared to lane 1).
High glucose regulates EGFR activity through GTPase
We further investigated the underlying mechanisms by which high glucose levels regulate EGFR activation. MDAMB231 cells transfected with siRNAs targeting Rac1 or Cdc42 were cultured under different conditions. As shown in Figure 4, high glucose-stimulated (25 mM glucose) EGFR phosphorylation under serum starvation condition, but had no effect when Rac1 expression was knocked-down (Figure 4A, panel 1, lanes 3 and 4, compared to lanes 1 and 2). The Rac1 and Cdc42 are molecular switches in EGFR signaling cascades. When high glucose directly activates EGFR, siRNAs targeting Rac1 should not be able to prevent high glucose from stimulating EGFR phosphorylation. Thus, the results suggest that high glucose indirectly activates EGFR through Rac1. In addition, this Rac1-mediated EGFR phosphorylation is independent of EGFR degradation (Figure 4A, panel 3). Similarly, together with serum, high glucose did not elevate the EGFR phosphorylation of MDAMB231 cells in which Cdc42 expression was knocked-down (Figure 4B, panel 1, lanes 3 and 4, compared to lanes 1 and 2). The results suggest that both Rac1 and Cdc42 are necessary for high glucose indirectly activating EGFR. However, this Cdc42-mediated EGFR phosphorylation depends on EGFR degradation (Figure 4B, panel 3). These conclusions are further supported by the phosphorylation of c-cbl in these MDAMB231 cells cultured in high glucose medium (Figure 4B, panel 4).
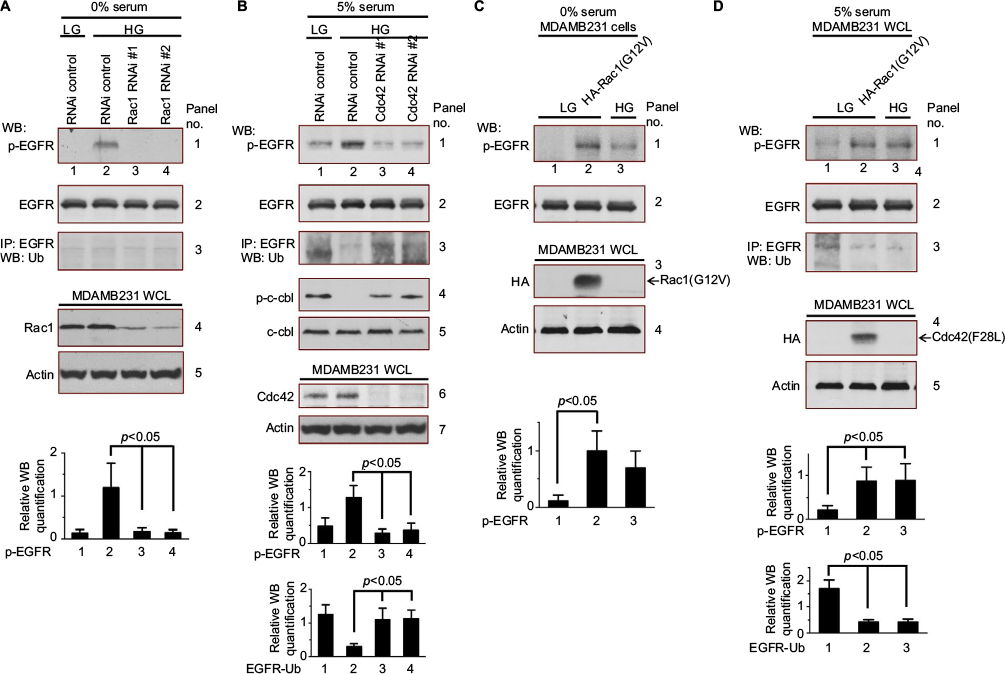 | Figure 4 High glucose regulates EGFR activity through GTPase. Notes: (A) Serum-deprived MDAMB231 cells expressing scrambled (lanes 1 and 2) or Rac1 RNAis (lanes 3 and 4) were cultured in low glucose condition (5 mM) for 12 hours and then exposed to high glucose (25 mM). After 6 hours, these cells were harvested for the measurement of EGFR phosphorylation, total EGFR levels and EGFR ubiquitination. The Western blot analysis of EGFR phosphorylation was quantified and presented by the plot at the bottom. (B) Serum-deprived MDAMB231 cells expressing scrambled (lanes 1 and 2) or Cdc42 RNAis (lanes 3 and 4) were cultured in low glucose condition (5 mM) for 12 hours and then exposed to high glucose (25 mM) with serum (5% FBS). After 6 hours, these cells were harvested for measurement of EGFR phosphorylation, total EGFR levels, the ubiquitination of EGFR, c-cbl phosphorylation, and total c-cbl levels. The Western blot analysis of EGFR phosphorylation and EGFR ubiquitination were quantified and presented by the plots at the bottom. (C) Serum-deprived MDAMB231 cells expressing HA-tagged Rac1 (G12V) (lane 2) were cultured in low glucose condition (5 mM) for 12 hours and then were harvested for measurement of EGFR phosphorylation and total EGFR levels. The Western blot analysis of EGFR phosphorylation was quantified and presented by the plot at the bottom. (D) Serum-deprived MDAMB231 cells expressing HA-tagged Cdc42 (F28L) (lane 2) were cultured in low glucose condition (5 mM) for 12 hours and then exposed to high glucose (25 mM, lane 3) with serum (5% FBS). After 6 hours, these cells were harvested for measurement of EGFR phosphorylation, total EGFR levels, and the ubiquitination of EGFR. The Western blot analysis of EGFR phosphorylation and EGFR ubiquitination were quantified and presented by the plots at the bottom. Abbreviations: EGFR, epidermal growth factor receptor; FBS, fetal bovine serum; HG, high glucose; LG, low glucose; WB, western blot; WCL, whole cell lysates. |
The constitutively active form of Rac1, Rac1(G12V), or the fast cycling Cdc42 mutant, Cdc42(F28L), were overexpressed in MDAMB231 cells. Rac1(G12V)-stimulated EGFR phosphorylation under low glucose and serum starvation conditions (Figure 4C, panel 1, lane 2, compared to lanes 1 and 3). In serum-containing low glucose medium, Cdc42 (F28L) promoted EGFR phosphorylation by blocking EGFR degradation (Figure 4D, panels 1 and 3, lane 2, compared to lanes 1 and 3). These experiments with Rac1 (G12V) or Cdc42 (F28L) further confirms that both Rac1 and Cdc42 are essential for the indirect activation of EGFR in high glucose.
High glucose-induced signaling regulates the proliferation of breast cancer cells
We examined the growth of MDAMB231 cells transfected with siRNAs targeting Rac1 or Cdc42 under low glucose (5 mM glucose, LG) or high glucose (25 mM glucose, HG) conditions. High glucose did not promote the growth of MDAMB231 cells in which Cdc42 or Rac1 expression was knocked-down (Figure 5A and B). In addition, cell proliferation of MDAMB231 cells overexpressing Cdc42 (F28L) or Rac1 (G12V) (Figure 5C and D) was also consistent with the cell signaling results shown in Figure 4C and D: Cdc42(F28L) and Rac1 (G12V) both promoted cell growth in low glucose medium. These results were further confirmed by BrdU incorporation assays using MDAMB231 cells that overexpress Cdc42 (F28L) or Rac1 (G12V) (Figure 5E).
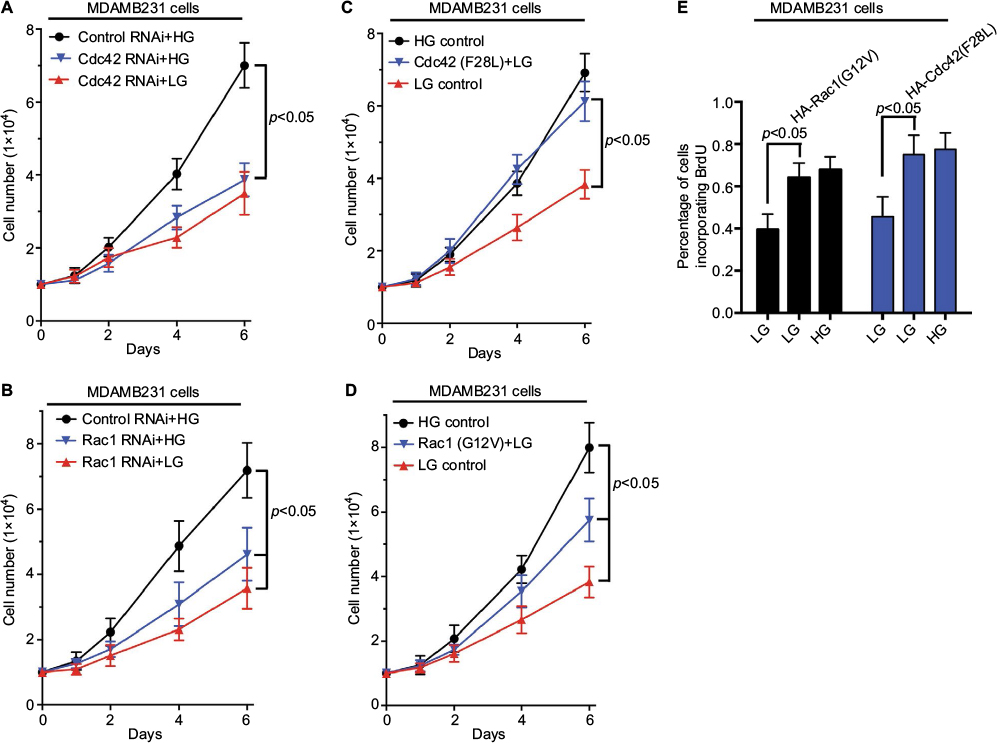 | Figure 5 High glucose-induced signaling regulates the proliferation of breast cancer cells. Notes: Cdc42 knockdown MDAMB231 cells (A), Rac1 knockdown MDAMB231 cells (B), MDAMB231 cells expressing HA-tagged Cdc42 (F28L) (C), or MDAMB231 cells expressing HA-tagged Rac1(G12V) (D) were seeded (1×104) in 12-well plates and cultured in DMEM supplemented with 2% FBS under low glucose (5 mM) or high glucose (25 mM) condition. The number of cells was counted at the indicated time points. (E) BrdU incorporation assays were performed on MDAMB231 cells expressing vector control (columns 1 and 4), HA-tagged Rac1 (G12V) (column 2), or HA-tagged Cdc42 (F28L) (column 5), which were cultured under low glucose (5 mM) or high glucose (25 mM, columns 3 and 6) condition. The percentage of cells incorporating BrdU after 12 hours was shown. Abbreviations: FBS, fetal bovine serum; HG, high glucose; LG, low glucose. |
Discussion
Hyperglycemia or diabetes is always linked to an increased risk of developing many types of cancers including breast cancer.1–5 Therefore, in this report, we investigated the underlying causality and corresponding mechanism between high glucose and the proliferation of some breast cancer cell lines. Our data indicate that high glucose (25 mM glucose) significantly increase the proliferation of breast cancer cells (MDAMB231, SKBR3 and MCF-7 cells) compared to low glucose condition (5 mM glucose). The stimulation of cell proliferation may be due to accelerate the cell cycle progression by modulating EGFR activation through GTPases Rac1 and Cdc42. In addition, Cdc42, rather than Rac1, promotes EGFR phosphorylation by blocking EGFR degradation mediated by Cbl proteins.
EGFR signaling is on the center stage of regulating cell proliferation. Aberrant EGFR signaling induces hyperproliferation and metastasis of cancer cells during tumor progression. Thus, EGFR signaling has been at the forefront of signaling research and key signaling proteins such as EGFR are attractive therapeutic targets.15–18 It has been shown that high glucose activates EGFR in pancreatic cancer cells.12,13 Similarly, we observed that high glucose stimulates EGFR phosphorylation in MDAMB231 cells. These results suggest that there is a pathway for cell proliferation in response to glucose stimulation. We found that high glucose (25 mM glucose) activates GTPase Rac1 and Rac1 knocked-down erases the glucose-stimulated EGFR phosphorylation. These data indicate the role of Rac1 in this signaling pathway. Rac1 is a critical regulator of insulin-induced glucose transport30 and it has also been reported that Rac1 is activated by glucose stimulation in podocytes.31 Further experiments are needed to uncover the detailed signaling pathway and examine whether similar signaling pathway exists in breast cancer cells. As a critical molecular switch in cell proliferation signaling, activated Rac1 may induce gene expression and cell secretion, which elevate the level of extracellular factors such as EGF and finally tranactivate EGFR.
Another GTPase, Cdc42, is also involved in the signaling pathways that respond to glucose-stimulated cell proliferation. Although Cdc42 knocked-down blocks the EGFR phosphorylation that should be stimulated by glucose, high glucose (25 mM glucose) does not activate Cdc42 (Figures 4B and 3C). As shown in this report, Cdc42 influences EGFR activation by reducing EGFR degradation. Cdc42 negatively regulates the Cbl-catalyzed ubiquitination of EGFR has been reported by several groups.6,23–25 However, there are subtle differences in the revealed underlying mechanism: Cdc42 regulates cbl through or independent of Cool/Pix proteins, which are guanine nucleotide exchange factors (GEFs) for Rho family GTPase.6,23–25,32 Furthermore, this regulatory signaling pathway is influenced by the phosphorylation/dephosphorylation cycle as well.25 These differences may be due to the different cells they studied or the different experimental setting they used. We found that the knocked-down of Cdc42 restored the phosphorylation of c-cbl and the ubiquitination of EGFR (Figure 4B). Without high glucose stimulation, Cdc42 (F28L) promoted EGFR phosphorylation by blocking EGFR degradation (Figure 4D). It is well known that Cdc42 (F28L) block EGFR degradation through cbl proteins.6,23–25 Thus, the data indicate that, in MDAMB231 cells, high glucose stimulates EGFR activation by GTPase Cdc42, which promotes EGFR phosphorylation by blocking Cbl protein-mediated EGFR degradation.
EGFR activation and degradation are carefully balanced by multiple factors and signals, including glucose stimulation. Among these various factors and signals, who takes the drive seat in the regulatory depends on cell type and the actual conditions. For breast cancer patients with diabetes or hyperglycemia, by stimulating GTPase Rac1 activity and the cycling of Cdc42, high glucose further unbalances the aberrant EGFR signaling in breast cancer cells and further promotes the proliferation of these cells. There is another signal pathway we should mention. Through reactive oxygen species (ROS), high glucose activates the tyrosine kinase, Src, which directly phosphorylates cbl and controls the phosphorylation/dephosphorylation cycle of Cool/Pix proteins.25,33 This signal pathway, which is beyond the scope of the discussion in this report, may be another important factor in high glucose-stimulated signaling and need to be further studied.
Disclosure
The authors report no conflicts of interest in this work.
References
Jee SH, Ohrr H, Sull JW, Yun JE, Ji M, Samet JM. Fasting serum glucose level and cancer risk in Korean men and women. JAMA. 2005;293(2):194–202. | ||
Srokowski TP, Fang S, Hortobagyi GN, Giordano SH. Impact of diabetes mellitus on complications and outcomes of adjuvant chemotherapy in older patients with breast cancer. J Clin Oncol. 2009;27(13):2170–2176. | ||
Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocr Relat Cancer. 2009;16(4):1103–1123. | ||
Peairs KS, Barone BB, Snyder CF, et al. Diabetes mellitus and breast cancer outcomes: a systematic review and meta-analysis. J Clin Oncol. 2011;29(1):40–46. | ||
Pandey A, Forte V, Abdallah M, et al. Diabetes mellitus and the risk of cancer. Minerva Endocrinol. 2011;36(3):187–209. | ||
Chen HY, Yang YM, Stevens BM, Noble M. Inhibition of redox/Fyn/c-Cbl pathway function by Cdc42 controls tumour initiation capacity and tamoxifen sensitivity in basal-like breast cancer cells. EMBO Mol Med. 2013;5(5):723–736. | ||
Zhuang Y, Chan DK, Haugrud AB, Miskimins WK. Mechanisms by which low glucose enhances the cytotoxicity of metformin to cancer cells both in vitro and in vivo. PLoS One. 2014;9(9):e108444. | ||
De Lorenzo MS, Baljinnyam E, Vatner DE, Abarzua P, Vatner SF, Rabson AB. Caloric restriction reduces growth of mammary tumors and metastases. Carcinogenesis. 2011;32(9):1381–1387. | ||
Lee C, Longo VD. Fasting vs dietary restriction in cellular protection and cancer treatment: from model organisms to patients. Oncogene. 2011;30(30):3305–3316. | ||
Seyfried TN, Kiebish MA, Marsh J, Shelton LM, Huysentruyt LC, Mukherjee P. Metabolic management of brain cancer. Biochim Biophys Acta. 2011;1807(6):577–594. | ||
Takatani-Nakase T, Matsui C, Maeda S, Kawahara S, Takahashi K. High glucose level promotes migration behavior of breast cancer cells through zinc and its transporters. PLoS One. 2014;9(2):e90136. | ||
Liu H, Ma Q, Li J. High glucose promotes cell proliferation and enhances GDNF and RET expression in pancreatic cancer cells. Mol Cell Biochem. 2011;347(1–2):95–101. | ||
Han L, Ma Q, Li J, et al. High glucose promotes pancreatic cancer cell proliferation via the induction of EGF expression and transactivation of EGFR. PLoS One. 2011;6(11):e27074. | ||
Okumura M, Yamamoto M, Sakuma H, et al. Leptin and high glucose stimulate cell proliferation in MCF-7 human breast cancer cells: reciprocal involvement of PKC-alpha and PPAR expression. Biochim Biophys Acta. 2002;1592(2):107–116. | ||
Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355–365. | ||
Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366(1):2–16. | ||
Libermann TA, Nusbaum HR, Razon N, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313(5998):144–147. | ||
Pavelic K, Banjac Z, Pavelic J, Spaventi S. Evidence for a role of EGF receptor in the progression of human lung carcinoma. Anticancer Res. 1993;13(4):1133–1137. | ||
Joazeiro CA, Wing SS, Huang H, Leverson JD, Hunter T, Liu YC. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science. 1999;286(5438):309–312. | ||
Levkowitz G, Waterman H, Ettenberg SA, et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell. 1999;4(6):1029–1040. | ||
Kozma R, Ahmed S, Best A, Lim L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol. 1995;15(4):1942–1952. | ||
Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81(1):53–62. | ||
Schmidt MH, Husnjak K, Szymkiewicz I, Haglund K, Dikic I. Cbl escapes Cdc42-mediated inhibition by downregulation of the adaptor molecule betaPix. Oncogene. 2006;25(21):3071–3078. | ||
Wu WJ, Tu S, Cerione RA. Activated Cdc42 sequesters c-Cbl and prevents EGF receptor degradation. Cell. 2003;114(6):715–725. | ||
Feng Q, Baird D, Peng X, et al. Cool-1 functions as an essential regulatory node for EGF receptor- and Src-mediated cell growth. Nat Cell Biol. 2006;8(9):945–956. | ||
Melkoumian ZK, Peng X, Gan B, Wu X, Guan JL. Mechanism of cell cycle regulation by FIP200 in human breast cancer cells. Cancer Res. 2005;65(15):6676–6684. | ||
Yamamoto M, Patel NA, Taggart J, Sridhar R, Cooper DR. A shift from normal to high glucose levels stimulates cell proliferation in drug sensitive MCF-7 human breast cancer cells but not in multidrug resistant MCF-7/ADR cells which overproduce PKC-betaII. Int J Cancer. 1999;83(1):98–106. | ||
Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267(5205):1782–1788. | ||
Saad S, Stevens VA, Wassef L, et al. High glucose transactivates the EGF receptor and up-regulates serum glucocorticoid kinase in the proximal tubule. Kidney Int. 2005;68(3):985–997. | ||
Satoh T. Rho GTPases in insulin-stimulated glucose uptake. Small GTPases. 2014;5:e28102. | ||
Lv Z, Hu M, Zhen J, Lin J, Wang Q, Wang R. Rac1/PAK1 signaling promotes epithelial-mesenchymal transition of podocytes in vitro via triggering beta-catenin transcriptional activity under high glucose conditions. Int J Biochem Cell Biol. 2013;45(2):255–264. | ||
Stevens BM, Folts CJ, Cui W, et al. Cool-1-mediated inhibition of c-Cbl modulates multiple critical properties of glioblastomas, including the ability to generate tumors in vivo. Stem cells. 2014;32(5):1124–1135. | ||
Taniguchi K, Xia L, Goldberg HJ, et al. Inhibition of Src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes. 2013;62(11):3874–3886. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.