Back to Journals » Lung Cancer: Targets and Therapy » Volume 7
High expression of AKR1C1 is associated with proliferation and migration of small-cell lung cancer cells
Authors Tian H, Li X, Jiang W, Lv C, Sun W, Huang C, Chen R
Received 18 June 2015
Accepted for publication 25 September 2015
Published 2 May 2016 Volume 2016:7 Pages 53—61
DOI https://doi.org/10.2147/LCTT.S90694
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Pan-Chyr Yang
He Tian,1 Xing Li,2 Wenli Jiang,3 Cuiting Lv,3 Weizhang Sun,4 Caiguo Huang,3 Ruohua Chen,5
1Department of Pediatrics of Changhai Hospital, Second Military Medical University, Shanghai, People's Republic of China; 2Department of Gynecology and Obstetrics of Changhai Hospital, Second Military Medical University, Shanghai, People's Republic of China; 3Department of Biochemistry and Molecular Biology, Second Military Medical University, Shanghai, People's Republic of China; 4PET Center, Chengdu Military Command Region General Hospital, Chengdu, People's Republic of China; 5VIP Clinic of Changhai Hospital, Second Military Medical University, Shanghai, People's Republic of China
Abstract: AKR1C1 is a member of the AKR1C family, which not only plays an important role in hormone metabolism but is believed to be involved in carcinogen metabolism. Our previous study demonstrated that AKR1C1 was highly expressed in lung tumor tissues as compared with the tumor-adjacent tissues. Small-cell lung cancer (SCLC) is a special type of lung cancer. Surgical treatment of SCLC is usually difficult due to the high degree of malignancy and early metastasis, and difficulty in obtaining clinical specimens. There is not much basic or clinical research on SCLC in the People's Republic of China even in recent years. To investigate the mechanism of AKR1C1 in the pathogenesis of SCLC, the present study used H446 cell line to see whether AKR1C1 could affect the proliferation or migration of SCLC cells, and used a lentivirus to build the AKR1C1 overexpression and under-expression cell lines. The results indicated that AKR1C1 was an important inducement in the proliferation and migration of H446 cells. AKR1C1 promoted cell proliferation and played a vital role in the migration of SCLC cells. These results were also verified in nude mice in vivo. In conclusion, AKR1C1 plays an important role in the development and progression of SCLC and may represent an independent biomarker for assessment of the primary prognosis and therapy of SCLC.
Keywords: AKR1C1, migration, NCI-H446, proliferation, small-cell lung cancer (SCLC)
Introduction
Lung cancer is one of the most common human cancers, accounting for approximately 14% and 13% of the overall male and female cancer cases, respectively. Lung cancer mortality accounts for 26% of total mortality in women and 28% in men, both in the first place in cancer mortality.1 Lung cancer is the primary cause of cancer deaths in many countries at present.1 Small-cell lung cancer (SCLC) is a type of neuroendocrine tumor, accounting for approximately 15%–25% of all malignant tumors of the lung and representing one of the most aggressive and lethal cancer types. It is difficult to predict the prognosis of SCLC at the primary stage due to rapid growth and early metastasis of the disease. The 5-year survival rate is only 10%–13% for limited-stage SCLC and 1%–2% for extensive-stage SCLC.2–4 Therefore, it is urgent to investigate the progression of SCLC.
NAD(P)(H)-dependent oxidoreductases of the AKR1C family play essential roles in the metabolism of all steroid hormones, conjugated steroids, neurosteroids, and bile acids.5 There are four AKR1C isoforms (AKR1C1–AKR1C4) existing in humans, whose genes are clustered on chromosome 10p14.6,7 Recently, a study8 demonstrated that the members of the AKR1C superfamily were involved in carcinogen metabolism. For instance, AKR1C3 was highly expressed in prostate and breast cancer tissues and could stimulate cancer cell proliferation, suggesting that AKR1C3 is a potential therapeutic target for a number of malignancies.9 AKR1C2 could serve as a biomarker to evaluate the efficacy of chemotherapy in patients with advanced non-SCLC.10 AKR1C4 is one of the susceptible genes for common familial colorectal cancer.11 AKR1C1 is a less well-known member of the AKR1C family, and was reported to be highly expressed in different cancer types such as prostate cancer,12 endometrial cancer,13 and non-SCLC.14 In contrast, recent studies demonstrated that low AKR1C1 expression was associated with poor survival rates in breast cancer patients.15 In addition, AKR1C1 expression was also related to drug resistance in several tumor types, such as human colon cancer and skin keratinocyte HaCaT cells.16–18 The low expression of AKR1C1 may slow down the progression of cancer.19 All these results suggest that AKR1C1 plays a complex role in carcinogenesis. However, the exact function and regulatory mechanism of AKR1C1 in carcinogenesis remain unclear. Additionally, it appears that none of the previous studies on AKR1C1 has been conducted in SCLC.
In the present study, we first investigated the differential expression of AKR1C1 in 60 pairs of lung cancer tissues including ten pairs of SCLC tissues, and then changed the expression level of AKR1C1 in SCLC cells and analyzed changes in H446 cells in a nude mouse model in vivo to explore the biological value of AKR1C1 in SCLC.
Materials and methods
Human tissue samples
Tissue microarrays (OD-CT-Rslug01-007, Xinchao Biotechnology Company, Shanghai, People’s Republic of China) were performed on 60 pairs of lung cancer and adjacent tissues from 45 clinical stage II~III male patients and 15 clinical stage I female patients according to the 2007 World Health Organization (WHO) classification system. The mean age of the 60 patients at the time of surgery was 62.9 years (Table 1). In addition, the AKR1C1 expression was also evaluated in ten pairs of SCLC and adjacent tissues from male patients, including five cases of clinical stage I and five cases of stage II~III according to the 2007 WHO classification system. The mean age of the ten patients at the time of surgery was 64.8 years (Table 2).
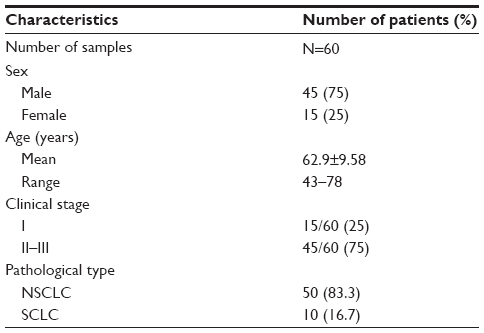 | Table 1 Clinical characteristics of the 60 lung cancer patients |
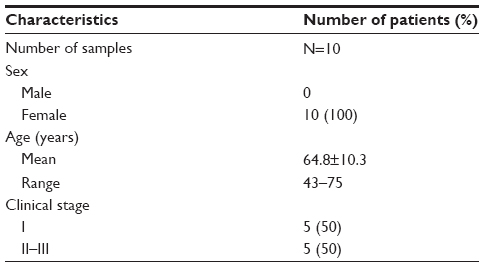 | Table 2 Clinical characteristics of the ten SCLC patients |
Cell line and culture
Human SCLC cell line NCI-H446 cells (Cell Bank Type Culture Collection of the Chinese Academy of Sciences, Shanghai, People’s Republic of China) were maintained in Roswell Park Memorial Institute (RPMI)-1640 (Hyclone, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific) in a humidified atmosphere with 5% CO2. Confluent cells were passaged with trypsin-ethylenediaminetetraacetic acid (EDTA) (0.05% trypsin and 0.53 mM tetrasodium EDTA).
Lentivirus transfection
AKR1C1 was silenced and overexpressed by AKR1C1 lentivirus transfection into NCI-H446 cells. The lentivirus with human AKR1C1 was chemically synthesized by GenePharma (Shanghai, People’s Republic of China) according to the published sequence on National Center for Biotechnology Information (Gen ID: 1645). The siRNA sequence was 5′- AAGCTTTAGAGGCCACCAAAT-3′. The effectiveness of AKR1C1 gene silencing and overexpression was confirmed by Western blotting as described later. To detect tumor metastasis in vivo, different groups of cells (overexpression group, silenced group, untreated group, and two negative control groups) were transfected stably with the lentivirus with luciferase gene (GenePharma, Shanghai, People’s Republic of China).
Cell proliferation assay
Cells were seeded into 96-well culture plates at a density of 5×103 cells in a final volume of 100 μL/well, and the untransfected cells were used as an untreated control. The cell proliferation rate was analyzed at different time points (24, 48, 72, and 96 hours) by addition of 50 μL MTT solution (Dojindo, Kumamoto, Japan) to each well and 4-hour incubation at 37°C. After that, the MTT solution was discarded. Finally, 150 μL dimethyl sulfoxide was added to each well. The absorbance was measured with a spectrophotometer (Thermo Fisher Scientific) at 570 nm. The mean absorbance values from six wells per group were calculated.
Colony formation assay
Cells were seeded into six-well plates at a density of 500 cells per well and cultured for 2 weeks at 37°C in a humidified incubator. The culture medium was refreshed every 3 days. Finally, the cells were washed with phosphate-buffered saline (PBS), fixed in 1 mL methanol for 30 minutes, and stained with 0.4% crystal violet for 20 minutes at room temperature. The number of colonies containing more than 50 cells was counted and averaged.
Migration assay
AKR1C1 H446 cell migration was evaluated in 24-well Transwell chambers (Corning Incorporated, Corning, NY, USA), as directed by the manufacturer. Briefly, in the Transwell chambers, the upper and lower culture compartments of each well were partitioned by a polycarbonate membrane (pore size 8 μm). A total of 1.0×104 cells were placed into the upper chambers with 100 μL RPMI-1640 medium supplemented with 1% FBS, and 600 μL RPMI-1640 medium supplemented with 20% FBS was added to the lower chambers. After 20-hour incubation in a humidified 5% CO2 atmosphere at 37°C, cells on the upper surfaces of the wells were removed, and those migrating to the lower surfaces were fixed in cold methanol for 10 minutes and stained with 0.4% crystal violet for 10 minutes at room temperature. Excess stain was removed with physiological saline, and the chambers were air-dried. For each experiment, five fields on the undersides of the membranes were randomly selected and photographed, and transmigrated cells were counted and averaged. The experiment was performed in triplicate.
Wound-healing assay
Cells were seeded into six-well plates at a density of 1×106 and incubated with growth medium as described earlier. After 24-hour incubation, confluent monolayer cells were wounded with a yellow sterile pipette tip. The cellular debris was removed with a PBS wash, and the wounded areas of the cell monolayers were imaged with an inverted microscope equipped with a digital camera (Olympus Corporation, Tokyo, Japan). Cells were then incubated in 5% FBS RPMI-1640 medium and migration of cells into the scratch wound was photographed and assessed at different time points. The distance of cell migration was compared with that of the untreated control cells by statistical analysis. The experiments were repeated in duplicate at least three times.
Western blotting analysis
Proteins were extracted from cells. Cell lysates were prepared with radioimmunoprecipitation assay buffer supplemented with PMSF (Beyotime, Shanghai, People’s Republic of China). The supernatants were collected, and the protein concentrations were determined with a BCA protein assay (Beyotime). An equal amount of the protein extract from each sample (50 μg per lane) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on a 12% or 10% polyacrylamide gel and electro-transferred onto nitrocellulose membranes. The membranes were incubated overnight at 4°C with polyclonal antibodies against anti-AKR1C1 (1:500 dilution, Abcam, Cambridge, UK) and anti-GAPDH (1:5,000 dilution, Bioworld Technology Inc., St Louis Park, MN, USA), and then incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody (1:5,000 dilution, Abgent, San Diego, CA, USA) at room temperature for 1.5 hours. The blots were treated with an enhanced chemiluminescence reagent (EMD Millipore, Billerica, MA, USA) and immunoreactive bands were detected by exposure to X-ray films.
Tumor growth study in nude mice
The tumorigenic capability of the AKR1C1 stably transfected cells along with control cells was assessed in 6-week-old male athymic nude mice. All animals were maintained in a sterile environment. Cages, bedding, food, and water were all autoclaved. All animals were maintained on a daily 12-hour light/12-hour dark cycle. Briefly, cells were grown to 80% confluence, harvested by trypsinization, washed twice in PBS (Thermo Fisher Scientific), and re-suspended to a final concentration of 1×106 cells/mL in sterile PBS. A 0.1 mL cell suspension was injected into the left flank of the 6-week-old mice. Six mice were inoculated per test group. The tumor volume (mm3) was calculated using the formula for the volume of a prolate ellipsoid, (w2/2)(L), in which w and l are the width and length of the tumor in millimeters, respectively. Tumors were harvested from the mice after 6-week growth and fixed in formaldehyde for further analysis.
Immunohistochemistry
The microarray was incubated with primary antibodies against AKR1C1. Sections were subsequently incubated with the Cell and Tissue Staining Kit HRP-DAB system (R&D Systems, Inc., Minneapolis, MN, USA) according to the manufacturer’s instructions. Formalin-fixed paraffin-embedded H446 tumors were cut with a microtome into 6 μm sections. Antigen retrieval was performed in 10 mM sodium citrate buffer at pH 6.0 for 16 minutes at 96°C–98°C. Slides were incubated with primary antibodies against AKR1C1, P-ERK, and EGFR. Sections were subsequently incubated with the Cell and Tissue Staining Kit HRP-DAB system (R&D Systems, Inc.), according to the manufacturer’s instructions. The positive expression of AKR1C1 was mainly located in the cytoplasm and stained brown. The staining intensity was scored as follows: 0= no color; 1= light yellow; 2= yellow; 3= brown; and 4= dark brown. Positive cells accounted for a percentage score standard: 0= no positive cells; 1=1%–25% positive cells; 2=26%–50% positive cells; 3=51%–75% positive cells; and 4= more than 75% positive cells. A total score of the two lower than 4 was defined as low expression, and a total score of the two higher than 4 was defined as high expression.
Statistical analysis
Statistical analyses were carried out using SPSS version 13.0 (SPSS Inc., Chicago, IL, USA), and significance was determined by two-tailed Student’s t-test. Data were considered to be statistically significant at P<0.05.
Ethics
The study protocols were approved by the Second Military Medical University Institutional Ethical Committee, People’s Republic of China. All animal-handling procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and followed the guidelines of the Animal Welfare Act.
Results
Expression of AKR1C1 in lung cancer
To evaluate the AKR1C1 expression in lung cancer, we analyzed 60 pairs of lung cancer and matched tumor-adjacent tissues by immunohistochemistry. The high-expression rate in the tumor tissues was 90% vs 10% in the tumor-adjacent tissues, indicating that AKR1C1 was highly expressed in lung cancer tumor tissues. Then, we evaluated the AKR1C1 expression in SCLC, and found that the high-expression rate in the tumor tissues was 60% vs 0% in the tumor-adjacent tissues, indicating that AKR1C1 was highly expressed in SCLC tumor tissues as compared with the tumor-adjacent tissues (Figure 1).
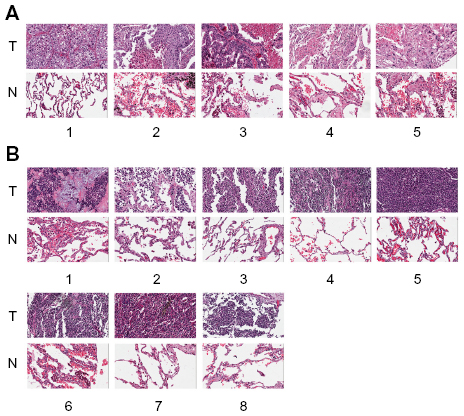 | Figure 1 Immunofluorescence shows that the expression of AKR1C1 in the tumor tissues is significantly higher than that in the tumor-adjacent tissues from the 60 lung cancer patients. |
AKR1C1 induces the proliferation and migration of SCLC cells
To uncover the function of AKR1C1 in SCLC, H446 cells were transfected with lentivirus to create four new cell lines: AKR1C1 overexpressed cell line, AKR1C1 silenced cell line, and two negative control cell lines. The effectiveness of the transfection was confirmed by Western blotting (Figure 2A).
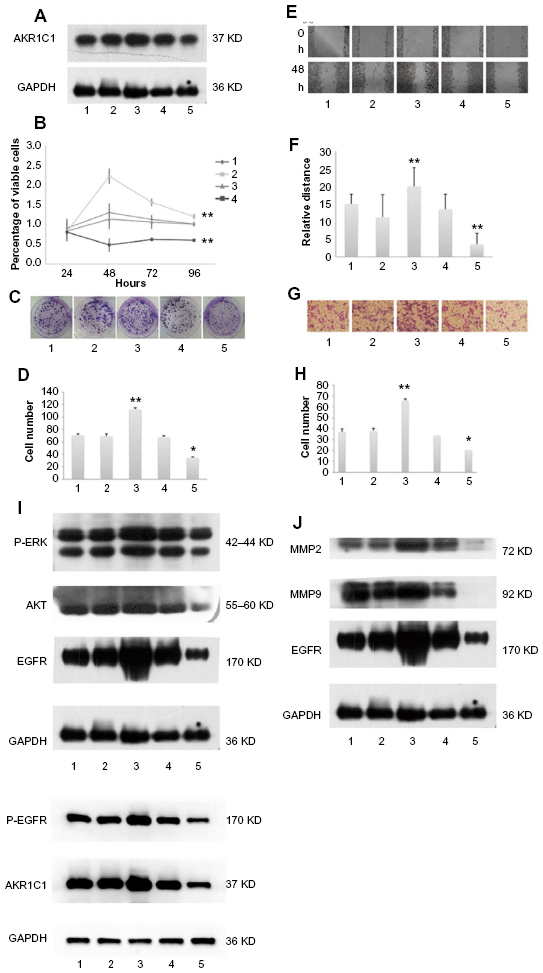 | Figure 2 Western blotting showing the expression levels of AKR1C1 in the five cell lines. |
Next, MTT and colony formation assays were performed to observe the function of AKR1C1 in the proliferation of H446 cells, and the percentage of viable cells was detected at four time points (24, 48, 72, and 96 hours). It was found that the percentage of viable cells in AKR1C1 overexpressed cells was significantly higher than that in untreated H446 cells, and the percentage of viable cells of the AKR1C1 silenced cells was the lowest of all five cell lines (AKR1C1 overexpressed cell line, AKR1C1 silenced cell line, two negative control cell lines, and the untreated H446 cell line). There was no significant difference between the two negative control groups as compared with the normal H446 cell group (Figure 2B). Similar results were obtained in the colony formation assay. After 2-week incubation of the five different cell lines, the number of colonies formed was counted. The highest number of colonies was observed in AKR1C1 overexpressed cell line, followed by the normal cell line, the AKR1C1 silenced cell line, and the two negative control cell lines (Figure 2C and D). The number of colonies was 70.7±2.05, 69.3±3.68, 112±2.50, 68.0±1.63, and 34.7±1.70 in the untreated control group, AKR1C1 overexpressed negative control group, AKR1C1 overexpressed group, AKR1C1 silenced negative control group, and AKR1C1 silenced group, respectively. Based on these results, we concluded that AKR1C1 was a key mediator in the proliferation of H446 cells.
Subsequently, the role of AKR1C1 in H446 cell migration was observed. As shown in Figure 2E and F, a wound-healing assay was used to examine the impact of AKR1C1 on H446 cell migration. The results showed that cells in AKR1C1 silenced group migrated more slowly than those in the two negative control groups and the untreated control groups, while the velocity of migration in AKR1C1 overexpressed group was the fastest of all. The relative distance of cell migration in the untreated control group, AKR1C1 overexpressed negative control group, AKR1C1 overexpressed group, AKR1C1 silenced negative control group, and AKR1C1 silenced group was 15±2.78, 11.16±6.45, 20±5.27, 13.5±4.27, and 3.5±3.12, respectively. At the same time, another experiment was performed to examine the impact of AKR1C1 on H446 cell migration, the number of cells migrating to the lower surface of the Transwells in AKR1C1 overexpressed group was significantly higher than that in normal H446 cell group (Figure 2G and H). The number of cells migrating to the lower surface of the Transwells in AKR1C1 silenced group was significantly lower than that in normal H446 cell group. The number of cells migrating to the lower surface in the two negative control groups was not significantly different from that in normal H446 cell group. The number of migrated cells was 37.3±3.05/5, 38±4.36/5, 66±2.65/5, 34±5/5, and 20.77±1.53/5 fields in untreated control group, AKR1C1 overexpressed negative control group, AKR1C1 overexpressed group, AKR1C1 silenced negative control group, and AKR1C1 silenced group, respectively. At the same time, a wound-healing assay was used to examine the impact of AKR1C1 on H446 cell migration. The results showed that cells in AKR1C1 silenced group migrated more slowly than those in the two negative control groups and the untreated control groups (Figure 2E and F), while the velocity of migration in AKR1C1 overexpressed group was the fastest of all. The relative distance of cell migration in the untreated control group, AKR1C1 overexpressed negative control group, AKR1C1 overexpressed group, AKR1C1 silenced negative control group, and AKR1C1 silenced group was 15±2.78, 11.16±6.45, 20±5.27, 13.5±4.27, and 3.5±3.12, respectively. These results indicate that AKR1C1 down-regulation not only affected the proliferation but reduced the migration ability of H446 cells. On the contrary, the up-regulation of AKR1C1 suppressed the proliferation and migration of H446 cells.
AKR1C1 regulates the expression of proliferation and migration-related proteins
The EGFR, MMP9, and MMP2 proteins play vital roles in cell invasion of SCLC cell lines.20–22 P-ERK, AKT, P-EGFR, and EGFR are reported to be related to cell proliferation and migration.20,22–24 Therefore, we tested the EGFR, P-EGFR, MMP9, MMP2, AKT, and P-ERK protein expression levels in H446 cells in AKR1C1 overexpressed, AKR1C1 overexpressed negative control, untreated, AKR1C1 silenced, and AKR1C1 silenced negative control groups (Figure 2H–J), and found that AKR1C1 induced the up-regulation of these migration-related proteins.
AKR1C1 affects tumor growth in nude mice in vivo
As shown before, we confirmed the role of AKR1C1 in an SCLC cell line. Next, we attempted to detect the biological function of AKR1C1 in SCLC in vivo. It was found that the rate of tumor growth in the AKR1C1 overexpression group was significantly higher than that in the untreated group. The same result was obtained in terms of the tumor volume and mass, showing that the tumor growing rate and tumor volume and mass were the lowest in the AKR1C1 silencing group (Figure 3A–C). The bodyweight of the nude mice of all three groups decreased as the tumors grew (Figure 3D). In addition, we used immunochemistry to support our result and found that the percentage of positive cells for AKR1C1, P-ERK, and EGFR in the AKR1C1 overexpression group was significantly higher than that in the untreated group, and the lowest in the AKR1C1 silenced group (Figure 3E).
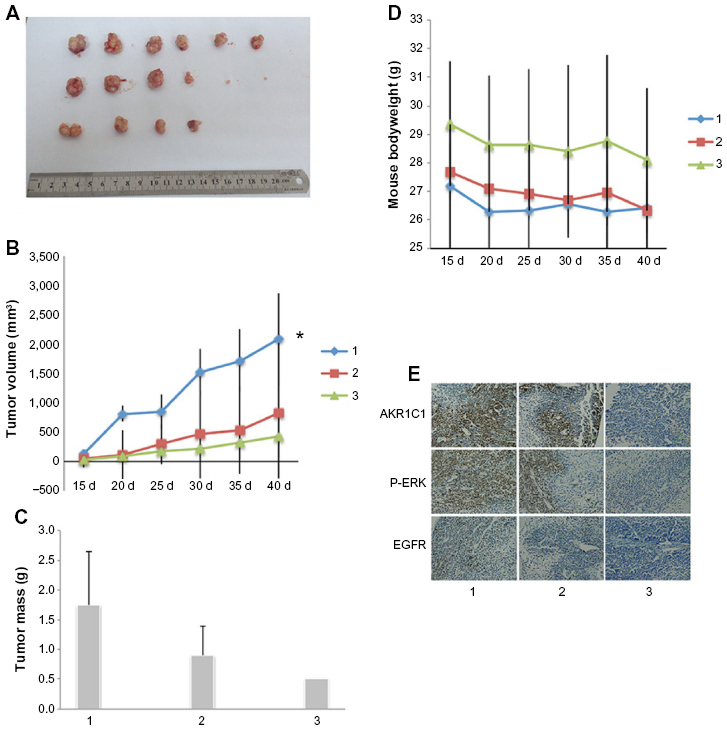 | Figure 3 AKR1C1 induced the proliferation in a mouse model in vivo. |
Discussion
Rapid growth and metastasis are the main reasons that explain why SCLC is the most aggressive form of cancer. The resistance to chemotherapy further increases the difficulty in the clinical treatment of SCLC.25 Therefore, it is an urgent task to identify an effective way to inhibit the proliferation and migration of SCLC cells. In the present study, we demonstrated that AKR1C1 was associated with the progression of SCLC. Our study on AKR1C1 might provide an early diagnostic marker and a treatment target to suppress the invasion and metastasis of SCLC. In the present study, we analyzed the differences in AKR1C1 expression levels between SCLC tumors and matched tumor-adjacent tissues and observed significantly higher expression levels of AKR1C1 in the SCLC tissues vs the matched tumor-adjacent tissues.
Many studies have evaluated the silencing of genes involved in the pathways that drive cancer, such as cell cycle and apoptosis, neoplastic transformation, and resistance to conventional therapies.26–31 To investigate the function of AKR1C1 in SCLC, we overexpressed and silenced AKR1C1 using a lentivirus. The Western blotting results showed that the AKR1C1 protein expression level underwent significant changes. First, we performed in vitro experiments and cell proliferation assays to evaluate the correlation between the AKR1C1 expression levels and H446 cell proliferation. Both MTT and colony formation assays supported the conclusion that AKR1C1 is a proliferation-related gene, and AKR1C1 suppressed the cell proliferation in SCLC cells. Second, our in vivo experiments showed that AKR1C1 also suppressed tumor growth, suggesting that AKR1C1 is a gene related to tumor growth, at least in SCLC. Knowing that cell migration plays a pivotal role in cancer progression and targeting migration is an alternative means of therapy,31 we performed both in vitro and in vivo experiments to investigate the role of AKR1C1 in tumor invasion, and found that AKR1C1 was not only a gene related to proliferation but affected metastasis in SCLC.
Additionally, EGFR is known to be highly expressed in several tumor cell lines, including SCLC cell line, and there is a correlation between the expression level of EGFR and invasion. AKT and P-ERK induced the proliferation in tumors.20,22 MMP2 and MMP9 are members of the family of zinc-binding endopeptidases, and reported to be associated with cancer cell invasion.32 It was found in our study that the silencing of AKR1C1 induced the down-regulation of MMP2, MMP9, EGFR, AKT, and P-ERK, further suggesting that AKR1C1 is associated with migration and proliferation in SCLC cells. The exact way in which AKR1C1 affects cell proliferation and invasion remains to be further verified. The results of the present study suggest that AKR1C1 might regulate AIF directly.14
To avoid differences between individuals, we used case-matched samples of SCLC and tumor-adjacent tissues from the same patient in the microarray. Obviously, the limitation of our study is the limited quantities of clinical samples. The results revealed that AKR1C1 was significantly more highly expressed in the tumor tissue. Because of the limited sample number, we were unable to detect a correlation between the expression levels of AKR1C1 and the stage of the SCLC, and also unable to detect a correlation between the expression level of AKR1C1 and condition of the patient. If AKR1C1 would be a marker to detect the patient’s condition in the future, a large sample examination will be needed. Perhaps the expression level of AKR1C1 could be used as a marker of early diagnosis and dynamic changes of SCLC.
Conclusion
In the present study we measured different expression levels of AKR1C1 in SCLC tissues and tumor-adjacent tissues. AKR1C1 may be related to the proliferation and metastasis of SCLC cells. SCLC is characterized by a short cell doubling time, a high rate of disease progression, early migration to become blood-borne, and lymph metastasis. Despite a relatively good initial response to chemotherapy and radiotherapy, multidrug resistance usually leads to relapse or progression of the disease.33–35 As AKR1C1 is reported to be related to drug resistance, targeting AKR1C1 might be an alternative therapy method for SCLC patients.14 Lower the expression of AKR1C1 could inhibit proliferation and migration at the same time, so we can reduce the occurrence of drug resistance. Silencing AKR1C1 could suppress tumor growth and invasion, and also avoid the resistance to regular chemotherapy and radiotherapy. We could consider a new therapy for SCLC, which is the combination of gene targeting therapy and chemotherapy or radiotherapy.
Acknowledgment
This research project was supported by the National Natural Science Foundation of China (number 81473239).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014,64(1):69–90. | |
Qiu X, Wang Z, Li Y, Miao Y, Ren Y, Luan Y. Characterization of sphere-forming cells with stem-like properties from the small cell lung cancer cell line H446. Cancer Lett. 2012;323(2):161–170. | |
Maurya P, Meleady P, Dowling P, Clynes M. Proteomic approaches for serum biomarker discovery in cancer. Anticancer Res. 2007;27(3A):1247–1255. | |
Albain KS, Crowley JJ, Livingston RB. Long-term survival and toxicity in small cell lung cancer. Expanded Southwest Oncology Group experience. Chest. 1991;99(6):1425–1432. | |
Rižner TL, Penning TM. Role of aldo-keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids. 2014;79:49–63. | |
Penning TM, Byrns MC. Steroid hormone transforming aldo-keto reductases and cancer. Ann N Y Acad Sci. 2009;1155:33–42. | |
Nishizawa M, Nakajima T, Yasuda K, et al. Close kinship of human 20alpha-hydroxysteroid dehydrogenase gene with three aldo-keto reductase genes. Genes Cells. 2000;5(2):111–125. | |
Burczynski ME, Lin HK, Penning TM. Penning. Isoform-specific Induction of a human aldo-keto reductase by polycyclic aromatic hydrocarbons (PAHs), electrophiles, and oxidative stress: implications for the alternative pathway of pah activation catalyzed by human dihydrodiol dehydrogenase. Cancer Res. 1999;59(3):607– 614. | |
Byrns MC, Steckelbroeck S, Penning TM. An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3alpha-HSD, type 5 17beta-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem Pharmacol. 2008;75(2):484–493. | |
Kuang P, Zhou C, Li X, et al. Proteomics-based identification of secreted protein dihydrodiol dehydrogenase 2 as a potential biomarker for predicting cisplatin efficacy in advanced NSCLC patients. Lung Cancer. 2012;77(2):427–432. | |
Gylfe AE, Katainen R, Kondelin J, et al. Eleven candidate susceptibility genes for common familial colorectal cancer. PLoS Genet. 2013;9(10):e1003876. | |
Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66(5):2815–2825. | |
Rizner TL, Smuc T, Rupreht R, Sinkovec J, Penning TM. AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Mol Cell Endocrinol. 2006;248(1–2):126–135. | |
Wang HW, Lin CP, Chiu JH, et al. Reversal of inflammation-associated dihydrodiol dehydrogenases (AKR1C1and AKR1C2) overexpression and drug resistance in non-small cell lung cancer cells by wogonin and chrysin. Int J Cancer. 2007;120(9):2019–2027. | |
Lewis MJ, Wiebe JP, Heathcote JG. Expression of progesterone metabolizing enzyme genes (AKR1C1, AKR1C2, AKR1C3, SRD5A1, SRD5A2) is altered in human breast carcinoma. BMC Cancer. 2004;4:27. | |
Matsunaga T, Hojo A, Yamane Y, Endo S, El-Kabbani O, Hara A. Pathophysiological roles of aldo–keto reductases (AKR1C1 and AKR1C3) in development of cisplatin resistance in human colon cancers. Chem Biol Interact. 2013;202(1–3):234–242. | |
Wsol V, Szotakova B, Martin HJ, Maser E. Aldo-keto reductases (AKR) from the AKR1C subfamily catalyze the carbonyl reduction of the novel anticancer drug oracin in man. Toxicology. 2007;238(2–3):111–118. | |
Gwinn MR, Whipkey DL, Tennant LB, Weston A. Differential gene expression in normal human mammary epithelial cells treated with malathion monitored by DNA microarrays. Environ Health Perspect. 2005;113(8):1046–1051. | |
Klippel S, Jakubikova J, Delmore J, et al. Methyljasmonate displays in vitro and in vivo activity against multiple myeloma cells. Br J Haematol. 2012;159(3):340–351. | |
Damstrup L, Rude Voldborg B, Spang-Thomsen M, Brünner N, Skovgaard Poulsen H. In vitro invasion of small-cell lung cancer cell lines correlates with expression of epidermal growth factor receptor. Br J Cancer. 1998;78(5):631–640. | |
Yang X, Di J, Zhang Y, et al. The Rho-kinase inhibitor inhibits proliferation and metastasis of small cell lung cancer. Biomed Pharmacother. 2012;66(3):221–227. | |
Schmid K, Bago-Horvath Z, Berger W, et al. Dual inhibition of EGFR and mTOR pathways in small cell lung cancer. Br J Cancer. 2010;103(5):622–628. | |
Irie HY, Pearline RV, Grueneberg D, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171(6):1023–1034. | |
Chin YR, Toker A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal. 2009;21(4):470–476. | |
Sarvi S, Mackinnon AC, Avlonitis N, et al. CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 2014;74(5):1554–1565. | |
Selga E, Noé V, Ciudad CJ. Transcriptional regulation of aldo-keto reductase 1C1 in HT29 human colon cancer cells resistant tomethotrexate: role in the cell cycle and apoptosis. Biochem Pharmacol. 2008; 75(2):414–426. | |
Chien CW, Ho IC, Lee TC. Induction of neoplastic transformation by ectopic expression of human aldo-ketoreductase 1C isoforms in NIH3T3 cells. Carcinogenesis. 2009;30(10):1813–1820. | |
Stoddard FR 2nd, Brooks AD, Eskin BA, Johannes GJ. Iodine alters gene expression in the MCF7 breast cancer cell line: evidence for an anti-estrogen effect of iodine. Int J Med Sci. 2008;5(4):189–196. | |
Wanichwatanadecha P, Sirisrimangkorn S, Kaewprag J, Ponglikitmongkol M. Transactivation activity of human papillomavirus type 16 E6*I on aldo-keto reductase genes enhances chemoresistance in cervical cancer cells. J Gen Virol. 2012;93(Pt 5):1081–1092. | |
Chen CC, Chu CB, Liu KJ, et al. Gene expression profiling for analysis acquired oxaliplatin resistant factors in human gastric carcinoma TSGH-S3 cells: the role of IL-6 signaling and Nrf2/AKR1C axis identification. Biochem Pharmacol. 2013;86(7):872–887. | |
Levin EG. Cancer therapy through control of cell migration. Curr Cancer Drug Targets. 2005;5(7):505–518. | |
Kim WY, Jang JY, Jeon YK, et al. Syntenin increases the invasiveness of small cell lung cancer cells by activating p38, AKT, focal adhesion kinase and SP1. Exp Mol Med. 2014;46:e90. | |
Califano R, Abidin AZ, Peck R, Faivre-Finn C, Lorigan P. Management of small cell lung cancer: recent developments for optimal care. Drugs. 2012;72(4):471–490. | |
Lawson MH, Cummings NM, Rassl DM, et al. Two novel determinants of etoposide resistance in small cell lung cancer. Cancer Res. 2011; 71(14):4877–4887. | |
Rezonja R, Knez L, Cufer T, Mrhar A. Oral treatment with etoposide in small cell lung cancer dilemmas and solutions. Radiol Oncol. 2013; 47(1):1–13. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.