Back to Journals » Drug Design, Development and Therapy » Volume 20
High-Dose Propofol Induces Cytotoxicity by Elevating Intracellular Ca2⁺ via GABAA Receptor and IP3R in HT22 Cells
Authors Zhou H, Zhou H, Tan X, Jin Y ![]() , Choi TY, Dong C
, Choi TY, Dong C ![]()
Received 3 December 2025
Accepted for publication 4 March 2026
Published 10 March 2026 Volume 2026:20 566272
DOI https://doi.org/10.2147/DDDT.S566272
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Tuo Deng
Hao Zhou,1 Huimin Zhou,1 Xi Tan,1 Yadong Jin,2 Tin Yin Choi,2 Chaoxuan Dong1
1Department of Anesthesiology, The First Affiliated Hospital of Jinan University, Guangzhou, Guangdong, People’s Republic of China; 2International School, Jinan University, Guangzhou, Guangdong, People’s Republic of China
Correspondence: Chaoxuan Dong, Department of Anesthesiology, The First Affiliated Hospital of Jinan University, 613 West Huangpu Ave., Guangzhou, Guangdong, 510630, People’s Republic of China, Tel +86-13354712423, Email [email protected]; [email protected]
Objective: Disruption of calcium (Ca2⁺) homeostasis has been implicated as a key pathological mechanism underlying propofol-induced neurodevelopmental and cognitive deficits. However, the mechanisms underlying propofol-induced intracellular Ca2⁺ dysregulation remain incompletely understood. Extending findings of anesthetic-induced metabolic disruptions in non-neuronal models to the central nervous system, this study aimed to elucidate the underlying mechanisms of the Ca2⁺ imbalance in neuronal cells, with implications for the safety of clinical anesthesia in pediatric populations.
Material and Methods: Mouse hippocampal neurons (HT22 cells) served as an in vitro model. Cell viability was assessed using the CCK-8 assay. Intracellular Ca2⁺ dynamics were evaluated using the Fluo-4 AM Ca2⁺ fluorescent probe to investigate the mechanisms underlying propofol-induced Ca2⁺ dysregulation.
Results: Propofol exposure at 10 μM and 50 μM across all time points (2, 6, or 24 hours) showed no significant impact on cell viability. Similarly, 100 μM propofol lacked toxicity at 2 or 6 hours, but survival significantly declined after 24 hours exposure (P < 0.0001). Furthermore, 200 μM propofol decreased cell viability after 2 hours of treatment (P < 0.01), with further reduction following prolonged exposure (P < 0.05). A rapid increase in intracellular Ca2⁺ concentration was observed with 200 μM propofol (P < 0.0001), which was entirely abolished by the inhibition of the γ-aminobutyric acid type A (GABAA) receptor. Conversely, inhibition of the inositol trisphosphate receptor (IP3R) alone partially mitigated the propofol-induced Ca2⁺ elevation (P < 0.0001). Notably, chelation of elevated intracellular Ca2⁺ using BAPTA-AM fully prevented the propofol-induced decrease in cell viability (P < 0.01).
Conclusion: Propofol induces cytotoxicity in HT22 cells in a concentration- or time-dependent manner. Notably, cytotoxicity at 100 μM propofol was observed only after 24 hours of exposure, whereas 200 μM propofol produced rapid cytotoxicity. This rapid toxicity is mediated by activation of GABAA receptor and IP3R, which triggers the endoplasmic reticulum (ER) Ca2⁺ release and elevating intracellular Ca2⁺ concentration.
Plain Language Summary: Propofol is a commonly used anesthetic, but its safety in the developing brain is unclear. In this study, high doses of propofol damaged mouse hippocampal neurons and caused a rapid rise in calcium inside the cells. This effect involved specific calcium-regulating pathways, and removing the extra calcium prevented the damage. These results suggest that high doses of propofol can disrupt calcium balance in developing neurons. They also highlight the need for careful dosing in pediatric anesthesia.
Keywords: propofol, general anesthesia, developmental neurotoxicity, Ca2⁺, HT22 cells
Introduction
In the United States, approximately 2–3 million children under 36 months and 1.5 million infants receive general anesthesia annually, with many infants requiring multiple or prolonged exposure.1 Since the initial report in 1999 that ketamine induces apoptosis in developing neurons,2 extensive studies in rodents and non-human primates have demonstrated that early postnatal anesthetic exposure impairs the developing nervous system, leading to behavioral and cognitive dysfunction.3–6 Clinical studies have further linked anesthesia and surgery to cognitive decline in young children, suggesting that prolonged or repeated exposure to general anesthesia may contribute to behavioral and memory deficits later in development.7–11
Propofol is a widely used intravenous anesthetic that exerts sedative-hypnotic effects via γ-aminobutyric acid (GABAA) receptor activation. Due to its rapid onset and favorable pharmacokinetic profile, propofol is extensively applied for anesthetic induction, maintenance, and intensive care unit (ICU) sedation - accounting for over 75% of surgical procedures across more than 50 countries.12,13 Preclinical studies indicate that propofol possesses potential developmental neurotoxicity and triggers adverse developmental events, such as neuronal apoptosis, abnormal neurogenesis, and reduced dendritic density, ultimately leading to persistent impairment of learning and memory functions.14–20 In 2016, the US Food and Drug Administration (FDA) issued a warning stating that exposure to propofol anesthesia for more than three hours or multiple times in late-pregnancy women and children under three years old may increase the risk of brain developmental defects, yet approximately 1–2% of pregnant women even receive anesthesia and surgery unrelated to childbirth, which includes procedures involving propofol.21,22 Robbins et al reported that general anesthesia for cesarean delivery doubled the odds of severe motor delay at age two, despite showing no association with overall neurodevelopmental delay.23 Clinical studies and case reports also suggested that infants and young children (5–14 years old) exposed to propofol developed progressive microcephaly and impaired memory, cognitive, and behavioral functions.24,25 However, confounding factors in clinical settings like surgical trauma and underlying medical conditions, make it difficult to isolate propofol’s direct neurotoxic effects in humans. Thus, in vitro experimental studies are required to elucidate the mechanisms underlying propofol-associated developmental neurotoxicity. HT22 cells, an immortalized mouse hippocampal neuronal cell line, have been widely used as neural precursor-like cells to investigate the developmental neurotoxicity of inhaled anesthetics,26 and thus represent a suitable in vitro model of anesthesia-induced neurotoxicity. Accordingly, HT22 cells were used in this study.
Propofol induces neurotoxic effects through mechanisms including calcium (Ca2⁺) signaling, mitochondrial energy metabolism, and inflammation.13 In addition to calcium-dependent mechanisms, emerging metabolomics-based studies indicate that intravenous anesthetics can profoundly disrupt amino acid and carnitine metabolism, reflecting mitochondrial stress not only in neuronal but also in renal and hepatic cell models.27,28 This bioenergetic failure triggers pathological crosstalk with intracellular Ca2⁺ signaling. Adenosine triphosphate (ATP) depletion leads to Ca2⁺ pump dysfunction and cytoplasmic Ca2⁺ overload, which further exacerbates mitochondrial energy metabolism disorders.29,30 Cytosolic Ca2⁺ concentration is maintained at 10–100 nM at rest and can increase to 500–1000 nM upon activation.31 This precise homeostatic regulation is cooperatively controlled by diverse Ca2⁺ channels and transporters localized on the plasma and organelle membranes.32–34 Cytoplasmic Ca2⁺ overload impairs cell survival, development, and normal function.35–37
Propofol disrupts intracellular Ca2⁺ homeostasis in immature neurons, leading to impaired cell survival and neurodevelopmental abnormalities.38,39 In developing neurons, propofol triggered the opening of L-type voltage-dependent Ca2⁺ channel (VDCC) through the activation of GABAA receptor, resulting in intracellular Ca2⁺ overload and subsequent neuronal apoptosis.17 In immature neurons, the activation of GABAA receptor induces membrane depolarization due to a reversed intracellular chloride gradient, which subsequently opens the L-type VDCCs and elevates cytoplasmic Ca2⁺.40 Beyond membrane-bound channels, Qiao et al demonstrated that propofol also stimulates IP3R and ryanodine receptor (RyR)-mediated Ca2⁺ release from the endoplasmic reticulum (ER). Such sustained Ca2⁺ elevations impair neurogenesis in neural stem cells (NSCs) and promote cell death.41 However, the molecular mechanisms linking propofol exposure to IP3R and RyR activation remain unclear. Notably, IP3R and RyR are ER Ca2⁺ release channels with shared structural and functional features. Although IP3R is activated by inositol 1,4,5-trisphosphate (IP3), both channels are also regulated by cytoplasmic Ca2⁺, with specific Ca2⁺ levels promoting their opening.42 Based on the results of previous studies, we hypothesized that propofol exposure may activate the opening of L-type VDCC through GABAA receptor, induce extracellular Ca2⁺ inward flow, and further promote IP3R-dependent Ca2⁺ release from the ER, which leads to intracellular Ca2⁺ overload and induces cytotoxicity.
Material and Methods
Reagents
Dulbecco’s modified Eagle’s medium (C11995500BT) and HBSS solution (H1020) were purchased from Saiguo Bio (Guangzhou, China). 10% fetal bovine serum (C0251), 1% penicillin-streptomycin solution (C0222) and Fluo-4 AM Ca2⁺ detection kit (S1061S) were purchased from Beyotime (Shanghai, China). Xestospongin C (GC14484), Nimodipine (GC16587) and BAPTA-AM (GC13517) were purchased from GLPBIO (Montclair, California, USA). CCK-8 assay (K1018/CK04) was purchased from APExBIO (Houston, USA). Propofol (HY-B0649, purity ≥ 99%) and Bicuculline (HY-N021) were purchased from MCE (New Jersey, USA). EGTA (SA8500) was purchased from Solarbio (Beijing, China).
Cell Culture
The HT22 cell line was commercially obtained from the National Collection of Authenticated Cell Cultures (Shanghai, China; certified by STR). HT22 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution. The cells were maintained in a thermostatic incubator (160i CR, Thermo Fisher, Waltham, Massachusetts, USA) at 37 °C with 5% CO2, and the culture medium was completely replaced every 72 hours.
Cytotoxicity Assay
CCK-8 assay was employed to assess cytotoxicity. HT22 single-cell suspensions were inoculated into 96-well culture plates at a density of 1 × 104 to 1 × 105 cells/mL, with 100 μL of the cell suspension added to each well (approximately 5,000 cells/well). Blank, control, and experimental groups were established. After 24 hours of incubation in a constant-temperature cell incubator, the medium was replaced with serum-free DMEM for 12 hours to achieve cell cycle synchronization. Then, different concentrations of propofol solution (10, 50, 100, 200 μM) were added to the experimental group. Incubation was continued for 2, 6, or 24 hours according to the respective treatment times. Following treatment, 10μL of CCK-8 reagent was added to each well, and the plates were incubated in a constant-temperature cell culture incubator, protected from light, for 1–4 hours. After incubation, the absorbance at 450 nm was measured using an enzyme reader (Varioskan LUX, Thermo Fisher, Waltham, Massachusetts, USA).
Intracellular Ca2⁺ Measurement
The Fluo-4 Ca2⁺ probe was utilized for real-time monitoring of intracellular Ca2⁺ concentration changes. HT22 cells were seeded into 96-well plates and cultured for 24 hours. On the following day, the medium was aspirated, and the cells were washed twice with Ca2⁺- and magnesium-free, phenol red-free HBSS solution. A 100 μL aliquot of Fluo-4 AM (1X) staining solution was added to each well, and the cells were incubated in a cell culture incubator, protected from light, for 20–30 minutes. The Fluo-4 AM staining solution was then aspirated, and the cells were washed three times with Ca2⁺- and magnesium-free, phenol red-free HBSS solution. For each experimental group, the cells were treated with one of the following solutions: HBSS (containing Ca2⁺ and magnesium), HBSS (containing Ca2⁺ and magnesium) + Bicuculline (100 μM), HBSS (containing Ca2⁺ and magnesium) + Xestospongin C (250 nM), HBSS (containing Ca2⁺ and magnesium) + Nimodipine (100 μM), HBSS (containing Ca2⁺ and magnesium) + EGTA (5 μM), or HBSS (containing Ca2⁺ and magnesium) + BAPTA-AM (5 μM). The cells were then incubated in a cell culture incubator for an additional 30 minutes. At the end of the incubation, baseline fluorescence (F0) changes were measured and recorded for 50 seconds under an inverted fluorescence microscope (Leica DMi1, Leica, Wetzlar, Germany). Propofol was applied at 50 seconds, and fluorescence (F) reflecting Ca2⁺ fluctuations were continuously recorded for 200 seconds, without interruption during drug administration. The total filming duration was 250 seconds. To minimize photobleaching, minimal laser power and short imaging duration were used. Data were background-subtracted and normalized to baseline (F/F0), with a parallel control group monitored under identical settings to account for residual fluorescence decay.
Statistical Methods
Ca2⁺ fluorescence image data were extracted using ImageJ (ImageJ Software, Bethesda, Maryland, USA). For Ca2⁺ imaging analysis, only cells with stable baseline fluorescence and clear intracellular dye loading were included. Cells showing abnormal morphology, loss of membrane integrity, or excessively low baseline fluorescence were excluded from analysis. Regions of interest (ROIs) were randomly selected from viable cells within each field of view. Data visualization and statistical analysis were performed using GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA) and Origin 2024 (Origin Software, Northampton, Massachusetts, USA). Data are presented as mean ± standard deviation (SD) from a minimum of five independent experiments. No formal a priori sample size calculation was performed, as this study was exploratory in nature and based on established in vitro experimental paradigms. Post-hoc power analysis indicated that the study was adequately powered to detect the observed treatment effects. Student’s t-test was employed for comparisons between two experimental groups. For comparisons involving multiple groups with a single independent variable, one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test was performed. To evaluate the effects of drug concentration and exposure time, two-way ANOVA followed by Tukey’s multiple comparisons test was applied. p-value < 0.05 was considered statistically significant.
Results
Effect of Propofol on the Activity of HT22 Cells
To investigate the effects of propofol on HT22 cell toxicity at various concentrations and exposure durations, cell viability was assessed using the CCK-8 assay. HT22 cells were exposed to different concentrations of propofol (10, 50, 100, and 200 μM) for 2, 6, or 24 hours. After treatment with 10 or 50 μM propofol for 2, 6, or 24 hours, there was no significant difference in the effect of propofol on cell survival rates at different exposure concentrations and times compared to the control group (Figure 1). Notably, 200 μM propofol significantly reduced cell viability as early as 2 hours (to 79.1% of control) (P < 0.01), with further declines to 60.9% at 6 hours and 45.4% at 24 hours (P < 0.0001) (Figure 1). In contrast, 100 μM propofol showed no significant toxicity at 2 hours and 6 hours, but decreased viability to 64.1% after 24 hours (Figure 1). The above results indicate that propofol induces cytotoxicity in a concentration- or time-dependent manner. 10 μM and 50 μM of propofol exposure did not significantly induce cytotoxicity; 100 μM propofol only markedly reduced cell viability after 24 hours of exposure; whereas 200 μM of propofol exhibited pronounced cytotoxic effects at nearly all exposure time points.
 |
Figure 1 Effect of propofol exposure on HT22 cell viability. HT22 cells were exposed to various concentrations of propofol (10, 50, 100, 200 μM) for 2, 6, or 24 hours. The results of the CCK-8 assays demonstrated that cell viability was not significantly affected by exposure to 10 or 50 μM propofol at any of the exposure times (2, 6, or 24 hours) compared to the control group. In contrast, exposure to 100 μM propofol did not affect cell viability with short-term exposure but induced cytotoxicity following 24-hour exposure. Moreover, treatment with 200 μM propofol resulted in significant cell damage at all exposure times, with a further reduction in cell survival as exposure time increased. Data are expressed as the mean ± SD in two-way ANOVA with Tukey’s test from five independent experiments. **P < 0.01 and ****P < 0.0001 vs control group; #P < 0.05 vs 200 μM propofol exposure for 2 hours group; ΔP < 0.05 vs 200 μM propofol exposure for 6 hours group. |
Effect of Propofol on Intracellular Ca2⁺ Homeostasis in HT22 Cells
Propofol Induced Elevated Cellular Ca2⁺ Concentration
To investigate the mechanism underlying Ca2⁺ homeostasis dysregulation in propofol-induced neural precursor cytotoxicity, we employed the Fluo-4 AM Ca2⁺ assay kit to assess the impact of propofol exposure on the intracellular Ca2⁺ concentration in HT22 cells. In previous experiments, we observed that exposure to 200 μM propofol for 2, 6, or 24 hours induced cytotoxicity, with the extent of toxicity increasing in a time-dependent manner. Therefore, we investigated the effect of propofol at 200 μM concentration on intracellular Ca2⁺ levels in HT22 cells. Propofol significantly increased the fluorescence intensity of intracellular Ca2⁺, while no such increase was observed in the control group treated with HBSS buffer (Figure 2A). The ratio of fluorescence intensity (F) to baseline fluorescence intensity (F0) was analyzed by subtracting the background fluorescence, and the change in this ratio was used to quantify intracellular Ca2⁺ concentration in both groups. Propofol significantly increased the peak intracellular Ca2⁺ fluorescence intensity by 75.3% compared to the control group (P < 0.0001) (Figure 2B and 2C). These results indicate that 200 μM propofol markedly induces elevated Ca2⁺ concentrations within HT22 cells.
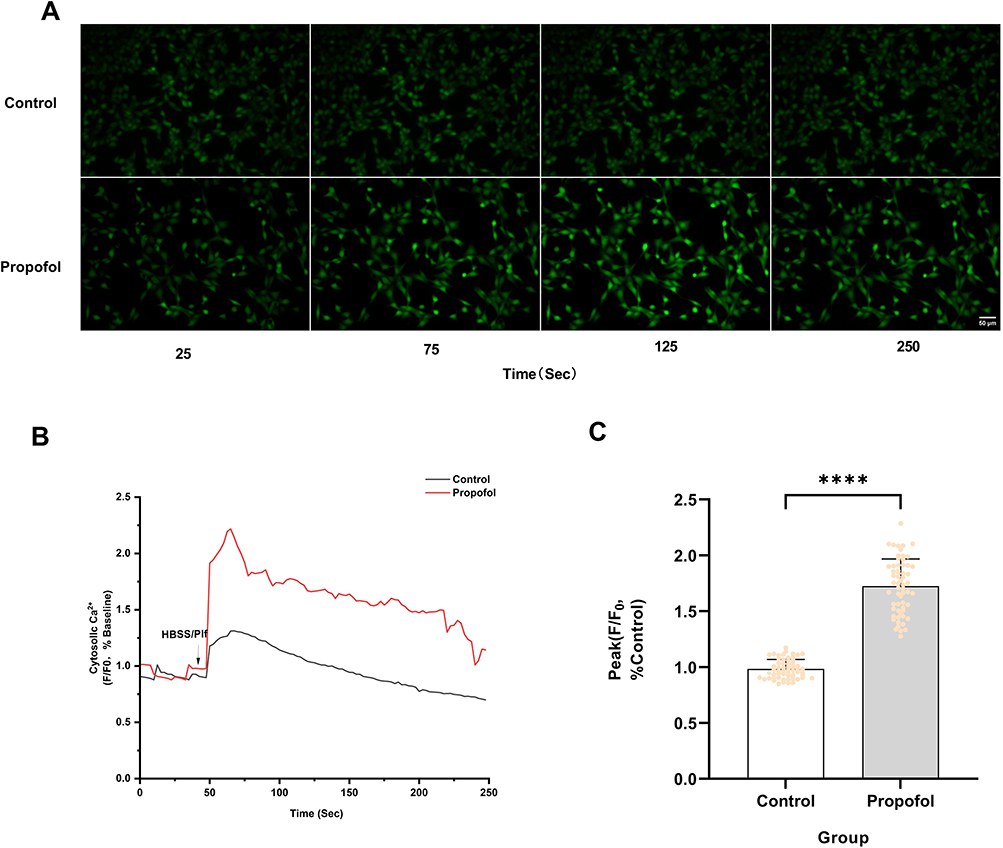 |
Figure 2 Effect of propofol on intracellular Ca2⁺ homeostasis in HT22 cells. (A) The fluorescence intensity of intracellular Ca2⁺ was significantly increased upon the addition of 200 µM propofol compared to the control group (scale bar=50 µm). (B) Ca2⁺ fluorescence response of HT22 cells in HBSS buffer (containing Ca2⁺) after treatment with HBSS buffer (Control) and 200 µM propofol (Plf) (indicated by arrows). The ratios, with background fluorescence subtracted and normalized to baseline, were used to represent changes in intracellular Ca2⁺ concentration. Data are shown as representative results from five independent experiments. (C) A significant increase in peak intracellular Ca2⁺ signaling was observed with 200 µM propofol compared to the blank control group. Data are expressed as the mean ± SD in Student’s t-test from 60 cells across five independent experiments. ****P < 0.0001 vs control group. Sec, Seconds. |
GABAA Receptor but Not L-Type VDCC is Involved in Propofol-Induced Intracellular Ca2⁺ Increase
Propofol may depolarize presynaptic neurons via GABAA receptor, leading to activation of L-type VDCC and mediating extracellular Ca2⁺ influx.17 To elucidate the potential role of GABAA receptor in propofol-induced Ca2⁺ elevation, we investigated the effect of the GABAA receptor antagonist Bicuculline (Bicu) on propofol-induced intracellular Ca2⁺ elevation. Treatment with 100 μM Bicu significantly inhibited the propofol-induced increase in intracellular fluorescence intensity (Figure 3A). Furthermore, Bicu significantly attenuated the propofol-induced peak intracellular Ca2⁺ fluorescence intensity by 78.6% (P < 0.0001) (Figure 3B and C). These findings indicate that Bicu suppresses propofol-induced increases in intracellular Ca2⁺ concentration. Next, we investigated the potential mechanism underlying propofol-induced Ca2⁺ elevation using the L-type VDCC antagonist Nimodipine (Nim). Treatment with 100 μM Nim failed to inhibit the propofol-induced increase in intracellular fluorescence intensity (Figure 3A). Furthermore, Nim did not significantly reduce the peak intracellular Ca2⁺ fluorescence intensity induced by propofol (Figure 3B and C). These findings indicate that Nim does not inhibit the propofol-induced increase in intracellular Ca2⁺ concentration. These results suggest that GABAA receptor rather than L-type VDCC may be involved in the propofol-induced increase in intracellular Ca2⁺ concentration in HT22 cells.
 |
Figure 3 Mechanism of propofol-induced increase in intracellular Ca2⁺ concentration in HT22 cells. (A) Pretreatment with 100 μM GABAA receptor antagonist Bicuculline (Bicu) significantly reduced the increase in intracellular fluorescence intensity induced by 200 μM propofol, while pretreatment with 100 μM L-type VDCC antagonist Nimodipine (Nim) did not significantly inhibit the increase in intracellular fluorescence intensity induced by 200 μM propofol (scale bar=50 µm). (B) Ca2⁺ fluorescence response of HT22 cells after the addition of 200 μM Propofol (Plf) (arrow) to HBSS buffer solution (containing Ca2⁺) or HBSS buffer solution (containing Ca2⁺) pretreated with 100 μM Bicu and 100 μM Nim. The ratio after subtracting the background fluorescence and normalizing to the baseline was used to represent the change in intracellular Ca2⁺ concentration. The data show representative data from five experiments. (C) Pretreatment with100 μM Bicu reduced the peak amplitude of propofol-induced Ca2⁺ signals, whereas pretreatment with 100 μM Nim did not significantly affect the peak amplitude of propofol-induced Ca2⁺ signals. Data are expressed as the mean ± SD in one-way ANOVA with Tukey’s test from 60 cells across five independent experiments. ****P < 0.0001 vs control group; ####P < 0.0001 vs propofol group; ns, no significant difference. Sec, Seconds. |
In addition, we also explored whether extracellular Ca2⁺ influx is involved in propofol-induced increase in intracellular Ca2⁺. We used the extracellular Ca2⁺ chelator EGTA to remove Ca2⁺ from the extracellular fluid. 5 μM EGTA pretreatment did not inhibit the propofol-induced increase in intracellular fluorescence intensity (Figure 4A). Furthermore, 5 μM EGTA pretreatment did not reduce the peak intensity of propofol-induced intracellular Ca2⁺ fluorescence (Figure 4B and C). This indicates that extracellular Ca2⁺ does not participate in propofol-induced increases in intracellular Ca2⁺ concentration.
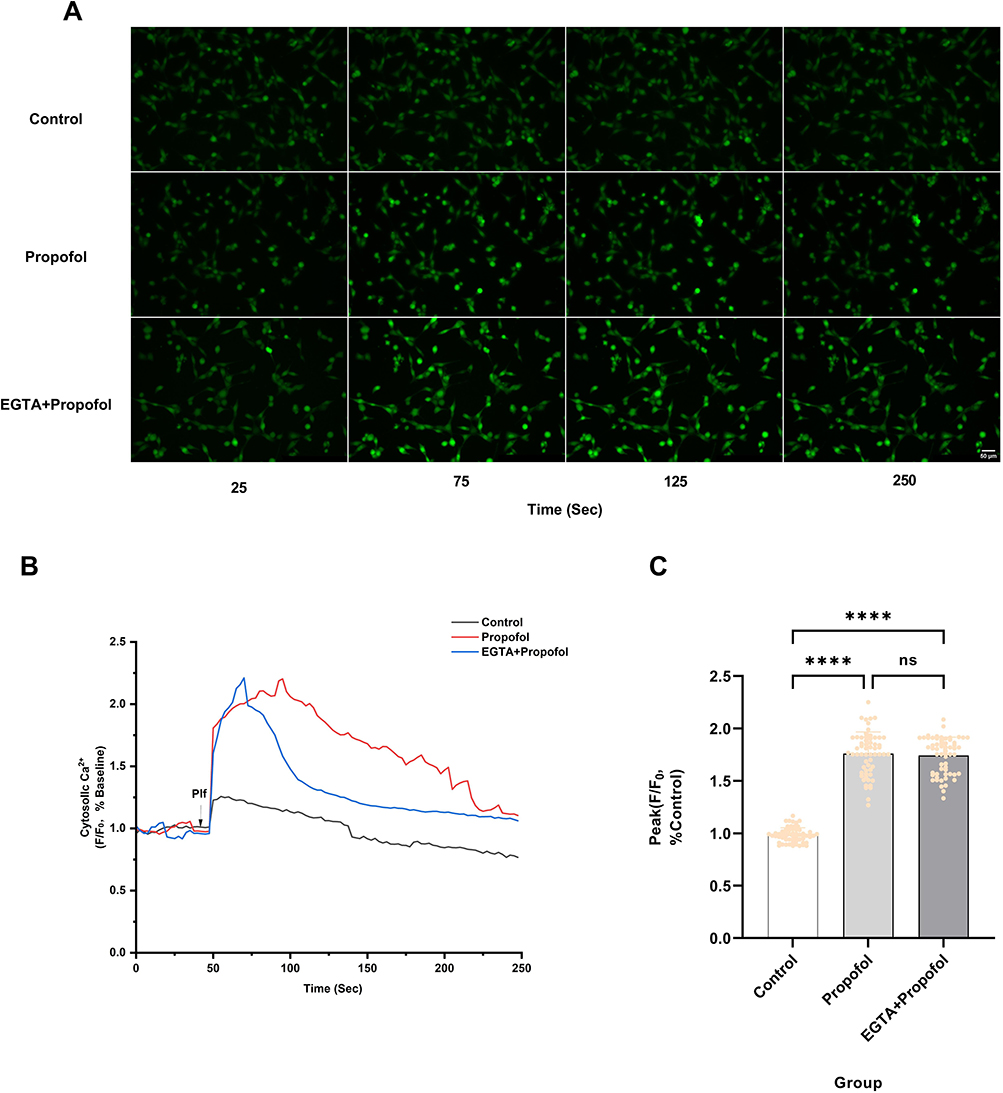 |
Figure 4 Extracellular Ca2⁺ does not participate in propofol-induced increases in intracellular Ca2⁺ concentration. (A) Pretreatment with 5 μM extracellular Ca2⁺ chelator EGTA did not reduce the increase in intracellular fluorescence intensity caused by 200 μM Propofol (scale bar=50 µm). (B) Ca2⁺ fluorescence response of HT22 cells after the addition of 200 μM Propofol (Plf) (arrow) in HBSS buffer solution (containing Ca2⁺) or HBSS buffer solution (containing Ca2⁺) pretreated with 5 μM EGTA. The ratio of background fluorescence subtracted and normalized to baseline was used to represent the change in intracellular Ca2⁺ concentration. The data show representative data from five experiments. (C) Pretreatment with 5 μM EGTA did not reduce the peak amplitude of Ca2⁺ signals induced by 200 μM propofol. Data are expressed as the mean ± SD in one-way ANOVA with Tukey’s test from 60 cells across five independent experiments. ****P < 0.0001 vs control group; ns, no significant difference. Sec, Seconds. |
IP3R Is Involved in Propofol-Induced Increase in Intracellular Ca2⁺
IP3R is a key receptor mediating Ca2⁺ release from the ER. Therefore, we examined the effect of Xestospongin C (Xc), an IP3R inhibitor, on propofol-induced Ca2⁺ elevation. Treatment with 250 nM Xc partially inhibited the propofol-induced increase in intracellular Ca2⁺ fluorescence intensity but did not completely suppress this elevation (Figure 5A). Furthermore, pretreatment with 250 nM Xc significantly attenuated the propofol-induced peak intracellular Ca2⁺ fluorescence intensity by 29.0% (P < 0.0001) (Figure 5B and C). However, the peak intensity in the Xc-pretreated group remained 34.2% higher than that of the control group (P < 0.0001) (Figure 5B and C). These results indicate that while Xc significantly mitigates propofol-induced Ca2⁺ overload, it does not fully restore Ca2⁺ homeostasis to baseline levels.
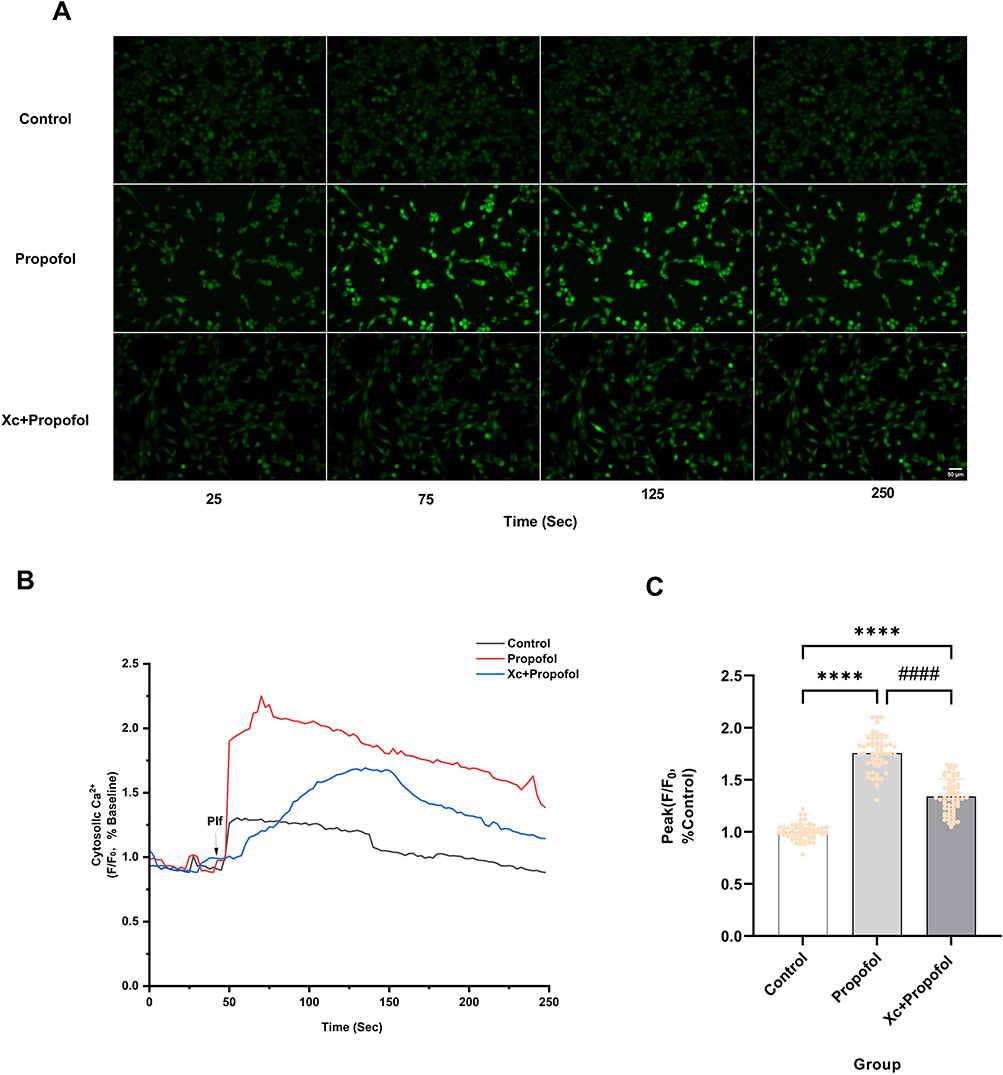 |
Figure 5 IP3R is involved in propofol-induced increases in intracellular Ca2⁺ concentration in HT22 cells. (A) Pretreatment with 250 nM IP3R inhibitor Xestospongin C (Xc) significantly reduced the propofol-induced increase in intracellular fluorescence intensity (scale bar=50 µm). (B) The Ca2⁺ response of HT22 cells was measured following the addition of 200 μM propofol (Plf) (indicated by arrowheads) to either HBSS solution containing Ca2⁺ or HBSS solution pretreated with 250 nM Xc (containing Ca2⁺). Changes in intracellular Ca2⁺ concentration were represented by ratios subtracting background fluorescence and normalizing to baseline, with representative data from five independent experiments. (C) Pretreatment with 250 nM Xc reduced the amplitude of the peak Ca2⁺ signal triggered by 200 μM propofol. Data are expressed as the mean ± SD in one-way ANOVA with Tukey’s test from 60 cells across five independent experiments. ****P < 0.0001 vs control group; ####P < 0.0001 vs propofol group. Sec, Seconds. |
Inhibition of Propofol-Induced Elevation of Ca2⁺ Concentration Increases Cell Viability
Propofol-induced elevation of intracellular Ca2⁺ concentration may contribute to cytotoxic effects. We pretreated cells with the intracellular Ca2⁺ chelator BAPTA-AM. We found that 5 μM BAPTA-AM pretreatment significantly suppressed propofol-induced increases in intracellular Ca2⁺ fluorescence (Figure 6A). Furthermore, 5 μM BAPTA-AM markedly reduced the propofol-induced peak intracellular Ca2⁺ fluorescence intensity by 74.2% (P < 0.0001) (Figure 6B and C), without compromising baseline intracellular Ca2⁺ homeostasis. This indicates that 5 μM BAPTA-AM suppresses propofol-induced increases in intracellular Ca2⁺ concentration.
 |
Figure 6 BAPTA-AM inhibits propofol-induced elevation of intracellular Ca2⁺ concentration. (A) Pretreatment with 5 μM intracellular Ca2⁺ chelator BAPTA-AM significantly reduced the enhancement of intracellular fluorescence intensity induced by 200 μM propofol (scale bar=50 µm). (B) Ca2⁺ response of HT22 cells after the addition of 200 μM propofol (Plf) (arrowheads) to HBSS solution containing Ca2⁺ (HBSS) or HBSS solution pretreated with 5 µM BAPTA-AM. The ratio of fluorescence intensity, with background fluorescence subtracted and normalized to baseline, represents changes in intracellular Ca2⁺ concentration. The data show representative results from five independent experiments. (C) Pretreatment with 5 µM BAPTA-AM reduced the amplitude of the peak Ca2⁺ signal triggered by propofol, Data are expressed as the mean ± SD in one-way ANOVA with Tukey’s test from 60 cells across five independent experiments. ****P < 0.0001 vs control group; ####P < 0.0001 vs propofol group; ns, no significant difference. Sec, Seconds. |
To directly assess the involvement of elevated intracellular Ca2⁺ in cytotoxicity, we pretreated cells with 5 µM BAPTA-AM and examined the effect of 200 µM propofol on HT22 cell viability after 2 hours using the CCK-8 assay. Pretreatment with 5 μM BAPTA-AM significantly attenuated propofol-induced cytotoxicity, restoring cell viability by 17.8% (P < 0.001) (Figure 7), indicating that elevated intracellular Ca2⁺ concentrations contribute to propofol-induced cytotoxicity.
 |
Figure 7 Inhibiting the increase in intracellular Ca2⁺ concentration can enhance the survival rate of HT22 cells. Pretreatment with 5 µM BAPTA-AM significantly enhanced the survival of cells exposed to 200 µM propofol, compared to the control group. Data are expressed as the mean ± SD in one-way ANOVA with Tukey’s test from five independent experiments. ****P < 0.0001 vs control group; ###P < 0.001 vs propofol group; ns, no significant difference. |
Discussion
This study investigates the effects of propofol on the toxicity of mouse hippocampal neural progenitor cells and the role of Ca2⁺ homeostasis in propofol-induced neurotoxicity. Propofol induces cytotoxicity in a concentration- or time-dependent manner. Exposure to clinically relevant concentrations (10 μM) of propofol did not result in significant toxic effects on HT22 cells. However, exposure to high concentrations (100 or 200 μM) of propofol led to a marked reduction in cell survival in a concentration- or time-dependent manner. Our findings also indicate that exposure to 200 μM propofol induces ER Ca2⁺ release through activation of GABAA receptor and IP3R, resulting in a significant increase in intracellular Ca2⁺ concentration. Furthermore, this elevated intracellular Ca2⁺ concentration correlates with propofol-induced cytotoxicity.
There have been a series of studies reporting on propofol-related developmental neurotoxicity, but the conclusions have been varied. A study has found that exposing immature rat hippocampal neurons to propofol at concentrations ranging from 0.1 to 1000 μM for 3 hours can significantly induce apoptosis and inhibit proliferation.18 In a study using neonatal rat models, exposure to 5 or 50 μM of propofol for 5 hours was shown to trigger acute neuronal death.17 Conversely, a study indicated that neuronal progenitor cell death only occurs when propofol concentrations exceed 7.1 μM (supratherapeutic doses), while clinically low doses do not affect cell viability.43 Additionally, one study found that exposing mouse NSCs to 100 μM propofol for 6 hours led to a significant decrease in cell viability.44 In a dose-gradient experiment (5–50 μM), it was demonstrated that propofol exerts a time- and dose-dependent inhibitory effect on NSCs proliferation.45 However, another study suggested that exposure to 50 μM propofol for 3 or 6 hours does not affect the viability of rat cortical NSCs, with cell death and proliferation inhibition only occurring after prolonged exposure (24 hours) or at high concentrations (300 or 600 μM).46 Contrary to these varied conclusions, our results indicate that treatment with 10 or 50 μM propofol for 2, 6, or 24 hours does not induce cytotoxicity. Only prolonged (24 hours) and high concentrations (100 and 200 μM) of propofol exposure led to a significant decrease in cell viability.
Propofol primarily binds to red blood cells and albumin in human blood, with only about 1–3% remaining as free propofol capable of diffusing and interacting with cell surface receptors. This makes it challenging to determine the appropriate doses of propofol used in in vitro experiments. Typically, the anesthetic doses of propofol refer to the concentration range in the brain maintained during surgical anesthesia, known as the clinical concentration range. Concentrations exceeding this range are considered supratherapeutic doses.22,43 However, there is currently no consensus among different studies regarding the definition of the clinical concentration range. Some studies suggest that the effective concentration of propofol in the brain is between 2 μg/mL and 6 μg/mL (approximately 11–33 μM),47 while others define the clinical effective concentration range as 1–20 μM,48 and some even report clinical brain concentrations of 4–20 μg/mL (approximately 22–110 μM).22 In this study, 10 μM of propofol was considered a clinically relevant concentration, while concentrations above 20 μM were regarded as supraclinical doses. Although the 200 μM concentration is considered supraclinical, previous studies in renal and hepatic cells have employed similar supratherapeutic concentrations in metabolomics toxicity models to simulate long-term or cumulative exposure scenarios.27,28 Propofol is highly lipophilic and can accumulate significantly in lipid-rich tissues like the brain during prolonged infusions, such as long-term sedation in the ICU or complex pediatric surgeries. Local tissue concentrations may exceed measured plasma levels, making the study of “supraclinical” doses relevant to these specific pathological or extreme scenarios (eg, Propofol Infusion Syndrome).49,50 From a clinical perspective, our results also demonstrated that 10 μM propofol did not induce significant cytotoxicity or calcium dysregulation, which aligns with clinical safety data of propofol for short-term anesthesia. However, the threshold-dependent toxicity at higher concentrations highlights a potential safety concern for pediatric anesthesiologists during prolonged infusions. These findings suggest that maintaining the lowest effective therapeutic concentration and limiting exposure duration could be critical strategies to mitigate developmental neurotoxicity risks in clinical practice.
The dose-response relationship concerning the developmental neurotoxicity of propofol anesthesia shows significant heterogeneity in existing studies. The discrepancies in conclusions may arise from the different in vitro cell models or the varying developmental time windows of the cells used. Neurotoxicity typically occurs during specific periods of brain development, defined as the brain growth spurt (BGS). For humans, this period begins in late pregnancy and continues until approximately three years postnatally. For rhesus monkeys, it starts around day 115 of gestation and lasts until 60 days postnatally. While for rodents, the BGS usually occurs during the first two weeks after birth.13 Neurogenesis continues during this phase, making it a vulnerable period for neurodevelopment. Therefore, conducting in vitro experiments on cells extracted from the brains of different species or at different developmental stages, even using similar drugs and concentrations, may lead to varying research conclusions.
Intracellular Ca2⁺ overload is one of the key factors leading to cell damage. Previous studies have shown that Ca2⁺ overload can induce apoptosis and autophagy to varying degrees, and can even directly lead to cell necrosis.36,51 Inhalational anesthetics (such as isoflurane and sevoflurane) and intravenous anesthetics (such as propofol and ketamine) have been reported to induce apoptosis or autophagy in neural cells by causing intracellular Ca2⁺ overload, thereby affecting cell survival.41,52–55 Studies indicate that a surge in cytosolic Ca2⁺ induces mitochondrial permeability transition pore (mPTP)-mediated mitochondrial Ca2⁺ overload, leading to structural and functional impairment of mitochondria and subsequently triggering programmed cell death. Notably, recent metabolomic evidence indicates that propofol significantly disrupts amino acid and carnitine metabolism in hepatocytes and HEK-293 cells, leading to mitochondrial dysfunction.27,28 Although the association between propofol-induced metabolic disruption and mitochondrial dysfunction remains a mechanistic hypothesis rather than a conclusive causal pathway, it indicates that propofol directly interferes with mitochondrial bioenergetics and fatty acid oxidation. In neurons, ATP deficiency—this energy emergency may impair the function of ATP-dependent Ca2⁺ pumps, such as the sarcoplasmic/ER Ca2⁺-ATPase (SERCA), thereby creating a vicious cycle that exacerbates the IP3R-mediated Ca2⁺ overload observed in this study.56 Ca2⁺ also plays a crucial role in regulating cell cycle processes, which in turn affects cell proliferation.57,58 Prior research has indicated that sevoflurane, isoflurane, and midazolam can inhibit the proliferation of NSCs by inducing excessive increases in intracellular Ca2⁺ ion concentrations.54,59,60 Our study results demonstrate that propofol increases intracellular Ca2⁺ concentrations in HT22 cells, leading to a decrease in cell viability. However, since the HT22 cell line is derived from the HT4 cell line (a clonal line of immortalized mouse hippocampal neural progenitor cells) and is capable of infinite proliferation in vitro, the effects of propofol on HT22 cell viability may result from either inducing cell death or inhibiting proliferation.
We undertook further investigations to elucidate the mechanisms underlying propofol-induced increases in intracellular Ca2⁺ concentration. Our experimental findings demonstrated that inhibiting GABAA receptor alone completely abolished propofol-induced changes in intracellular Ca2⁺ concentrations, whereas inhibiting L-type VDCC with nimodipine alone did not significantly attenuate propofol-induced increases in intracellular Ca2⁺ concentration. Subsequently, we chelated extracellular Ca2⁺ and observed that their elimination did not significantly affect propofol-induced increases in intracellular Ca2⁺ concentration. Furthermore, we examined the impact of ER Ca2⁺ release and IP3R activation on intracellular Ca2⁺ homeostasis. Inhibition of IP3R alone significantly reduced propofol-induced increases in intracellular Ca2⁺ concentration. However, in contrast to GABAA receptor inhibition, IP3R blockade failed to restore Ca2⁺ levels to control values, as the fluorescence intensity remained significantly higher than the baseline.
GABAA receptor is recognized as a primary pharmacological receptor mediating the sedative-hypnotic effects of propofol. Propofol binding to this receptor induces Cl− influx, leading to neuronal hyperpolarization, suppression of afferent stimuli transmission, and consequently producing sedative-hypnotic effects.12 Unlike mature neurons, neural precursor cells express Na⁺/K⁺/2Cl− cotransporter protein 1 (NKCC1) on their membranes, which maintains elevated intracellular Cl− levels. Consequently, GABAA receptor activation induces Cl− efflux, resulting in cellular membrane depolarization.61 This depolarizing effect of GABAA receptor activation plays a crucial role in early neurodevelopment. It contributes to maintaining the NSCs pool, supporting their proliferation and differentiation, and facilitating the migration of newborn neurons.62–65 This regulatory process is closely linked to intracellular Ca2⁺ transients. GABAA receptor activation-induced depolarization opens VDCC, triggering extracellular Ca2⁺ influx. This influx regulates neural precursor cell proliferation and neuronal differentiation.64,66 However, our study demonstrated that L-type VDCC are not involved in propofol-induced intracellular Ca2⁺ accumulation. The increase in cytoplasmic Ca2⁺ concentration is not dependent on extracellular Ca2⁺ influx but is primarily due to Ca2⁺ release from the ER via IP3R activation. Although IP3R inhibition significantly attenuated propofol-induced cytoplasmic Ca2⁺ increases, it did not fully abolish the effect, as Ca2⁺ levels remained significantly elevated compared to the control group, suggesting other Ca2⁺ channels may also be involved. Previous studies have suggested that RyR on the ER may be one of the targets of propofol.41,67 In addition to propofol, many inhalational anesthetics, such as sevoflurane and isoflurane, may also induce the release of Ca2⁺ from the ER by activating IP3R or RyR, leading to elevated intracellular Ca2⁺ levels in neural cells.59,68–71 However, the precise pathway through which propofol triggers RyR activation remains to be elucidated.
The findings of the present study suggest that GABAA receptor activation may act upstream of IP3R-mediated Ca2⁺ release. While the exact coupling mechanism remains to be fully elucidated, the complete blockade of Ca2⁺ elevation by GABAA receptor inhibition highlights its primary role in propofol-induced Ca2⁺ signaling in HT22 cells. Admittedly, as the direct interaction between GABAA receptor and IP3R was not explicitly validated in this study, our findings currently serve as a preliminary mechanistic hypothesis. Beyond Ca2⁺ signaling mechanisms, it is essential to recognize that propofol-induced neurotoxicity is a complex, multifactorial process involving several interconnected pathways. Neuroinflammatory responses, characterized by microglial activation and the release of pro-inflammatory cytokines, represent another critical mechanism of propofol-induced injury.13 Other regulatory layers, such as altered mitochondrial fission dynamics and epigenetic modulations,44,72 further illustrate the multifaceted nature of this neurotoxicity.
This study has several limitations. First, although we observed that propofol-induced calcium overload reduces HT22 cell viability, the specific modes of cell death (eg, apoptosis, necrosis, or autophagy) were not investigated. Because our conclusions are based solely on CCK-8 assay data, we cannot determine whether the reduced viability reflects true cell death or inhibition of cell proliferation. Further studies are needed to clarify the precise mechanisms underlying propofol-induced cytotoxicity. Second, our investigation into the mechanisms underlying propofol-induced alterations in intracellular Ca2⁺ homeostasis within HT22 cells is incomplete. Although our experiments demonstrated that inhibiting IP3R significantly reduced propofol-induced increases in cytoplasmic Ca2⁺ concentration, this effect was not completely eliminated, suggesting the involvement of other Ca2⁺ channels. Furthermore, the specific downstream mechanism by which propofol induces neurotoxicity through disruption of intracellular Ca2⁺ homeostasis remains unknown. As a second messenger, Ca2⁺ regulates numerous downstream signaling factors and interact with other pathways, together forming the intracellular Ca2⁺ signaling network that governs cellular activities. Finally, the inherent limitations of employing the HT22 cell line as the primary model must be acknowledged. As an immortalized mouse hippocampal neuronal line, HT22 cells may differ from primary neural progenitor cells in the developing brain, particularly regarding Ca2⁺ signaling regulation, receptor expression profiles, and metabolic states. These discrepancies may constrain the extent to which our findings can be extrapolated to simulate complex “developmental neurotoxicity” in vivo. Therefore, while this study provides significant mechanistic insights, the conclusions should be interpreted with caution.
To address the limitations of this study, future research should involve further validation using primary neural progenitor cells in combination with in vivo animal models. Additionally, multidimensional experimental approaches should be employed to investigate the interference of propofol with intracellular Ca2⁺ signaling mechanisms at multiple levels. These include potentially involved Ca2⁺ channel receptors, alterations in intracellular Ca2⁺ dynamics, downstream transcription factors related to Ca2⁺ signaling, and the target genes they regulate. Understanding these mechanisms of propofol-induced neurotoxicity is crucial for advancing the safe use of anesthetics, particularly during critical periods of neural development.
Conclusions
Propofol induces cytotoxicity in HT22 cells in a concentration- or time-dependent manner. Notably, cytotoxicity at 100 μM propofol was observed only after 24 hours of exposure, whereas 200 μM propofol produced rapid cytotoxicity. This rapid toxicity is mediated by activation of GABAA receptor and IP3R, which triggers the ER Ca2⁺ release and elevating intracellular Ca2⁺ concentration.
Abbreviations
GABAA, γ-aminobutyric acid type A; Ca2+, Calcium; VDCC, Voltage dependent Ca2+ channel; IP3R, Inositol-1,4,5-triphosphate receptor; RyR, Ryanodine receptor; ER, Endoplasmic reticulum; ICU, Intensive care unit; FDA, Food and Drug Administration; ATP, Adenosine triphosphate; NSCs, Neural stem cells; ROIs, Regions of interest; SERCA, Sarcoplasmic/ER Ca2+ ATPase; NKCC1, Na⁺/K⁺/2Cl− cotransporter protein 1; IP3, Inositol 1,4,5-trisphosphate; BGS, Brain growth spurt; year, Mitochondrial permeability transition pore.
Acknowledgments
We would like to extend our sincere gratitude to all the authors who contributed to this research, as well as to the staff of the Central Laboratory at the First Affiliated Hospital of Jinan University, for their invaluable support and assistance.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (81503167); Science and Technology Projects in Guangzhou (2023A03J0619); Foreign Expert Project of the Ministry of Science and Technology of China (DL2023199003L); National Innovation and Entrepreneurial Training Program for Undergraduate Students (202310559035 and 801677).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sun L. Early childhood general anaesthesia exposure and neurocognitive development. Brit J Anaesth. 2010;105:I61–16. doi:10.1093/bja/aeq302
2. Ikonomidou C, Bosch F, Miksa M, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283(5398):70–74. doi:10.1126/science.283.5398.70
3. Raper J, De Biasio JC, Murphy KL, Alvarado MC, Baxter MG. Persistent alteration in behavioural reactivity to a mild social stressor in rhesus monkeys repeatedly exposed to sevoflurane in infancy. Brit J Anaesth. 2018;120(4):761–767. doi:10.1016/j.bja.2018.01.014
4. Song SY, Meng XW, Xia Z, et al. Cognitive impairment and transcriptomic profile in hippocampus of young mice after multiple neonatal exposures to sevoflurane. Aging (Albany NY). 2019;11(19):8386–8417. doi:10.18632/aging.102326
5. Fehr T, Janssen WGM, Park J, Baxter MG. Neonatal exposures to sevoflurane in rhesus monkeys alter synaptic ultrastructure in later life. iScience. 2022;25(12):105685. doi:10.1016/j.isci.2022.105685
6. Diana P, Joksimovic SM, Faisant A, Jevtovic-Todorovic V. Early exposure to general anesthesia impairs social and emotional development in rats. Mol Neurobiol. 2020;57(1):41–50. doi:10.1007/s12035-019-01755-x
7. Wilder RT, Flick RP, Sprung J, et al. Early Exposure to Anesthesia and Learning Disabilities in a Population-based Birth Cohort. Anesthesiology. 2009;110(4):796–804. doi:10.1097/01.anes.0000344728.34332.5d
8. Reighard C, Junaid S, Jackson WM, et al. Anesthetic Exposure During Childhood and Neurodevelopmental Outcomes A Systematic Review and Meta-analysis. JAMA Network Open. 2022;5(6):17427. doi:10.1001/jamanetworkopen.2022.17427
9. Flick RP, Katusic SK, Colligan RC, et al. Cognitive and Behavioral Outcomes After Early Exposure to Anesthesia and Surgery. Pediatrics. 2011;128(5):E1053–E1061. doi:10.1542/peds.2011-0351
10. Song JY, Cha HR, Lee SW, Ha EK, Kim JH, Han MY. Association Between Receipt of General Anesthesia During Childhood and Attention Deficit Hyperactive Disorder and Neurodevelopment. Article. J Korean Med Sci. 2023;38(6):e42. doi:10.3346/jkms.2023.38.e42
11. Xin A, Grobler A, Bell G, et al. Neurodevelopmental Outcomes after Multiple General Anesthetic Exposures before 5 Years of Age: a Cohort Study. Anesthesiology. 2025;142(2):308–319. doi:10.1097/aln.0000000000005293
12. Chidambaran V, Costandi A, D’Mello A. Propofol: a Review of its Role in Pediatric Anesthesia and Sedation. Cns Drugs. 2015;29(7):543–563. doi:10.1007/s40263-015-0259-6
13. Bosnjak ZJ, Logan S, Liu YA, Bai XW. Recent Insights Into Molecular Mechanisms of Propofol-Induced Developmental Neurotoxicity: implications for the Protective Strategies. Anesth Analg. 2016;123(5):1286–1296. doi:10.1213/Ane.0000000000001544
14. Creeley C, Dikranian K, Dissen G, Martin L, Olney J, Brambrink A. Propofol-induced apoptosis of neurones and oligodendrocytes in fetal and neonatal rhesus macaque brain. Brit J Anaesth. 2013;110:29–38. doi:10.1093/bja/aet173
15. Xiong M, Li J, Alhashem H, et al. Propofol Exposure in Pregnant Rats Induces Neurotoxicity and Persistent Learning Deficit in the Offspring. Brain Sci. 2014;4(2):356–375. doi:10.3390/brainsci4020356
16. Yu D, Xiao R, Huang J, et al. Neonatal exposure to propofol affects interneuron development in the piriform cortex and causes neurobehavioral deficits in adult mice. Psychopharmacology. 2019;236(2):657–670. doi:10.1007/s00213-018-5092-4
17. Kahraman S, Zup SL, McCarthy MM, Fiskum G. GABAergic Mechanism of Propofol Toxicity in Immature Neurons. J Neurosurg Anesth. 2008;20(4):233–240. doi:10.1097/ANA.0b013e31817ec34d
18. Zhong YL, Liang YB, Chen J, et al. Propofol inhibits proliferation and induces neuroapoptosis of hippocampal neurons via downregulation of NF-κB p65 and Bcl-2 and upregulation of caspase-3. Cell Biochem Funct. 2014;32(8):720–729. doi:10.1002/cbf.3077
19. Pearn ML, Schilling JM, Jian M, et al. Inhibition of RhoA reduces propofol-mediated growth cone collapse, axonal transport impairment, loss of synaptic connectivity, and behavioural deficits. Brit J Anaesth. 2018;120(4):745–760. doi:10.1016/j.bja.2017.12.033
20. Xian F, Li QF, Chen ZP. Overexpression of phosphoprotein enriched in astrocytes 15 reverses the damage induced by propofol in hippocampal neurons. Mol Med Rep. 2019;20(2):1583–1592. doi:10.3892/mmr.2019.10412
21. Kuczkowski KM. Central & peripheral nervous system - The safety of anaesthetics in pregnant women. Expert Opin Drug Saf. 2006;5(2):251–264. doi:10.1517/14740338.5.2.251
22. Li Y, Zhang J. Propofol-Induced Developmental Neurotoxicity: from Mechanisms to Therapeutic Strategies. Acs Chem Neurosci. 2023;14(6):1017–1032. doi:10.1021/acschemneuro.2c00755
23. Robbins LS, Blanchard CT, Biasini FJ, et al. General anesthesia for cesarean delivery and childhood neurodevelopmental and perinatal outcomes: a secondary analysis of a randomized controlled trial. Int J Obstet Anesth. 2021;45:34–40. doi:10.1016/j.ijoa.2020.08.007
24. Meyer P, Langlois C, Soëte S, et al. Unexpected neurological sequelae following propofol anesthesia in infants: three case reports. Brain Dev. 2010;32(10):872–878. doi:10.1016/j.braindev.2009.11.011
25. Millar K, Bowman AW, Burns D, et al. Children’s cognitive recovery after day-case general anesthesia: a randomized trial of propofol or isoflurane for dental procedures. Pediatr Anesth. 2014;24(2):201–207. doi:10.1111/pan.12316
26. Bai YF, Li WJ, Ji YW, An LX, Zhang L, Li JF. Sevoflurane induces neurotoxic effects on developing neurons through the WNK1/NKCC1/Ca/Drp-1 signalling pathway. Clin Exp Pharmacol. 2023;50(5):393–402. doi:10.1111/1440-1681.13755
27. Pehlivan VF, Pehlivan B, Duran E, et al. Propofol and Thiopental Disrupt Amino Acid and Carnitine Metabolism in HEK-293 Cells: insights into Mitochondrial Toxicity. Harran Üniversitesi Tip Fakültesi Dergisi. 2025;22(4):624–634. doi:10.35440/hutfd.1719754
28. Pehlivan VF, Pehlivan B, Duran E, Koyuncu I, Erdogdu H. Anesthetic-Induced Disruption of Amino Acid and Carnitine Profiles: a Metabolomic Comparison of Propofol and Thiopental in Hepatocytes. Pharmaceuticals. 2025;18(8):1221. doi:10.3390/ph18081221
29. Zhou H, Dai Z, Li J, et al. TMBIM6 prevents VDAC1 multimerization and improves mitochondrial quality control to reduce sepsis-related myocardial injury. Metabolism. 2023;140:155383. doi:10.1016/j.metabol.2022.155383
30. Truong TT, Singh AA, Bang NV, et al. Mitochondria-Associated Membrane Dysfunction in Neurodegeneration and Its Effects on Lipid Metabolism, Calcium Signaling, and Cell Fate. Membranes. 2025;15(9):263. doi:10.3390/membranes15090263
31. Patergnani S, Danese A, Bouhamida E, et al. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int J Mol Sci. 2020;21(21):8323. doi:10.3390/ijms21218323
32. Brini M, Calì T, Ottolini D, Carafoli E. Neuronal calcium signaling: function and dysfunction. Cell Mol Life Sci. 2014;71(15):2787–2814. doi:10.1007/s00018-013-1550-7
33. Raffaello A, Mammucari C, Gherardi G, Rizzuto R. Calcium at the Center of Cell Signaling: interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem Sci. 2016;41(12):1035–1049. doi:10.1016/j.tibs.2016.09.001
34. Giorgi C, Marchi S, Pinton P. Publisher Correction: the machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol. 2018;19(11):746. doi:10.1038/s41580-018-0066-2
35. Parone PA, James D, Martinou JC. Mitochondria: regulating the inevitable. Biochimie. 2002;84(2–3):105–111. doi:10.1016/s0300-9084(02)01380-9
36. Marchi S, Patergnani S, Missiroli S, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69:62–72. doi:10.1016/j.ceca.2017.05.003
37. Zhao Q, Lu D, Wang J, et al. Calcium dysregulation mediates mitochondrial and neurite outgrowth abnormalities in SOD2 deficient embryonic cerebral cortical neurons. Cell Death Differ. 2019;26(9):1600–1614. doi:10.1038/s41418-018-0230-4
38. Tao T, Zhao Z, Hao L, Gu M, Chen L, Tang J. Propofol promotes proliferation of cultured adult rat hippocampal neural stem cells. J Neurosurg Anesthesiol. 2013;25(3):299–305. doi:10.1097/ANA.0b013e31828baa93
39. Lu J, Yao XQ, Luo X, et al. Monosialoganglioside 1 may alleviate neurotoxicity induced by propofol combined with remifentanil in neural stem cells. Neural Regen Res. 2017;12(6):945–952. doi:10.4103/1673-5374.208589
40. Young SZ, Platel JC, Nielsen JV, Jensen NA, Bordey A. GABA(A) Increases Calcium in Subventricular Zone Astrocyte-Like Cells Through L- and T-Type Voltage-Gated Calcium Channels. Front Cell Neurosci. 2010;4:8. doi:10.3389/fncel.2010.00008
41. Qiao H, Li Y, Xu Z, et al. Propofol Affects Neurodegeneration and Neurogenesis by Regulation of AutophagyviaEffects on Intracellular Calcium Homeostasis. Anesthesiology. 2017;127(3):490–501. doi:10.1097/aln.0000000000001730
42. Woll KA, Van Petegem F. Calcium-Release Channels: structure and Function of Ip Receptors and Ryanodine Receptors. Physiol Rev. 2022;102(1):209–268. doi:10.1152/physrev.00033.2020
43. Sall Jeffrey W, Stratmann G, Leong J, Woodward E, Bickler PE. Propofol at Clinically Relevant Concentrations Increases Neuronal Differentiation But Is Not Toxic to Hippocampal Neural Precursor Cells In Vitro. Anesthesiology. 2012;117(5):1080–1090. doi:10.1097/ALN.0b013e31826f8d86
44. Zhang W, Liu Q, Zhu H, et al. Propofol induces the apoptosis of neural stem cells via microRNA-9-5p / chemokine CXC receptor 4 signaling pathway. Bioengineered. 2022;13(1):1062–1072. doi:10.1080/21655979.2021.2017590
45. Liang C, Sun M, Zhong J, Miao C, Han X. The Role of Pink1-Mediated Mitochondrial Pathway in Propofol-Induced Developmental Neurotoxicity. Neurochem Res. 2021;46(9):2226–2237. doi:10.1007/s11064-021-03359-1
46. Liu F, Rainosek SW, Sadovova N, et al. Protective effect of acetyl-L-carnitine on propofol-induced toxicity in embryonic neural stem cells. Neurotoxicology. 2014;42:49–57. doi:10.1016/j.neuro.2014.03.011
47. Hu Q, Huang L, Zhao C, et al. Ca(2+)-PKCalpha-ERK1/2 signaling pathway is involved in the suppressive effect of propofol on proliferation of neural stem cells from the neonatal rat hippocampus. Brain Res Bull. 2019;149:148–155. doi:10.1016/j.brainresbull.2019.04.005
48. Vutskits L, Gascon E, Tassonyi E. Kiss Jozsef Z. Clinically Relevant Concentrations of Propofol but Not Midazolam Alter In Vitro Dendritic Development of Isolated γ-Aminobutyric Acid-positive Interneurons. Anesthesiology. 2005;102(5):970–976. doi:10.1097/00000542-200505000-00016
49. Krajcova A, Waldauf P, Andel M, Duska F. Propofol infusion syndrome: a structured review of experimental studies and 153 published case reports. Crit Care. 2015;19:398. doi:10.1186/s13054-015-1112-5
50. Van Hese L, Theys T, Absalom AR, Rex S, Cuypers E. Comparison of predicted and real propofol and remifentanil concentrations in plasma and brain tissue during target-controlled infusion: a prospective observational study. Anaesthesia. 2020;75(12):1626–1634. doi:10.1111/anae.15125
51. Sukumaran P, Da Conceicao VN, Sun YY, et al. Calcium Signaling Regulates Autophagy and Apoptosis. Cells-Basel. 2021;10(8):2125. doi:10.3390/cells10082125
52. Slikker Jr W, Liu F, Rainosek SW, et al. Ketamine-Induced Toxicity in Neurons Differentiated from Neural Stem Cells. Mol Neurobiol. 2015;52(2):959–969. doi:10.1007/s12035-015-9248-5
53. Sinner B, Friedrich O, Zink W, Zausig Y, Graf BM. The toxic effects of s(+)-ketamine on differentiating neurons in vitro as a consequence of suppressed neuronal Ca2+ oscillations. Anesth Analg. 2011;113(5):1161–1169. doi:10.1213/ANE.0b013e31822747df
54. Zeng S, Zhu R, Wang Y, et al. Role of GABA(A) receptor depolarization-mediated VGCC activation in sevoflurane-induced cognitive impairment in neonatal mice. Front Cell Neurosci. 2022;16:964227. doi:10.3389/fncel.2022.964227
55. Zhao Y, Liang G, Chen Q, et al. Anesthetic-induced neurodegeneration mediated via inositol 1,4,5-trisphosphate receptors. J Pharmacol Exp Ther. 2010;333(1):14–22. doi:10.1124/jpet.109.161562
56. Alberti P, Semperboni S, Cavaletti G, Scuteri A. Neurons: the Interplay between Cytoskeleton, Ion Channels/Transporters and Mitochondria. Cells-Basel. 2022;11(16):2499. doi:10.3390/cells11162499
57. Kapur N, Mignery GA, Banach K. Cell cycle-dependent calcium oscillations in mouse embryonic stem cells. Am J Physiol Cell Physiol. 2007;292(4):C1510–8. doi:10.1152/ajpcell.00181.2006
58. Capiod T. Cell proliferation, calcium influx and calcium channels. Biochimie. 2011;93(12):2075–2079. doi:10.1016/j.biochi.2011.07.015
59. Zhao X, Yang Z, Liang G, et al. Dual effects of isoflurane on proliferation, differentiation, and survival in human neuroprogenitor cells. Anesthesiology. 2013;118(3):537–549. doi:10.1097/ALN.0b013e3182833fae
60. Zhao S, Zhu Y, Xue R, Li Y, Lu H, Mi W. Effect of midazolam on the proliferation of neural stem cells isolated from rat hippocampus. Neural Regen Res. 2012;7(19):1475–1482. doi:10.3969/j.issn.1673-5374.2012.19.005
61. Kolbaev SN, Mohapatra N, Chen RQ, et al. NKCC-1 mediated Cl uptake in immature CA3 pyramidal neurons is sufficient to compensate phasic GABAergic inputs. Sci Rep. 2020;10(1):18399. doi:10.1038/s41598-020-75382-1
62. Song J, Zhong C, Bonaguidi MA, et al. Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature. 2012;489(7414):150–154. doi:10.1038/nature11306
63. Bao H, Asrican B, Li W, et al. Long-Range GABAergic Inputs Regulate Neural Stem Cell Quiescence and Control Adult Hippocampal Neurogenesis. Cell Stem Cell. 2017;21(5):604–617e5. doi:10.1016/j.stem.2017.10.003
64. Tozuka Y, Fukuda S, Namba T, Seki T, Hisatsune T. GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron. 2005;47(6):803–815. doi:10.1016/j.neuron.2005.08.023
65. Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002;3(9):728–739. doi:10.1038/nrn920
66. Xiuxin Liu QW, Haydar TF, Bordey A. Nonsynaptic GABA signaling in postnatal subventricular zone controls GFAP-expressing progenitor proliferation. Nat Neurosci. 2005;8(9):1179–1187. doi:10.1038/nn1522
67. Ren G, Zhou Y, Liang G, et al. General Anesthetics Regulate Autophagy via Modulating the Inositol 1,4,5-Trisphosphate Receptor: implications for Dual Effects of Cytoprotection and Cytotoxicity. Sci Rep. 2017;7(1):12378. doi:10.1038/s41598-017-11607-0
68. Wang QJ, Li KZ, Yao SL, Li ZH, Liu SS. Different effects of isoflurane and sevoflurane on cytotoxicity in primary cortical neurons of rats. Chinese Med J. 2008;121(4):341–346. doi:10.1097/00029330-200802020-00012
69. Wang H, Dong Y, Zhang J, et al. Isoflurane induces endoplasmic reticulum stress and caspase activation through ryanodine receptors. Brit J Anaesth. 2014;113(4):695–707. doi:10.1093/bja/aeu053
70. Chai D, Jiang H, Li Q. Isoflurane neurotoxicity involves activation of hypoxia inducible factor-1alpha via intracellular calcium in neonatal rodents. Brain Res. 2016;1653:39–50. doi:10.1016/j.brainres.2016.10.014
71. Zhang Q, Li YA, Wang XP, et al. Sevoflurane exposure causes neuronal apoptosis and cognitive dysfunction by inducing ER stress via activation of the inositol 1, 4, 5-trisphosphate receptor. Front Aging Neurosci. 2022;14:990679. doi:10.3389/fnagi.2022.990679
72. Twaroski DM, Yan Y, Zaja I, Clark E, Bosnjak ZJ, Bai X. Altered Mitochondrial Dynamics Contributes to Propofol-induced Cell Death in Human Stem Cell-derived Neurons. Anesthesiology. 2015;123(5):1067–1083. doi:10.1097/ALN.0000000000000857
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Comparative Study About Different Doses of Remimazolam in Short Laparoscopic Surgery: A Randomized Controlled Double-Blind Trial
Luo L, Jiang J, Zhang M, Guo Z, Zhang X, Wang F, Yang F
Therapeutics and Clinical Risk Management 2023, 19:829-837
Published Date: 20 October 2023
Effect of Remimazolam on Induction and Maintenance of General Anesthesia in Kidney Transplant Patients
Chen L, Qin W, Wu J, Zhao G, Jiang X, Li M, Huang Z, Du X
International Journal of General Medicine 2024, 17:2455-2463
Published Date: 28 May 2024
Ciprofol in Children Undergoing Adenoidectomy and Adenotonsillectomy: A Retrospective Cohort Study
Zeng C, Li L, Wang M, Xiong J, Pang W, Yu H, He J, Wang X, Chen Y, Sun Y
Drug Design, Development and Therapy 2024, 18:4017-4027
Published Date: 6 September 2024
The Impact of Bispectral Index Monitoring on Outcomes in Spinal Cord Stimulation for Chronic Disorders of Consciousness
Chen X, Qin X, Zhuang Y, Li Z, Liang Z, Zhang H, Yao L, Li X, He J, Guo X
Therapeutics and Clinical Risk Management 2024, 20:677-687
Published Date: 27 September 2024
Clinical Use of Fospropofol Disodium: Evaluation of Pharmacology, Safety, and Efficacy
Zou C, Chen H, Qiu Y
Drug Design, Development and Therapy 2025, 19:5453-5460
Published Date: 26 June 2025