")
Back to Journals » International Journal of General Medicine » Volume 15
Hereditary Transthyretin Amyloidosis with Polyneuropathy: Monitoring and Management
Authors Vélez-Santamaría V, Nedkova-Hristova V, Morales de la Prida M , Casasnovas C
Received 21 October 2022
Accepted for publication 12 December 2022
Published 20 December 2022 Volume 2022:15 Pages 8677—8684
DOI https://doi.org/10.2147/IJGM.S338430
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Valentina Vélez-Santamaría,1– 3 Velina Nedkova-Hristova,1 Moisés Morales de la Prida,1 Carlos Casasnovas1– 3
1Neuromuscular Unit, Neurology Department, Bellvitge University Hospital, Barcelona, Spain; 2Neurometabolic Diseases Laboratory, Bellvitge Biomedical Research Institute (IDIBELL), Barcelona, Spain; 3Centre for Biomedical Research on Rare Diseases (CIBERER), Instituto de Salud Carlos III, Madrid, Spain
Correspondence: Valentina Vélez-Santamaría, Neurometabolic Diseases Laboratory, IDIBELL, Hospital Duran i Reynals, Gran Via 199, 08908 L’Hospitalet de Llobregat, Barcelona, Spain, Tel +34 932607343, Fax +34 932607414, Email [email protected]
Abstract: Our aim in this review is to discuss current treatments and investigational products and their effect on patients with hereditary transthyretin amyloidosis with polyneuropathy (ATTRv-PN) and provide suggestions for monitoring disease progression and treatment efficacy.
Keywords: polyneuropathy, transthyretin, amyloid, transthyretin stabilizers, gene silencers, CRISPR/Cas9
Introduction
Hereditary amyloidogenic transthyretin amyloidosis with polyneuropathy (ATTRv-PN) is an adult-onset, autosomal dominant disease produced by mutations in the TTR gene, which encodes the transthyretin (TTR) protein.1 ATTRv-PN was thought to be endemic to Portugal,2 Sweden,3 and Japan,4 however, an expanding number of cases, frequently adult-onset and sporadic, has been observed globally. According to a study from 2018,5 there are 10,186 cases of ATTRv-PN worldwide, with a range of 5526–38,468 cases. With greater clinical knowledge of the disease and an increased use of genetic testing, especially in areas where it is not endemic, the incidence of ATTRv-PN is predicted to increase.5
TTR, also known as prealbumin, is a homo-tetrameric carrier protein that transports thyroid hormones (thyroxine) in the plasma and cerebrospinal fluid. It is also involved in the transport of retinol (vitamin A) by associating with retinol-binding protein.6 The tetramer conformation is thermodynamically or kinetically destabilized by mutations in the TTR gene, which cause the tetramer to split into erratic monomers that misfold and clump together to form mature amyloid fibrils that lead to tissue injury by microangiopathy, cytotoxicity or direct compression.7 Moreover, fibrillar (monomers and oligomers) TTR might induce tissue damage through increased cytotoxicity.8 The peripheral nervous system, heart, and kidneys are the most commonly damaged organ systems.
A point mutation in the TTR gene, a four-exon gene located on chromosome 18, that results in the substitution of methionine for valine at position 30 of the mature protein causes the majority of cases of ATTRv-PN (standardized nomenclature now begins at the methionine initiation codon and the mutation is technically known as p.ATTRVal50Met, although Val30Met continues to be widely used in the literature).9 However, over 130 mutations have been identified in this gene, the majority of which are pathogenic.10 ATTRv-PN manifests commonly as a progressive peripheral neuropathy with a predominantly sensory involvement. Autonomic neuropathy and gastrointestinal symptoms are prominent in Val30Met early-onset, whereas motor neuropathy and cardiac involvement were more common in late-onset patients.11 Furthermore, characteristics of amyloid deposits are distinct between Val30Met early and late-onset patients.12 The penetrance of mutations in the TTR gene is highly variable. In endemic regions, this can reach 100%, but outside these regions, incomplete penetrance is more common.13
Prior to 1990, the available treatments for ATTRv-PN were to manage symptoms and the condition was traditionally considered incurable. However, in the past three decades, various disease-modifying therapies have been developed. Current treatments and investigational therapies will be described and discussed here, and suggestions for monitoring disease progression and treatment effectiveness will be provided.
To treat ATTRv-PN, the main strategies focus on one of the following (Figure 1): (1) suppression of amyloidogenic TTR synthesis (TTR silencers) and liver transplantation, (2) stabilization of TTR tetramers to prevent misfolding (TTR stabilizers), and (3) removal of existing TTR amyloid fibrils (TTR disrupters).
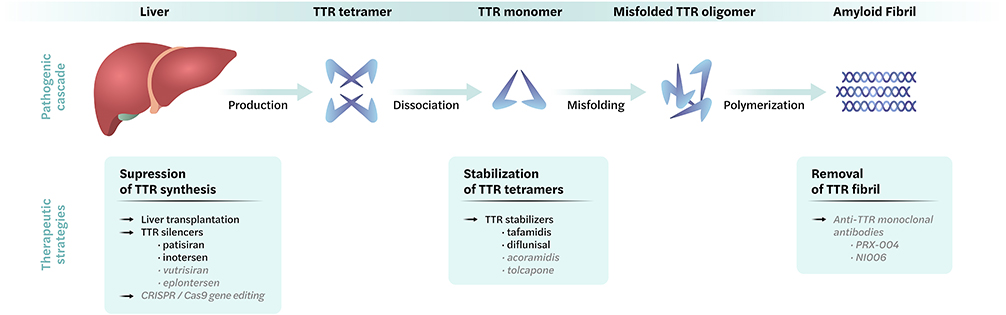 |
Figure 1 Overview of therapeutic strategies in hereditary transthyretin amyloidosis with polyneuropathy. Top: Pathogenic process, bottom: therapeutic approaches available in clinic (black) or clinical trials (grey). |
Available Disease-Modifying Therapies
Liver Transplantation
The first liver transplantation for ATTRv-PN was performed in Sweden in 1993.14 Since then, thousands of patients have undergone this treatment to replace the main source of mutant TTR for a more stable wild-type variant and slow the progression of the disease, since wild type TTR is less amyloidogenic. It is well acknowledged that transplanting patients with ATTRv-PN dramatically lengthens their life expectancy;15,16 however, neuropathy and cardiomyopathy,17 as well as ocular and central nervous system amyloidosis, can progress after liver transplantation.18,19
A long follow-up (more than 20 years) to collect outcome data and predictors indicated that late onset (after the age of 50 years) in male patients as well as non-Val30Met patients showed a less post-transplant benefit. On the other hand, patients with < 7 years since the debut of disease had a better prognosis.16 Nowadays, with the implementation of effective pharmacotherapy, liver transplantation for ATTRv-PN is considered exceptional.
TTR Silencers
Patisiran
Small interfering RNAs (siRNAs) are non-coding, double-stranded RNA molecules that trigger RNA-induced silencing complexes that bind to their complementary mRNA. By doing this, translation is hindered and protein synthesis is stopped.20 Patisiran (Onpattro) is a first-generation siRNA that was approved in 2018 for the treatment of ATTRv-PN, based on positive results in the Phase III trial APOLLO A that involved administering intravenous patisiran (0.3 mg/kg) or placebo once every three weeks to 225 patients with ATTRv-PN.21 The primary endpoint was the change from baseline at 18 months in the modified Neuropathy Impairment Score + 7 (mNIS + 7),22 a variation of the Neuropathy Impairment Score (NIS) that includes quantitative sensory and autonomic items and nerve conduction studies, achieving a greater detection of disease progression.23–25 The least-squares mean conversion was −6.0 versus 28.0 (p<0.001) and there were no differences between the subgroups with respect to age or variant. The majority of adverse events were mild or moderate, while the incidence of severe and serious adverse effects was similar in both arms. Other clinical assessments, including the Norfolk Quality of Life–Diabetic Neuropathy (Norfolk QOL-DN) questionnaire, the 10-meter walk test (10-MWT), and the COMPASS 31 autonomic score, also showed improvements from baseline.21 The open-label extension (OLE) trial included 211 subjects who had finished the phase III APOLLO study or Phase II OLE parent studies, and demonstrated a sustained improvement at 12 months.26
Remarkably, mortality and disability remained higher in the patients who had received patisiran at a late stage of the disease, highlighting the importance of early recognition and treatment of ATTRv-PN.
Inotersen
Inotersen (Tegsedi) is an antisense oligonucleotide designed to bind to TTR mRNA, triggering its degradation through nuclear ribonuclease H1 (RNaseH1) and resulting in a decreased protein expression. It was approved in 2018 for the treatment of ATTRv-PN based on a phase III trial (NEURO-TTR) that included 172 patients who had been randomized to receive intravenous inotersen (300 mg) weekly or placebo.27 The primary goals of improvements in the NIS and quality of life measurements were achieved. The difference in the least-squares mean change from baseline to week 66 between the two groups was −19.7 points.
The most frequent serious adverse events in the inotersen group were glomerulonephritis in three patients (3%) and severe thrombocytopenia in four patients (4%), with one caused by an intracranial hemorrhage associated with profound thrombocytopenia. Four more deaths occurred in the treated arm (vs 0 deaths in the placebo arm) that were related to disease progression. Thereafter, treatment with inotersen has required ongoing laboratory monitoring of these adverse effects.27
TTR Stabilizers
Tafamidis
Tafamidis is a selective TTR stabilizer that binds to one of the thyroxine-binding sites at the TTR tetramer, therefore inhibiting the first dissociation step of the amyloidogenic cascade. A phase III clinical trial with 125 Val30Met-related ATTRv-PN patients randomized to tafamidis (20 mg daily) or placebo for 18 months did not show statistically significant differences in the two primary outcome measures, the Neuropathy Impairment Score in the Lower Limbs (NIS-LL) and the Norfolk QOL-DN total score, in the intent-to-treat (ITT) population. However, in an “as treated” analysis, significantly more patients on tafamidis than those on placebo were NIS-LL responders (60.0% vs 38.1%; p = 0.041), with significant differences in most secondary endpoints, including changes in neurological function, nutritional status, and TTR stabilization that favored tafamidis.28 Based on these data, tafamidis was approved for the treatment of ATTR-PN in Europe, Asia and South America; however, it was rejected by the Food and Drug Administration (FDA) of the United States of America. In an observational study, the response to tafamidis in Val30Met Portuguese patients was evaluated after 6 years of treatment, beholding that patients with early disease, specially female patients, presented the best response to treatment (NIS 10).29
The Transthyretin Amyloidosis Cardiomyopathy Clinical Trial (ATTR-ACT) was a phase III study that had enrolled 441 patients with transthyretin amyloidosis with cardiomyopathy (ATTR-CM) who had been randomized to receive 80 mg, 20 mg, or placebo by mouth daily. Patients with ATTR-CM can be further classified into variant (ATTRv-CM) or wild-type (ATTRwt-CM) disease according to the presence of genetic mutations in the TTR gene; in this study both ATTR-CM and ATTRwt-CM patients were included. At 30 months, patients treated with tafamidis demonstrated a 13.4% absolute reduction in all-cause mortality when compared to those on placebo, as well as fewer cardiovascular-related hospitalizations, a lower decline in functional capacity, and a lower decline in quality of life. Based on these favorable results, tafamidis was ratified to treat both ATTRwt-CM and ATTRv-CM.30
Diflunisal
Diflunisal, a non-steroidal anti-inflammatory drug (NSAID), is a salicylic acid–derived medication that was developed as an analgesic. It stabilizes TTR by interfering with the binding site at the dimer–dimer interface of TTR tetramers. A randomized, placebo-controlled, double-blind, multicenter, international study conducted on 130 patients with ATTRv-PN showed a reduction in disease progression (measured as the progression scores on the NIS + 7 nerve tests and the NIS score at 2 years after treatment) in patients who received diflunisal compared to those who received placebo. However, 52% of the patients had discontinued treatment as a result of disease progression and liver transplantation, suggesting that this TTR stabilizer is insufficient to stop rapid progression.31 The study excluded patients who were at an increased risk with NSAID therapy, including those with severe congestive heart failure, renal insufficiency, and ongoing anticoagulation, which are common in this pool of patients in clinical practice.
Strategies Under Development
TTR Silencers
Second-Generation Gene Silencers
Vutrisiran (previously ALN-TTRsc02) is a second-generation RNA interference (RNAi) therapeutic that targets a sequence within the TTR mRNA, inhibiting the formation of TTR. Conjugation of vutrisiran with a triantennary GalNAc ligand allows subcutaneous dosing at a lower frequency of administration compared to patisiran treatment.32 Two Phase III, randomized, open-label studies are currently being conducted to estimate the safety and efficacy of vutrisiran in 164 patients with ATTRv-PN (projected finalization date in May 2024) and 665 patients with ATTRv-CM (estimated completion date in June 2025), who have been randomized to receive either vutrisiran or patisiran.33,34 Based on the Phase I trial with vutrisiran that demonstrated a safe profile in 80 healthy patients,32 the FDA has granted fast-track designation to vutrisiran as a potential therapy for ATTRv-PN.
Eplontersen is a subcutaneously administered single-stranded antisense oligonucleotide with the addition of a receptor ligand protein that allows direct binding to hepatocytes, leading to decreases in the amount of dose and interval of administration (every 4 weeks compared with previous weekly dosing) as well as reductions in adverse effects and dose increment. The estimated primary completion date of the study assessing the safety and efficacy of eplontersen is in July 2024.35
CRISPR/Cas9 Gene Editing
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) gene editing cuts DNA and lets natural DNA repair processes take over, disrupting, deleting, or inserting a specific DNA sequence. With this therapy, the TTR gene can be actually knocked out in those patients with ATTRv-PN after one single administration.
In a phase I study conducted on patients with ATTRv-PN, NTLA-2001, the first systemically delivered CRISPR/Cas9 therapy, was well tolerated and reached a drop in TTR levels.36
TTR Stabilizers
Acoramidis
Acoramidis (previously known as AG10), is a highly selective TTR stabilizer. Is orally bioavailable and was designed to mimic the structure of the protective Thr119Met mutation, which prevents tetramer dissociation by promoting hydrogen bond formation between serine residues.37 In a randomized, double-blind, placebo-controlled, multicenter phase II clinical trial in 49 patients with symptomatic ATTRv-CM and ATTRwt-CM, acoramidis was well tolerated, with no clinically significant safety concerns.38 Moreover, there was a statistically significant increase in the mean serum TTR concentration, a prognostic indicator of survival, in patients treated with acoramidis compared to those on placebo.38 An Open-Label Extension and Safety Monitoring Study with acoramidis in patients with symptomatic ATTRv-CM is being carried out, with the estimated study completion date being in May 2028.39
Tolcapone
Tolcapone, a well-known drug approved for the treatment of Parkinson’s disease, stabilizes TTR tetramers by binding both thyroxine-binding pockets of the TTR tetramer.40 An open-label, phase IIA proof-of-concept study in asymptomatic ATTR amyloidosis patients (ATTRwt, N = 6; Val30Met ATTRv, N = 11) demonstrated TTR stabilization, suggesting a potential role for tolcapone in ATTR amyloidosis.40 Moreover, tolcapone can cross the blood–brain barrier, thus, it could be an option for the treatment of leptomeningeal manifestations of ATTRv amyloidosis.41 A proof-of-concept study that evaluated tolcapone efficacy to stabilize TTR in the cerebrospinal fluid and plasma of leptomeningeal ATTRv patients was finished in 2019,42 but the results are not available yet. Therapy with tolcapone requires extensive monitoring for potential hepatotoxicity; therefore, the indication for treatment with this drug will have to be carefully evaluated.
TTR Disrupters
Monoclonal Antibody Therapy
Monoclonal antibodies can selectively target dissociative monomers, oligomers or TTR aggregates to suppress fibrillogenesis and further aggregation or they can target amyloid deposits to trigger their removal by phagocytic mechanisms, sparing the normal tetrameric TTR.43 In a phase I clinical trial, PRX004, a potential therapeutic monoclonal antibody targeting the TTR epitope comprising residues 89–97, was well-tolerated and safe at all dose levels. In this study, 21 patients received PRX004 intravenously once every 28 days for up to three infusions. All the evaluable patients experienced an improvement or slower progression versus the natural history of disease, at month 9.44 NI006 is another human antibody targeting TTR amyloid deposits that is currently being evaluated in a phase I trial in patients with hereditary or wild-type ATTR-CM.45
Monitoring Response to Therapy
Careful monitoring of the multiple signs of disease progression is necessary to address the challenges associated with the initiation and adjustment of disease-modifying therapies. The monitoring of a patient should begin immediately after diagnosis to establish a baseline assessment. Thereafter, periodic assessments of peripheral neuropathy symptoms, including sensory-motor and autonomic manifestations, as well as the monitoring of ocular and central nervous system involvement should be performed.46 Common symptomatology of peripheral neuropathy includes pain, paresthesia, walking difficulties, balance disorders, and difficulties with fine dexterity. The NIS, combining motor function, sensory function, and tendon reflexes, has been used in clinical trials in patients with ATTRv-PN31,47 and reflects the severity of the neuropathy.48,49 Others composite scales based in NIS as the mentioned mNIS+7 are often avoided in clinical practice due their complexity and time consumption.
The polyneuropathy disability (PND) score, as well as the six-minute walk test (6-MWT) and the timed 10-MWT, evaluates the impact of neuropathy on ambulation. Furthermore, 6MWT and 10MWT are functional exercise-based tests that correlate with the cardio-pulmonary function and provide prognostic information in patients with cardiac involvement.50 Other relevant neurological tests include nerve conduction studies in the four extremities to examine large, myelinated nerve fibers, which are mandatory in mild and moderate neuropathy. Investigations of unmyelinated and small nerve fibers, like the assessment of laser-evoked potentials, temperature quantitative sensory testing or silent period for small fiber sensory neuropathy assessment, are recommended when available.51
Autonomic manifestations are present in 75% of patients,52 therefore, a systematic clinical screening should be performed. The COMPASS 31 questionnaire, provides a broad assessment of the severity and extent of a range of autonomic symptoms, including vasomotor, secretomotor, gastrointestinal, and bladder dysfunction. It has shown efficacy in monitoring longitudinal changes in autonomic function.53 Additional assessments include sudomotor function tests, postural hypotension tests, heart rate variability tests, and the modified body mass index (mBMI), which can be used as an indirect marker of gastrointestinal dysautonomia.52
Finally, the Rasch-built Overall Disability Scale (R-ODS) can be used to assess disability, since it measures the effect of the disease on activities of daily living in patients with peripheral neuropathy.54 Table 1 summarizes the common minimum set of evaluations that should be used to monitor the progression of neuropathy.46,55 The recommended frequency of assessment in general is every 6 or 12 months, depending on the disease course, the severity of the polyneuropathy, and the response to treatment.52
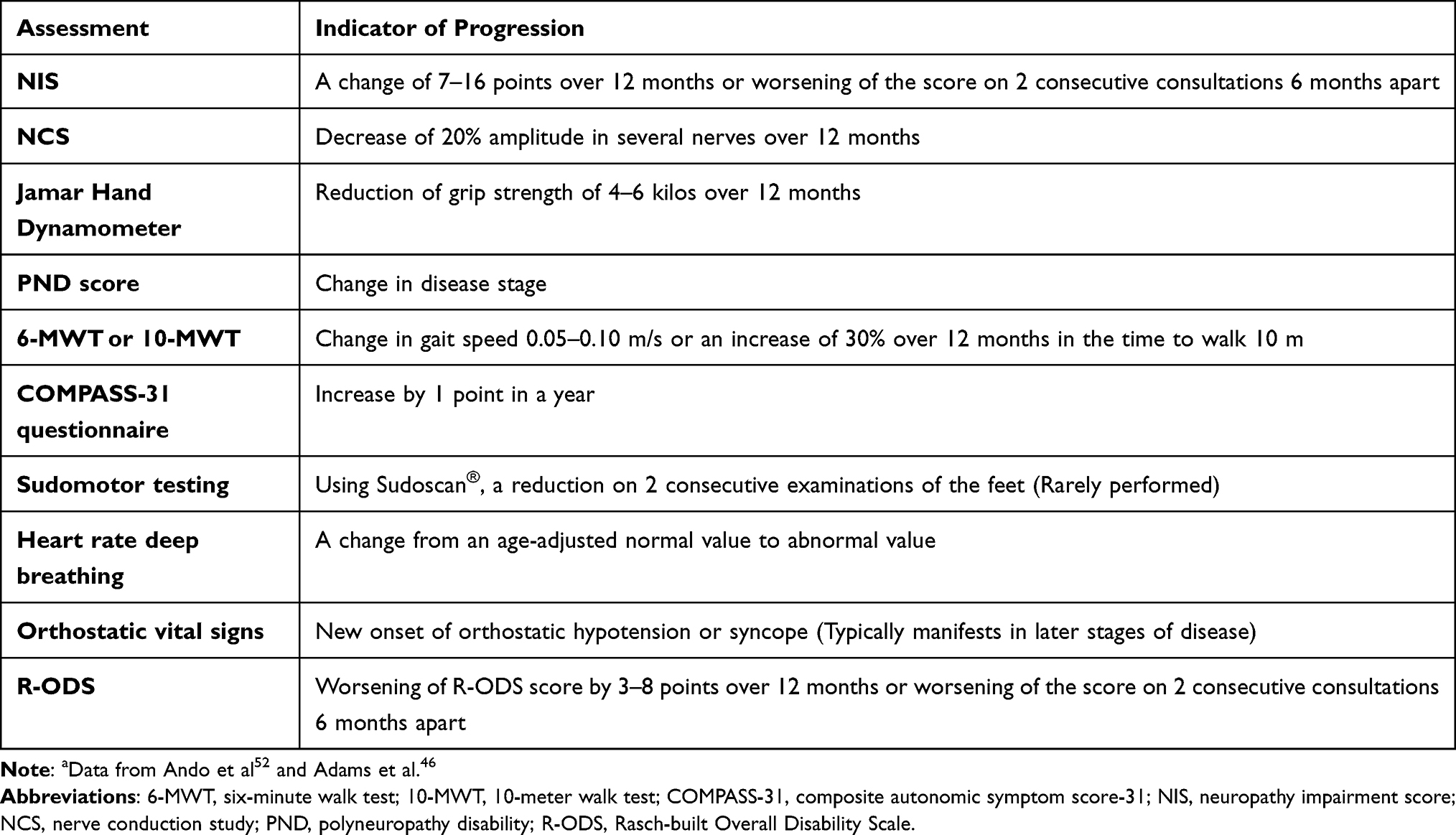 |
Table 1 Recommended Set of Evaluation to Monitor Progression of Neuropathya |
Conclusions
ATTRv-PN is a progressive and lethal disease in which early therapeutic intervention is key for better patient outcomes. Recently approved disease-modifying therapies have changed the course of the disease, highlighting the need for guidance on managing ATTRv-PN. To monitor the disease course, clinicians should undertake detailed assessments of the multiple symptoms and signs of neuropathy at baseline and during follow-up.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15(7):387–404. doi:10.1038/s41582-019-0210-4
2. Sousa A, Coelho T, Barros J, Sequeiros J. Genetic epidemiology of familial amyloidotic polyneuropathy (FAP)-type I in Póvoa do Varzim and Vila do Conde (north of Portugal). Am J Med Genet. 1995;60(6):512–521. doi:10.1002/ajmg.1320600606
3. Sousa A, Andersson R, Drugge U, Holmgren G, Sandgren O. Familial amyloidotic polyneuropathy in Sweden: geographical distribution, age of onset, and prevalence. Hum Hered. 1993;43(5):288–294. doi:10.1159/000154146
4. Kato-Motozaki Y, Ono K, Shima K, et al. Epidemiology of familial amyloid polyneuropathy in Japan: identification of a novel endemic focus. J Neurol Sci. 2008;270(1–2):133–140. doi:10.1016/j.jns.2008.02.019
5. Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2018;57(5):829–837. doi:10.1002/mus.26034
6. Richardson SJ. Cell and molecular biology of transthyretin and thyroid hormones. Int Rev Cytol. 2007;258:137–193. doi:10.1016/S0074-7696(07)58003-4
7. Sekijima Y, Wiseman RL, Matteson J, et al. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121(1):73–85. doi:10.1016/j.cell.2005.01.018
8. Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN. Tissue damage in the amyloidoses: transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci. 2004;101(9):2817–2822. doi:10.1073/pnas.0400062101
9. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 nomenclature guidelines. Amyloid. 2016;23(4):209–213. doi:10.1080/13506129.2016.1257986
10. Rowczenio DM, Noor I, Gillmore JD, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014;35(9):E2403–E2412. doi:10.1002/humu.22619
11. Dispenzieri A, Coelho T, Conceição I, et al. Clinical and genetic profile of patients enrolled in the Transthyretin Amyloidosis Outcomes Survey (THAOS): 14-year update. Orphanet J Rare Dis. 2022;17(1):236. doi:10.1186/s13023-022-02359-w
12. Koike H, Ando Y, Ueda M, et al. Distinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathy. J Neurol Sci. 2009;287(1–2):178–184. doi:10.1016/j.jns.2009.07.028
13. Plante-Bordeneuve V. Genetic study of transthyretin amyloid neuropathies: carrier risks among French and Portuguese families. J Med Genet. 2003;40(11):120e–120 . doi:10.1136/jmg.40.11.e120
14. Holmgren G, Steen L, Suhr O, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341(8853):1113–1116. doi:10.1016/0140-6736(93)93127-M
15. Adams D, Samuel D, Goulon-Goeau C, et al. The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation. Brain. 2000;123(Pt 7):1495–1504. doi:10.1093/brain/123.7.1495
16. Ericzon B-G, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis. Transplantation. 2015;99(9):1847–1854. doi:10.1097/TP.0000000000000574
17. Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277–282. doi:10.1080/13506120701614032
18. Beirão JM, Malheiro J, Lemos C, et al. Impact of liver transplantation on the natural history of oculopathy in Portuguese patients with transthyretin (V30M) amyloidosis. Amyloid. 2015;22(1):31–35. doi:10.3109/13506129.2014.989318
19. Salvi F, Pastorelli F, Plasmati R, et al. Brain microbleeds 12 years after orthotopic liver transplantation in Val30Met amyloidosis. J Stroke Cerebrovasc Dis. 2015;24(6):e149–e151. doi:10.1016/j.jstrokecerebrovasdis.2015.02.015
20. Dana H, Chalbatani GM, Mahmoodzadeh H, et al. Molecular mechanisms and biological functions of siRNA. Int J Biomed Sci. 2017;13(2):48–57.
21. Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. doi:10.1056/NEJMoa1716153
22. Adams D, Suhr OB, Dyck PJ, et al. Trial design and rationale for APOLLO, a phase 3, placebo-controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurol. 2017;17(1):181. doi:10.1186/s12883-017-0948-5
23. Suanprasert N, Berk JL, Benson MD, et al. Retrospective study of a TTR FAP cohort to modify NIS + 7 for therapeutic trials. J Neurol Sci. 2014;344(1–2):121–128. doi:10.1016/j.jns.2014.06.041
24. Dyck PJB, González-Duarte A, Obici L, et al. Development of measures of polyneuropathy impairment in hATTR amyloidosis: from NIS to mNIS + 7. J Neurol Sci. 2019;405:116424. doi:10.1016/j.jns.2019.116424
25. Dyck PJ, Kincaid JC, Dyck PJB, et al. Assessing mNIS+7 Ionis and international neurologists’ proficiency in a familial amyloidotic polyneuropathy trial. Muscle Nerve. 2017;56(5):901–911. doi:10.1002/mus.25563
26. Adams D, Polydefkis M, González-Duarte A, et al. Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol. 2021;20(1):49–59. doi:10.1016/S1474-4422(20)30368-9
27. Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. doi:10.1056/NEJMoa1716793
28. Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–792. doi:10.1212/WNL.0b013e3182661eb1
29. Monteiro C, Mesgazardeh JS, Anselmo J, et al. Predictive model of response to tafamidis in hereditary ATTR polyneuropathy. JCI Insight. 2019;4(12). doi:10.1172/jci.insight.126526
30. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi:10.1056/NEJMoa1805689
31. Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy. JAMA. 2013;310(24):2658. doi:10.1001/jama.2013.283815
32. Habtemariam BA, Karsten V, Attarwala H, et al. Single‐dose pharmacokinetics and pharmacodynamics of transthyretin targeting N‐acetylgalactosamine–small interfering ribonucleic acid conjugate, vutrisiran, in healthy subjects. Clin Pharmacol Ther. 2021;109(2):372–382. doi:10.1002/cpt.1974
33. Alnylam Pharmaceuticals. HELIOS-A: a study of vutrisiran (ALN-TTRSC02) in patients with hereditary transthyretin amyloidosis (hATTR amyloidosis). Available from: https://clinicaltrials.gov/ct2/show/NCT03759379.
34. Alnylam Pharmaceuticals. HELIOS-B: a study to evaluate vutrisiran in patients with transthyretin amyloidosis with cardiomyopathy. Available from: https://clinicaltrials.gov/ct2/show/NCT04153149.
35. Ionis Pharmaceuticals I. A study to assess the long-term safety and efficacy of eplontersen (formerly known as ION-682884, IONIS-TTR-LRx and AKCEA-TTR-LRx) in patients with hereditary transthyretin-mediated amyloid polyneuropathy. Available from: https://clinicaltrials.gov/ct2/show/NCT05071300?term=ION682884&draw=2&rank=2.
36. Gillmore JD, Gane E, Taubel J, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385(6):493–502. doi:10.1056/NEJMoa2107454
37. Judge DP, Heitner SB, Falk RH, et al. Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J Am Coll Cardiol. 2019;74(3):285–295. doi:10.1016/j.jacc.2019.03.012
38. Masri A, Aras M, Falk RH, et al. Long-term safety and tolerability of acoramidis (AG10) in symptomatic transthyretin amyloid cardiomyopathy: updated analysis from an ongoing phase 2 open-label extension study. J Am Coll Cardiol. 2022;79(9):227. doi:10.1016/S0735-1097(22)01218-9
39. Eidos Therapeutics a B Company. Open-label safety study of acoramidis (AG10) in symptomatic ATTR participants. Available from: https://clinicaltrials.gov/ct2/show/NCT04988386?cond=acoramidis&draw=2&rank=2.
40. Gamez J, Salvadó M, Reig N, et al. Transthyretin stabilization activity of the catechol- O -methyltransferase inhibitor tolcapone (SOM0226) in hereditary ATTR amyloidosis patients and asymptomatic carriers: proof-of-concept study. Amyloid. 2019;26(2):74–84. doi:10.1080/13506129.2019.1597702
41. Pinheiro F, Varejão N, Esperante S, et al. Tolcapone, a potent aggregation inhibitor for the treatment of familial leptomeningeal amyloidosis. FEBS J. 2021;288(1):310–324. doi:10.1111/febs.15339
42. Boston University CTI. Short-term effects of TOLCAPONE on transthyretin stability in subjects with leptomeningeal TTR amyloidosis. Available from: https://clinicaltrials.gov/ct2/show/NCT03591757.
43. Hosoi A, Su Y, Torikai M, et al. Novel antibody for the treatment of transthyretin amyloidosis. J Biol Chem. 2016;291(48):25096–25105. doi:10.1074/jbc.M116.738138
44. Suhr O, Grogan M, Martins de Silva A. Neurological and cardiac improvements with PRX004 in amyloidosis patients: results of a Phase 1 study, 2021 emerging science abstracts. AAN Annu Meet Abstr Neurol. 2021;96(22):e2783–e2788.
45. Neurimmune AG. First-in-human study of NI006 in patients with amyloid transthyretin cardiomyopathy. Available from: https://clinicaltrials.gov/ct2/show/NCT04360434?cond=NI006&draw=2&rank=1.
46. Adams D, Algalarrondo V, Polydefkis M, Sarswat N, Slama MS, Nativi-Nicolau J. Expert opinion on monitoring symptomatic hereditary transthyretin-mediated amyloidosis and assessment of disease progression. Orphanet J Rare Dis. 2021;16(1):411. doi:10.1186/s13023-021-01960-9
47. Cortese A, Vita G, Luigetti M, et al. Monitoring effectiveness and safety of Tafamidis in transthyretin amyloidosis in Italy: a longitudinal multicenter study in a non-endemic area. J Neurol. 2016;263(5):916–924. doi:10.1007/s00415-016-8064-9
48. Bril V. NIS-LL: the primary measurement scale for clinical trial endpoints in diabetic peripheral neuropathy. Eur Neurol. 1999;41(Suppl. 1):8–13. doi:10.1159/000052074
49. Dyck PJ, Melton LJ, O’Brien PC, Service FJ. Approaches to improve epidemiological studies of diabetic neuropathy: insights from the Rochester diabetic neuropathy study. Diabetes. 1997;46(Supplement_2):S5–S8. doi:10.2337/diab.46.2.S5
50. Balke B. A simple field test for the assessment of physical fitness. Rep 63-6. Rep Civ Aeromed Res Inst US. 1963;1963:1–8.
51. Cambieri C, Libonati L, Moret F, et al. The silent period for small fiber sensory neuropathy assessment in a mixed cohort of transthyretin-mediated amyloidosis. Biomedicines. 2022;10(9):2073. doi:10.3390/biomedicines10092073
52. Ando Y, Adams D, Benson MD, et al. Guidelines and new directions in the therapy and monitoring of ATTRv amyloidosis. Amyloid. 2022;29(3):143–155. doi:10.1080/13506129.2022.2052838
53. Sletten DM, Suarez GA, Low PA, Mandrekar J, Singer W. COMPASS 31: a refined and abbreviated composite autonomic symptom score. Mayo Clin Proc. 2012;87(12):1196–1201. doi:10.1016/j.mayocp.2012.10.013
54. Pruppers MHJ, Merkies ISJ, Faber CG, Da Silva AM, Costa V, Coelho T. The Val30Met familial amyloid polyneuropathy specific Rasch-built overall disability scale (FAP-RODS©). J Peripher Nerv Syst. 2015;20(3):319–327. doi:10.1111/jns.12120
55. Escolano-Lozano F, Geber C, Barreiros AP, Birklein F. Follow-up in transthyretin familial amyloid polyneuropathy: useful investigations. J Neurol Sci. 2020;413:116776. doi:10.1016/j.jns.2020.116776
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.