Back to Journals » International Journal of Nanomedicine » Volume 11
Hepatitis C virus E2 protein encapsulation into poly D, L-lactic-co-glycolide microspheres could induce mice cytotoxic T-cell response
Authors Roopngam P, Liu K, Mei L, Zheng Y, Zhu X, Tsai HI, Huang L
Received 23 March 2016
Accepted for publication 12 July 2016
Published 14 October 2016 Volume 2016:11 Pages 5361—5370
DOI https://doi.org/10.2147/IJN.S109081
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Piyachat Roopngam,1,2 Kewei Liu,1,2 Lin Mei,2 Yi Zheng,2 Xianbing Zhu,1,2 Hsiang-I Tsai,1,2 Laiqiang Huang1,2
1School of Life Sciences, Tsinghua University, Beijing, People’s Republic of China; 2The Shenzhen Key Laboratory of Gene and Antibody Therapy, Center for Biotechnology and Biomedicine, State Key Laboratory of Health Sciences and Technology, Division of Life and Health Sciences, Graduate School at Shenzhen, Tsinghua University, Shenzhen, People’s Republic of China
Abstract: Hepatitis C virus (HCV) is known to cause hepatitis and hepatocellular carcinoma. E2 envelope glycoprotein of HCV type (HCV-E2) has been reported to bind human host cells and is a major target for developing anti-HCV vaccines. However, the therapeutic vaccine for infected patients still needs further development. The vaccine aims to provide cytotoxic T-cells to eliminate infected cells and hepatocellular carcinoma. Currently, there is no effective HCV therapeutic vaccine because most chronically infected patients rarely generate cytotoxic T-cells, even though they have high levels of neutralizing antibodies. Therefore, the adjuvant must be applied to enhance the efficacy of the therapeutic vaccine. In this study, we constructed HCV1b-E2 recombinant protein, a truncated form of peptide, to combine with an effective vaccine adjuvant and delivery system by using poly D, L-lactic-co-glycolide (PLGA) microspheres. HCV1b-E2 protein was effectively encapsulated into PLGA microspheres (HCV1b-E2-PLGA) as a strategy to deliver an insoluble form of HCV1b-E2 protein. The size and shape of PLGA microspheres were generated properly to carry an insoluble form of viral peptide in vivo. The encapsulated viral protein was slowly and continuously released from PLGA microspheres, which indicated the property of the adjuvant. HCV1b-E2-PLGA can trigger a cell-mediated immune response by inducing an expression of mice CD8+ T-cells. Our results demonstrated that HCV1b-E2-PLGA-immunized mice have a significantly increased CD8+ T-cell number, whereas HCV1b-E2-immunized mice have a lower number of CD8+ T-cells. Moreover, HCV1b-E2-PLGA could induce a specific antibody to viral protein, and the immune cells could secrete IFN-γ, which is a significant cytokine for viral response. Thus, HCV1b-E2-PLGA is shown to have adjuvant property and efficacy in the murine model, which is a good strategy to develop HCV prophylactic and therapeutic vaccines.
Keywords: hepatitis C virus, HCV, therapeutic vaccine, PLGA microspheres, HCV-E2 peptide, cytotoxic T-cells, hepatitis, hepatocellular carcinoma
Introduction
Hepatitis C virus (HCV) is a major causative agent of both community-acquired and transfusion-associated non-A, non-B hepatitis.1,2 Most patients develop chronic infection, and this may lead to hepatocellular carcinoma (HCC).3–5 Currently, there is no effective vaccine against HCV infection. The standard of treatment is the combined use of nucleoside analog ribavirin and pegylated interferon (IFN)-α; however, the efficacy of this treatment is limited and depends on the viral genotypes.5–7 HCV has been classified into the genus of the Flaviviridae family with enveloped plus-strand RNA virus.8 The HCV genome contains 3,010 amino acids composed of structural and nonstructural proteins. For HCV core protein, the nucleocapsid component is the structural protein, whereas envelope proteins 1 and 2 (E1 and E2) are the viral membrane proteins.9–11 E2 envelope glycoprotein of HCV has been reported to bind human host cells and has been applied to generate anti-HCV vaccines’ immunological characterization and application of Escherichia coli (E. coli)-derived hepatitis.12 However, there is still no effective HCV therapeutic vaccine because most chronically infected patients lack cytotoxic T-cell responses, even though they have high levels of neutralizing antibodies.13,14 Therefore, there is a need to develop an HCV-effective prophylactic and therapeutic vaccine in order to reduce a worldwide epidemic.12,13 In particular, a therapeutic vaccine to treat HCC is more challenging to produce than a prophylactic vaccine. However, the peptide vaccine has several disadvantages, with poor immunogenicity of protein, low stability, delivery, and inefficient antigen presentation among them.15,16 Therefore, the adjuvants or immunomodulator molecules need intensive investigation to increase the potency of the vaccine.16
In this study, the vaccine adjutants have been applied to enhance cellular immune responses. Poly D,L-lactic-co-glycolide (PLGA) particles have been applied to viral vaccines to enhance the cytosolic delivery of antigens and have been shown to significantly increase antigen presentation to the major histocompatibility complex class I and activate IL-2 secretion by T-cells in comparison with soluble antigens and antigen-coated latex beads, respectively.17,18 For the viral therapeutic vaccine, an interesting study in HPV16-E6 and -E7 peptides combined with PLGA microspheres has shown a higher efficacy when compared with peptides alone in murine models.19 Therefore, PLGA particles could have a great potential as protein carriers and adjuvant for an HCV subtype 1b therapeutic vaccine. In this study, we generate an HCV genotype 1b protein because this genotype plays an important role in HCC development, especially in patients with early-stage liver disease.20 Moreover, the HCV genotype 1b is reported as a major risk factor associated with HCC in patients with cirrhosis, who have a statistically significantly higher risk of developing HCC.21 We constructed HCV1b-E2 recombinant protein because of its advantages as an inexpensive source and high antigenicity.22 However, HCV1b-E2 (181 aa) is hydrophobic, and it is a recombinant protein from an E. coli-expressed system, which could provide only a truncated form as insoluble protein.22 The solubility of protein was resolved by using 10% dimethyl sulfoxide (DMSO) for mice injections. However, the high concentration of DMSO could induce apoptosis.23 The solubilized protein in urea is also not suitable for human immunization. Thus, we applied PLGA microspheres to deliver the insoluble protein. The hydrophobic protein was dissolved in DMSO with PLGA and encapsulated inside PLGA microspheres. An in vitro releasing protein data indicated that the soluble protein could be released from PLGA microspheres. Mice immunizations with encapsulated HCV1b-E2 proteins into PLGA microspheres were investigated for specific humoral immunity and an expression of cytotoxic T-cells. The prophylactic and therapeutic vaccines have been enhanced by triggering the humoral and cell-mediated immune responses to target HCV1b-E2-specific antibody and cytotoxic T-cells, which would be very useful for a therapeutic approach.24 Therefore, a PLGA microsphere has performed a strategy as an adjuvant and effective delivery system. This system will provide a new alternative to the development of HCV prophylactic and therapeutic vaccines.
Materials and methods
Mice
Female BALB/c mice (8 weeks of age) were purchased from Guangzhou Traditional Chinese Medicine University (Guangzhou, People’s Republic of China) and were housed in filter-top cages under specific pathogen-free conditions. The animal studies were conducted with ethical approval from, and in accordance with, the protocols and guidelines of the Animal Care and Use Committee of Tsinghua University.
Construction of recombinant expression plasmids
pET28a is a histidine-tagged fusion expression vector containing E2-coding sequences of HCV subtype 1b (Genbank AJ288799.1), which was prepared from attenuated HCV. The pET28a-E2 gene was subjected to the DNA sequence analysis, as shown in Figure 1. Polymerase chain reaction and recombinant cloning were performed according to standard protocols. DNA sequence coding for E2 (384-564aa) was cloned into pET28a between the BamHI and EcoRI sites to create pET28a-E2 by using primer sequences, as shown in Table 1.
 | Figure 1 Whole sequence of truncated form HCV1b-E2 gene. |
 | Table 1 Sequences of primer used for PCR amplification of the HCV1b-E2 gene |
Expression conditions and purification
Recombinant HCV1b-E2 protein was prepared in E. coli strain DH5γ. Recombinant proteins with His-Tag were expressed upon induction with 1 mmol/L isopropyl-β-D-thiogalactopyranoside (Sigma-Aldrich Co., St Louis, MO, USA). E. coli was sonicated and subjected to protein extraction by 8 M urea, then purified on Ni-NTA agarose column (QIAGEN, Hilden, Germany). The column was washed with 30 mM imidazole in 8 M urea and then eluted with 100 mM imidazole in 8 M urea. Purified proteins were dialyzed against phosphate buffered saline (PBS, pH 7.4) and lyophilized. HCV1b-E2 recombinant protein molecular weight was determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) analysis, and gels were coomassie blue stained.
Preparation of encapsulated HCV1b-E2 into PLGA microspheres
PLGA (50:50, MW 50,000) with a carboxylic end group was purchased from Sigma-Aldrich Co. HCV1b-E2 protein encapsulated into PLGA microspheres (HCV1b-E2-PLGA) was prepared by the dialysis method initially described by Jeon et al17 with modification. Briefly, 0.5 mg HCV1b-E2 protein was dissolved and emulsified in 1 mL solution of polymer in DMSO (0.5 mg/mL) by sonication for 15 seconds (output 2) and then dialyzed by using 14 K cutoff dialysis membranes against PBS. The solvents were reduced by evaporation under reduced pressure without heating. The HCV1b-E2-PLGA pellet was collected by centrifugation at 3,000 rpm for 3 minutes to quantify for the protein.
Characterization of HCV1b-E2-PLGA microspheres
HCV1b-E2-PLGA microsphere sizes were subjected to ultrasonication and measured by Malvern Mastersizer 2000 (Zetasizer Nano ZS90, Malvern Instruments, Malvern, UK). Before measurement, the microspheres were freshly prepared and appropriately diluted. All measurements were carried out at room temperature after 10 minutes of equilibration. HCV1b-E2-PLGA microspheres were measured in triplicate and were further observed by transmission electron microscopy (TEM, Tecnai G2 20, FEI Company, Hillsboro, OR, USA). To prepare samples for the TEM, samples were dropped onto carbon-coated lacey support films. The support films were allowed to dry before characterization.
HCV1b-E2-PLGA-encapsulated efficiency
HCV1b-E2-PLGA microspheres (200 μL) were digested into 1 mL of 0.02 N NaOH containing 1% (weight/volume [w/v]) SDS. The solution was then adjusted to pH 7.0 by 1 M HCl and centrifuged at 10,000 rpm for 5 minutes. The HCV1b-E2 protein from a PLGA pellet was then quantified in triplicate by a UV-visible spectrophotometer at 595 nm as per the standard protocol of the Bradford assay (Bio-Rad Laboratories Inc., Hercules, CA, USA), and 1.4, 2.8, and 5.6 mg of bovine serum albumin (BSA) were measured to set a standard curve.
In vitro release studies
In vitro release studies were carried out by suspending 200 mg of microspheres in 60 mL of PBS containing 0.02% sodium azide as a bacteriostatic agent and 0.01% Tween 80 to prevent the microparticles from aggregation in the culture plate. The samples were collected and centrifuged for 5 minutes. The supernatant was assayed in triplicate for the protein release using a UV-visible spectrophotometer at 595 nm as per the standard protocol of the Bradford assay (Bio-Rad Laboratories Inc.) on days 1, 7, 14, and 21.
Mice antibody response and CD8+ T-cell analysis
Mice were divided into four groups of three for immunization, and each group received four subcutaneous injections (50 μL per injection). Injections were with complete Freud’s adjuvant on day 0, and with incomplete Freud’s adjuvant on day 14 for all groups. The first group was administered PBS to serve as a saline control. The second group was administered 5 μg/μL of PLGA microspheres in PBS per animal per injection. The third group was administered 5 μg/μL of HCV1b-E2 protein in 10% DMSO per animal per injection. The fourth group was administered 5 μg/μL of HCV1b-E2-PLGA microspheres per animal per injection. After day 28, a sample of 400 μL of blood was taken from each mouse’s heart, and the heparinized blood was measured for antibody response and cytotoxic T-cells. Mouse serum was evaluated for antibodies against HCV1b-E2 by indirect ELISA. The microtiter plates were coated overnight at 4°C with 25 μg/well of HCV1b-E2 protein in 2 M urea and then blocked for 2 hours at 37°C with 200 μL of PBS containing 5% nonfat dry milk and washed three times with PBS. Then, mouse serum was diluted 1:20 in 100 μL of PBS with 5% nonfat dry milk and incubated for 2 hours with HCV1b-E2 antigen coated on the microtiter plates. After incubation, the plates were washed three times with PBS. Antibodies bound to the coated antigen were detected by goat anti-mouse IgG-conjugated horseradish peroxidase (e-Bioscience, San Diego, CA, USA) in PBS by incubating for 2 hours at 37°C. Following washes, the plates were developed with TMB substrate (Bio-Rad Laboratories Inc.) and acid stop solution. The plates were measured for optical density (OD) at 450 nm with an ELISA reader (BioTek, Chaoyang, Beijing, People’s Republic of China). The mouse blood was subjected to cytotoxic T-cell surface marker (CD8+ T-cell) measurement. The heparinized blood was prepared for mononuclear cells separation by Ficoll-Hypaque density gradient centrifugation (Ficoll-PaquePREMIUM 1.084, Sigma-GE). The mononuclear cells were incubated with the specific monoclonal antibody for 30 minutes in PBS/1% BSA (w/v), and then unspecific binding was washed with PBS/1% BSA (w/v). CD8+ T-cells were labeled using fluorescein isothiocyanate (FITC) anti-mouse CD8 antibody (e-Bioscience) and counted by flow cytometry analysis. A percentage of mice CD8+ T-cells were analyzed by the FACSort™ flow cytometer (Becton Dickinson, NJ, USA).
IFN-γ ELISA assay
The intracellular IFN-γ was produced mainly from the activated antigen-specific cytotoxic T-cells, and other lymphocytes also secreted a low level of IFN-γ and were harvested from culture supernatant of mice lymphocytes at 72 hours. Mice lymphocytes from the HCV1b-E2- and HCV1b-E2-PLGA-treated groups were cultured with Roswell Park Memorial Institute-1640 (Gibco/BRL) supplemented with 10% fetal calf serum, 25 mM HEPES, 2 mM L-glutamine, 1 mM sodium pyruvate, and 100 μg/mL of penicillin/streptomycin. The culture supernatants were lyophilized and resuspended in 300 μL of PBS. The levels of IFN-γ in culture supernatant of mice lymphocytes that were immunized with HCV1b-E2 protein and HCV1b-E2-PLGA microspheres were compared using the IFN-γ ELISA kit25 (Dakewe Biotech Co. Ltd., Beijing, People’s Republic of China) according to the manufacturer’s instructions. Mice IFN-γ standard proteins at 40, 20, 10, and 5 pg were measured as the standard point of OD at 450 nm. Data are representative of three independent assays.
Statistical analysis
The significant differences between the mean values were determined by a two-tailed Student’s t-test. P-values <0.05 were considered to be statistically significant.
Results
HCV1b-E2 protein antigen was presented at a molecular mass of 20 kDa
HCV1b-E2 protein was generated in E. coli to express the truncated form of protein. The expressed protein was purified and collected for eluate fractions from the column. HCV1b-E2 eluate fractions of proteins were made concentrated by lyophilization and then resolved in 12% SDS–PAGE under reducing conditions. The resolved proteins were visualized using Coomassie Blue stain. Consistent with the measurement of protein content, it was found that HCV1b-E2 was presented predominantly at a molecular mass of 20 kDa from the eluate fractions obtained from Ni-NTA agarose column as shown in Figure 2.
 | Figure 2 SDS–PAGE of HCV1b-E2 band protein with a molecular weight of 20 kDa. |
HCV1b-E2-PLGA microspheres morphology and sizes
The protein was further subjected to encapsulation into PLGA microspheres. As shown in Figure 3A, HCV1b-E2-PLGA microspheres observed by TEM are spherical in shape, as expected. The size of the HCV1b-E2-PLGA microspheres was measured by Malvern Mastersizer 2000 (Malvern Instruments), and the microspheres are in colloidal systems with a size of 941.3 nm (Figure 3B). The polydispersity index (PDI) of HCV1b-E2-PLGA microspheres is 0.265.
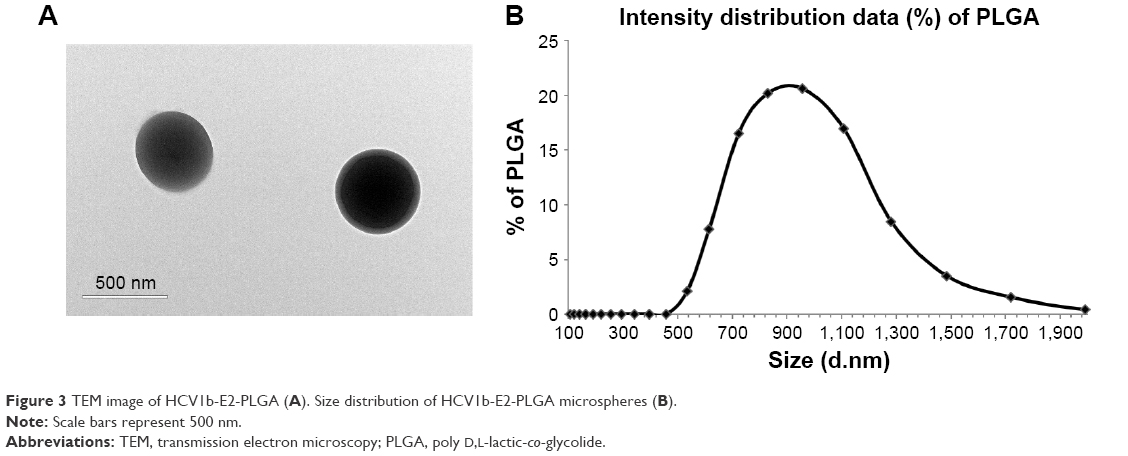 | Figure 3 TEM image of HCV1b-E2-PLGA (A). Size distribution of HCV1b-E2-PLGA microspheres (B). |
Encapsulation efficacy
To demonstrate that HCV1b-E2 protein was actually encapsulated into PLGA microspheres, the HCV1b-E2-PLGA microspheres were quantified for the protein amount using the Bradford assay, as shown in Figure 4. The BSA standard curve was measured for the standard protein point of OD at 595 nm. The protein quantification of HCV1b-E2 before encapsulation was 0.5 mg (OD =0.055). The encapsulated HCV1b-E2 protein from PLGA was 0.45 mg (OD =0.0495). This result indicated that the encapsulated protein is a 90% yield of encapsulated efficacy (Figure 4).
 | Figure 4 HCV1b-E2 protein at 0.5 mg was dissolved into DMSO and quantitated by the Bradford assay before encapsulation. |
HCV1b-E2 protein released from PLGA microspheres
HCV1b-E2 protein was further determined for the release from PLGA microspheres and quantified by the Bradford assay on days 1, 7, 14, and 21. The released amounts of protein were 0.040, 0.145, 0.285, and 0.407 mg, respectively. These data showed that HCV1b-E2 protein released on days 1, 7, 14, and 21 was 0.088%, 32%, 63%, and 90%, respectively, as shown in Figure 5. This result indicated that the microspheres released HCV1b-E2 protein slowly, and the releasing efficacy is 90%.
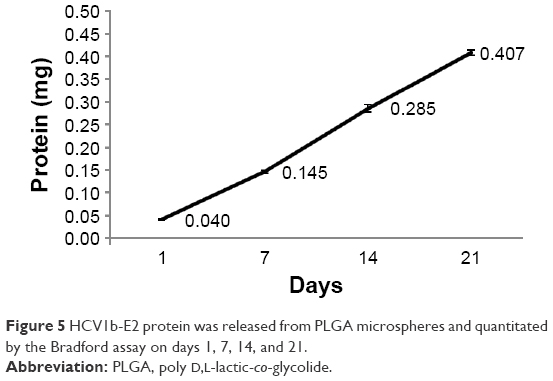 | Figure 5 HCV1b-E2 protein was released from PLGA microspheres and quantitated by the Bradford assay on days 1, 7, 14, and 21. |
An expression of mice cytotoxic T-cells
An expansion of cytotoxic T-cells induced by HCV1b-E2-PLGA microspheres was evaluated in vivo for mice CD8+ T-cells as shown in Figure 6. The saline control group (PBS-immunized mice) and PLGA control have no lymphocyte expansion. Some of the lymphocyte numbers from these control groups are too low to count with flow cytometry. The saline control group has no CD8+ T-cells (Figure 6A). For the PLGA control group of three mice, only one showed an expression of CD8+ T-cells in 1.65% (Figure 6B). The expression of mice CD8+ T-cells was compared by flow cytometry analysis data between the two groups of mice immunized with HCV1b-E2 (Figure 6C) and HCV1b-E2-PLGA microspheres (Figure 6D). The higher expansion of lymphocytes and cytotoxic T-cells was observed in an HCV1b-E2-PLGA microspheres-treated group from the increased positive events of flow cytometry analysis.
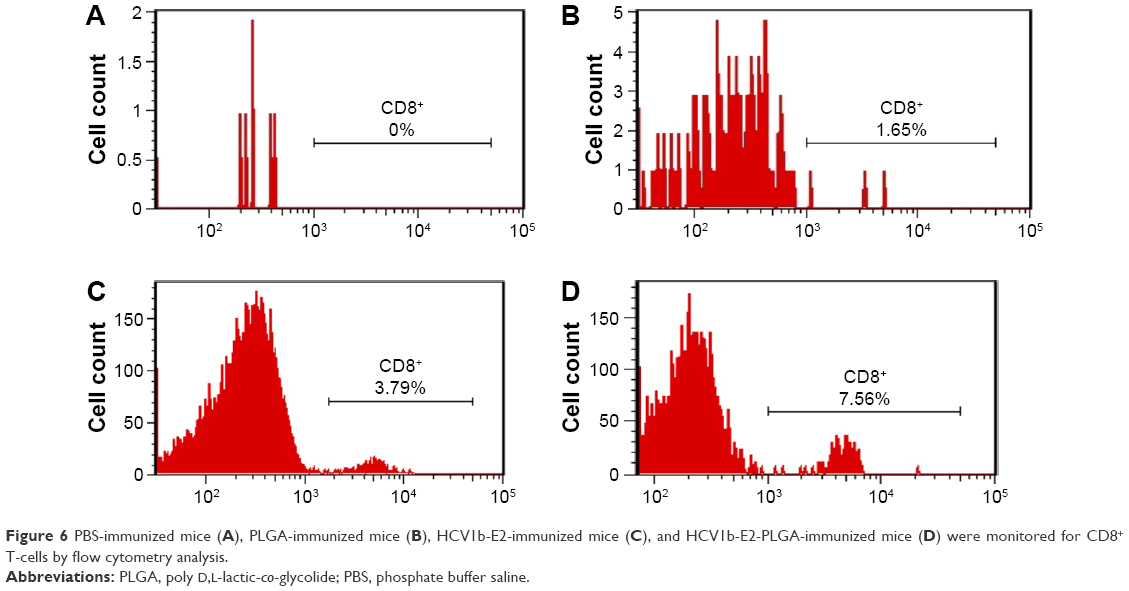 | Figure 6 PBS-immunized mice (A), PLGA-immunized mice (B), HCV1b-E2-immunized mice (C), and HCV1b-E2-PLGA-immunized mice (D) were monitored for CD8+ T-cells by flow cytometry analysis. |
To investigate a statistically significant expression of mice, CD8+ T-cells among the two groups of mice were immunized with HCV1b-E2 and HCV1b-E2-PLGA microspheres, as shown in Figure 7A. The HCV1b-E2-immunized mice expressed a lower number of CD8+ T-cells (4.427%±0.992%), whereas HCV1b-E2-PLGA-immunized mice showed a significant increase in CD8+ T-cell population (8.257%±1.502%).
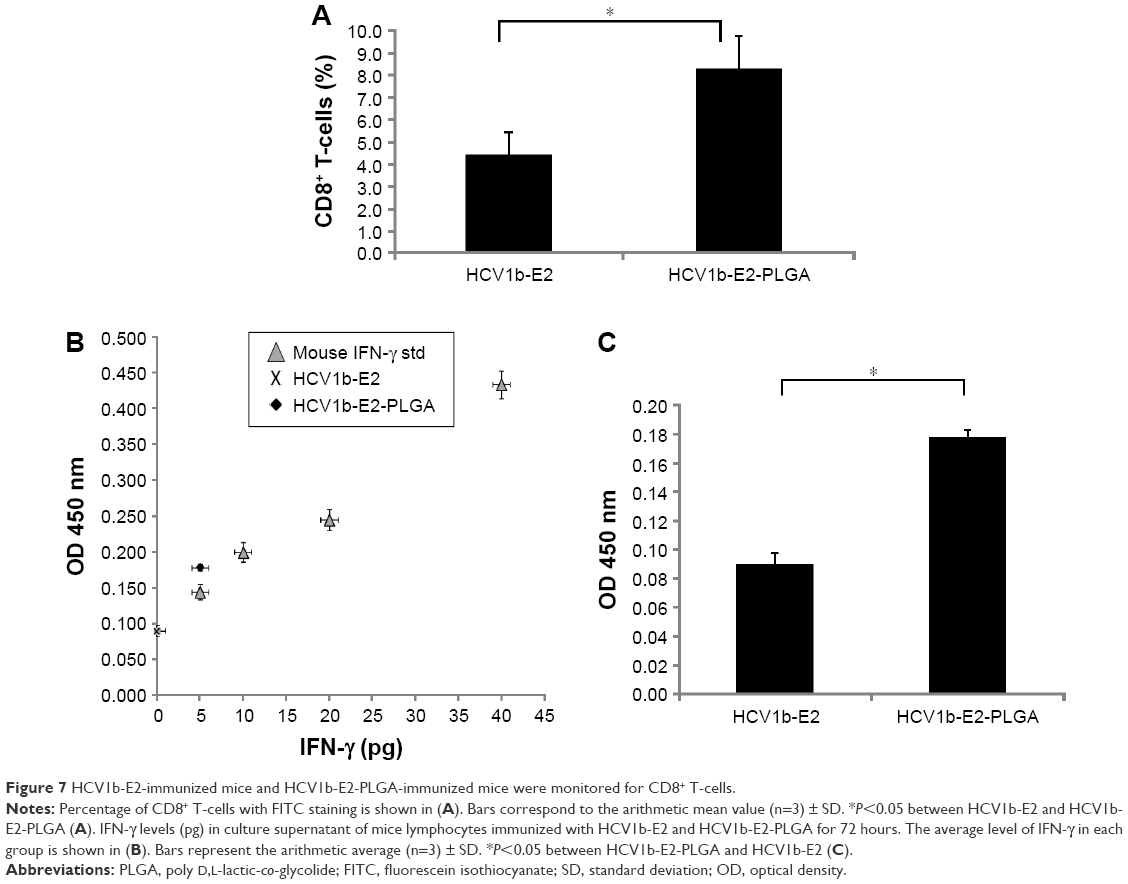 | Figure 7 HCV1b-E2-immunized mice and HCV1b-E2-PLGA-immunized mice were monitored for CD8+ T-cells. |
HCV1b-E2-PLGA microspheres could induce IFN-γ production
IFN-γ levels in cultured supernatant of lymphocytes culture, HCV1b-E2 and HCV1b-E2-PLGA microspheres-treated groups, were compared. The levels of IFN-γ from HCV1b-E2 and HCV1b-E2-PLGA microspheres-treated groups were 0 and 5 pg, respectively, as shown in Figure 7B. The group of HCV1b-E2-PLGA microspheres-treated mice showed a significantly higher OD point of IFN-γ secretion (0.089±0.08) when compared with the group of HCV1b-E2 microspheres-treated mice (0.177±0.004) (n=3, P<0.05, Figure 7C).
HCV1b-E2-PLGA could induce humoral immune response
The antigenicity of HCV1b-E2-PLGA was evaluated by mice immune response, as mice carry the HCV1b-E2 antibody. We observed that the mice have no antibody in the saline control and PLGA microspheres-treated group. The mice into which HCV1b-E2 and HCV1b-E2-PLGA were administered produced significantly higher levels of antibody (n=3, P<0.001) than PBS- and PLGA-administered mice, as shown in Figure 8. These data indicate that HCV1b-E2- and HCV1b-E2-PLGA-administered mice could induce adaptive immunity and humoral immune response by producing specific IgG to the protein.
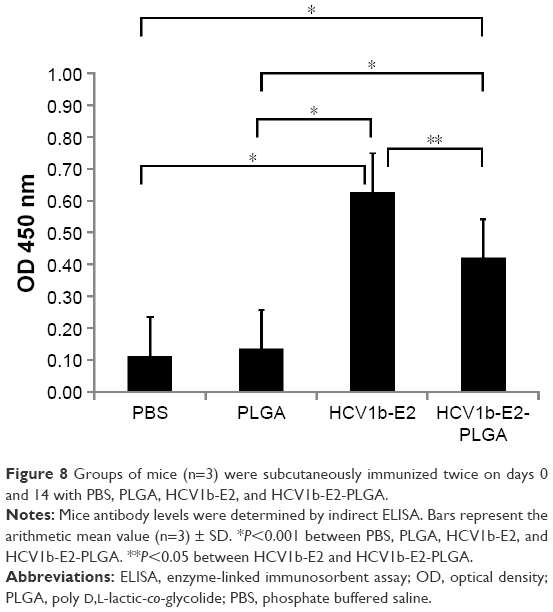 | Figure 8 Groups of mice (n=3) were subcutaneously immunized twice on days 0 and 14 with PBS, PLGA, HCV1b-E2, and HCV1b-E2-PLGA. |
Discussion
PLGA particles have been known to increase the potency of a vaccine formulation26,27 and are considered to be among several potential vaccine delivery systems. In general, antigen-presenting cells (APCs) have been known to prefer uptake antigens and immune stimulators in particulate form rather than the soluble form. The particulate nature of PLGA particles performed a rapid internalization, perhaps because of its pathogen-mimicking size and charge profile.28 The studies by Newman et al29 showed that PLGA particles are able to induce and enhance immune responses of poor immunogens. For a viral vaccine, this very interesting study showed that HIV p24 protein adsorbed onto the surface of surfactant-free anionic poly(D,L-lactide) or poly lactic acid nanoparticles was efficiently taken up by mouse dendritic cells, inducing the maturation of dendritic cells. Furthermore, PLGA particles not only provide a delivery system but also act as an adjuvant. They possess the capacity to present antigens to both major histocompatibility complex classes I and II pathways, inducing both humoral and cellular responses because of HIV vaccines requiring the induction of both kinds of the immune system.24 Therefore, biodegradable nanoparticles vaccine carrying viral antigens is a good strategy for viral vaccine.
Here, HCV1b-E2 protein was generated and effectively encapsulated to PLGA microspheres, which not only provide a delivery system but also act as an adjuvant. Moreover, for a therapeutic vaccine, PLGA microspheres have successfully demonstrated adjuvant activity to enhance immune responses to human papillomavirus type 16 (HPV16) protein in mice.19 PLGA particles have been shown to effectively process the antigens in vitro to murine bone marrow-derived APCs because of their slow degradation rate before the internalization in APCs.30,31 This suggests that PLGA particles are effective and useful for immunotherapy. Therefore, biodegradable particle vaccine carrying viral antigen is a good strategy for a viral vaccine. For this study, PLGA microspheres have demonstrated the adjuvant capacity to induce cellular immune responses by an expression of CD8+ T-cells, which is the therapeutic vaccine’s purpose, to generate cytotoxic T-cells.32 HPV1b-E2-PLGA microspheres were determined to induce cell-mediated immune responses in mice. Here, HPV1b-E2 antigen protein was effectively encapsulated into PLGA microspheres by the Bradford assay, a promising method to quantify protein into PLGA. The release of protein antigens is slow, which is a positive adjuvant property. In particular, HCV1b-E2 protein is a truncated form of insoluble protein, which can be effectively delivered in vivo and uptaken by APCs to trigger the cell-mediated immune system. The antigenicity of HCV1b-E2 and HCV1b-E2-PLGA was evaluated by mice antibody response. To study the viral vaccine, the humoral and cell-mediated immune responses were intensively focused on. The prophylactic antibody to HCV1b-E2 was studied in immunized mice, and it was shown that HCV1b-E2-PLGA could induce the specific antibody response. However, the antibody of the HCV1b-E2 protein alone treated group has a significantly higher level than that of the HCV1b-E2-PLGA group. This is because an encapsulated protein is slowly released from PLGA, which is a critical property of adjuvant to induce life-protective immunity.33 Therefore, HCV1b-E2-PLGA is potent to enhance the humoral immune response and is recommended to apply as the vaccine to prevent HCV. In addition, we generated a spherical shape of PLGA microspheres visualized by TEM and showed the size of 500 nm in a single particle. However, the microspheres are aggregated and show a size of 941 nm in the colloidal system. This is important because a PLGA particle with a size of approximately 1,000 nm has been reported to be the most potent immunomodulator for inducing immune response.34 Therefore, the size of microspheres in our study has potential as an effective immunomodulator. These results clearly indicate that an encapsulated viral protein antigen into PLGA microspheres effectively induces an expression of CD8+ T-cells. The expansion of lymphocytes and cytotoxic T-cells was observed in an HCV1b-E2-PLGA microspheres-treated group, showing increasing positive lymphocytes population events in flow cytometry analysis. The expression of mice cytotoxic T-cells treated by HCV1b-E2-PLGA microspheres is significantly increased. Moreover, the group of lymphocytes treated with HCV1b-E2-PLGA microspheres produced a high level of IFN-γ. This result is related to the previous report that PLGA microspheres can induce higher IFN-γ in murine models.19 IFN-γ, one of the key regulatory cytokines in the host immune response against viral infections, is secreted by effector CD8+ T-cells, Th1 CD4+ T-cells, and NK and NK T-cells.35,36 An indirect effect of IFN-γ is to enhance cytotoxic T-cells activity36 and to activate NK cells besides helping to differentiate CD4+ T-cells toward a Th1 response to promote cytotoxic T-cell differentiation.37 Therefore, increasing cytotoxic T-cell activity to kill the cervical cancer cells will be enhanced by synergism of increasing IFN-γ and effective cellular immune responses.
Our research shows, for the first time, that an HCV1b-E2-truncated form of protein can effectively encapsulate into PLGA microspheres, an effective strategy for delivering insoluble protein for vaccine immunization. The development of polymer chemistry and molecular immunology helps to create a successful vaccine delivery system and can lead to the next generation of vaccines. This novel strategy can be a potential therapeutic vaccination in HCV1b-infected patients.
Conclusion
HCV1b-E2-PLGA microspheres can enhance a humoral immunity and cell-mediated immune response to viral-caused hepatitis and HCC, and it can perform the delivery and adjuvant property in murine models. HCV1b-E2 encapsulated into PLGA microspheres is a very good strategy to develop a therapeutic HCV vaccine.
Acknowledgments
We thank Mr Wugen Zhan for his valuable help with flow cytometry analysis and other members of Huang Lab for suggestions and assistance. This work was supported by funding from Shenzhen Municipal Science, Technology & Innovation Commission’s Science & Technology Research Grants and the Municipal Program for Building State and Shenzhen Key Laboratories (2006464, 200712, SG200810150043A, CXB201005260070A, CXB201104220043A, ZDSY20120616222747467, JCYJ20130402145002438, ZDSYS20140509172959975, GJHZ20140416153844269, and JCYJ20140418112611757), and in part from Shenzhen Municipal and Nanshan District Science & Technology Grants (JCYJ20140718171607436 and KC2014JSCX0023A).
Author contributions
LH and PR conceived and designed the research; PR conducted most of the research; all authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Science. 1989;244(4902):359–362. | ||
Choojitr W, Ruangkris T. Surgical wound infections in gynaecology at Rajvithi Hospital 1989–1990. J Med Assoc Thai. 1995;78(Suppl 2):S78–S80. | ||
Seeff LB. Natural history of hepatitis C. Am J Med. 1999;107(6B):10S–15S. | ||
Seeff LB. Dilemma of the natural history of hepatitis C. J Gastroenterol Hepatol. 1999;14(3):199–201. | ||
Halliday J, Klenerman P, Barnes E. Vaccination for hepatitis C virus: closing in on an evasive target. Expert Rev Vaccines. 2011;10(5): 659–672. | ||
Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19(7):837–849. | ||
Wendt A, Adhoute X, Castellani P, et al. Chronic hepatitis C: future treatment. Clin Pharmacol. 2014;6:1–17. | ||
Weiner AJ, Brauer MJ, Rosenblatt J, et al. Variable and hypervariable domains are found in the regions of HCV corresponding to the flavivirus envelope and NS1 proteins and the pestivirus envelope glycoproteins. Virology. 1991;180(2):842–848. | ||
Houghton M, Weiner A, Han J, Kuo G, Choo QL. Molecular biology of the hepatitis C viruses: implications for diagnosis, development and control of viral disease. Hepatology. 1991;14(2):381–388. | ||
Grakoui A, Wychowski C, Lin C, Feinstone SM, Rice CM. Expression and identification of hepatitis C virus polyprotein cleavage products. J Virol. 1993;67(3):1385–1395. | ||
Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM. A second hepatitis C virus-encoded proteinase. Proc Natl Acad Sci U S A. 1993;90(22):10583–10587. | ||
Liu C, Liu W, Yang J, Fang D. [HCV core protein activates expression of vascular endothelial growth factor in HepG(2) cells]. Zhonghua Gan Zang Bing Za Zhi. 2001;9(4):214–216. Chinese. | ||
Feinstone SM, Hu DJ, Major ME. Prospects for prophylactic and therapeutic vaccines against hepatitis C virus. Clin Infect Dis. 2012;55(Suppl 1):S25–S32. | ||
Arichi T, Saito T, Major ME, et al. Prophylactic DNA vaccine for hepatitis C virus (HCV) infection: HCV-specific cytotoxic T lymphocyte induction and protection from HCV-recombinant vaccinia infection in an HLA-A2.1 transgenic mouse model. Proc Natl Acad Sci U S A. 2000;97(1):297–302. | ||
Hung CF, Ma B, Monie A, Tsen SW, Wu TC. Therapeutic human papillomavirus vaccines: current clinical trials and future directions. Expert Opin Biol Ther. 2008;8(4):421–439. | ||
Le Corre P, Rytting JH, Gajan V, Chevanne F, Le Verge R. In vitro controlled release kinetics of local anaesthetics from poly(D,L-lactide) and poly(lactide-co-glycolide) microspheres. J Microencapsul. 1997;14(2):243–255. | ||
Jeon HJ, Jeong YI, Jang MK, Park YH, Nah JW. Effect of solvent on the preparation of surfactant-free poly(D,L-lactide-co-glycolide) nanoparticles and norfloxacin release characteristics. Int J Pharm. 2000;207(1–2):99–108. | ||
Shen H, Ackerman AL, Cody V, et al. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology. 2006;117(1):78–88. | ||
Sharma C, Khan MA, Mohan T, Shrinet J, Latha N, Singh N. A synthetic chimeric peptide harboring human papillomavirus 16 cytotoxic T lymphocyte epitopes shows therapeutic potential in a murine model of cervical cancer. Immunol Res. 2014;58(1):132–138. | ||
Raimondi S, Bruno S, Mondelli MU, Maisonneuve P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: a meta-analysis. J Hepatol. 2009;50(6):1142–1154. | ||
Bruno S, Crosignani A, Maisonneuve P, Rossi S, Silini E, Mondelli MU. Hepatitis C virus genotype 1b as a major risk factor associated with hepatocellular carcinoma in patients with cirrhosis: a seventeen-year prospective cohort study. Hepatology. 2007;46(5):1350–1356. | ||
Liu J, Zhu L, Zhang X, et al. Expression, purification, immunological characterization and application of Escherichia coli-derived hepatitis C virus E2 proteins. Biotechnol Appl Biochem. 2001;34(Pt 2):109–119. | ||
Lin CK, Kalunta CI, Chen FS, Nguyen TT, Kaptein JS, Lad PM. Dimethyl sulfoxide suppresses apoptosis in Burkitt’s lymphoma cells. Exp Cell Res. 1995;216(2):403–410. | ||
Aline F, Brand D, Pierre J, et al. Dendritic cells loaded with HIV-1 p24 proteins adsorbed on surfactant-free anionic PLA nanoparticles induce enhanced cellular immune responses against HIV-1 after vaccination. Vaccine. 2009;27(38):5284–5291. | ||
Zheng Y, Zhang Y, Ma Y, Wan J, Shi C, Huang L. Enhancement of immunotherapeutic effects of HPV16E7 on cervical cancer by fusion with CTLA4 extracellular region. J Microbiol. 2008;46(6):728–736. | ||
Katare YK, Panda AK. Immunogenicity and lower dose requirement of polymer entrapped tetanus toxoid co-administered with alum. Vaccine. 2006;24(17):3599–3608. | ||
Diwan M, Elamanchili P, Cao M, Samuel J. Dose sparing of CpG oligodeoxynucleotide vaccine adjuvants by nanoparticle delivery. Curr Drug Deliv. 2004;1(4):405–412. | ||
Demento SL, Siefert AL, Bandyopadhyay A, Sharp FA, Fahmy TM. Pathogen-associated molecular patterns on biomaterials: a paradigm for engineering new vaccines. Trends Biotechnol. 2011;29(6):294–306. | ||
Newman KD, Sosnowski DL, Kwon GS, Samuel J. Delivery of MUC1 mucin peptide by Poly(D,L-lactic-co-glycolic acid) microspheres induces type 1 T helper immune responses. J Pharm Sci. 1998;87(11):1421–1427. | ||
Hamdy S, Haddadi A, Hung RW, Lavasanifar A. Targeting dendritic cells with nano-particulate PLGA cancer vaccine formulations. Adv Drug Deliv Rev. 2011;63(10–11):943–955. | ||
Kovacsovics-Bankowski M, Clark K, Benacerraf B, Rock KL. Efficient major histocompatibility complex class I presentation of exogenous antigen upon phagocytosis by macrophages. Proc Natl Acad Sci U S A. 1993;90(11):4942–4946. | ||
Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39(1):38–48. | ||
Alonso MJ, Gupta RK, Min C, Siber GR, Langer R. Biodegradable microspheres as controlled-release tetanus toxoid delivery systems. Vaccine. 1994;12(4):299–306. | ||
Oyewumi MO, Kumar A, Cui Z. Nano-microparticles as immune adjuvants: correlating particle sizes and the resultant immune responses. Expert Rev Vaccines. 2010;9(9):1095–1107. | ||
Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. | ||
Whitmire JK, Tan JT, Whitton JL. Interferon-gamma acts directly on CD8+ T cells to increase their abundance during virus infection. J Exp Med. 2005;201(7):1053–1059. | ||
Mocikat R, Braumuller H, Gumy A, et al. Natural killer cells activated by MHC class I(low) targets prime dendritic cells to induce protective CD8 T cell responses. Immunity. 2003;19(4):561–569. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.