Back to Journals » Journal of Hepatocellular Carcinoma » Volume 10
Hepatitis B Virus-Encoded MicroRNA (HBV-miR-3) Inhibits FIH-1 Expression to Promote Tumor Angiogenesis in HBV-Related Hepatocellular Carcinoma
Authors Chen H, Cao D ![]() , Han N, Zhang M, Jiang W, Wang X, Zeng Q, Tang H
, Han N, Zhang M, Jiang W, Wang X, Zeng Q, Tang H
Received 24 August 2023
Accepted for publication 8 December 2023
Published 27 December 2023 Volume 2023:10 Pages 2337—2353
DOI https://doi.org/10.2147/JHC.S436926
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr David Gerber
Han Chen,1,2 Dan Cao,1,2 Ning Han,1,2 Mingming Zhang,1,2 Wei Jiang,1,2 Xin Wang,1,2 Qinmin Zeng,1,2 Hong Tang1,2
1Center of Infectious Diseases, West China Hospital of Sichuan University, Chengdu, Sichuan, People’s Republic of China; 2Division of Infectious Diseases, State Key Laboratory of Biotherapy and Center of Infectious Diseases, West China Hospital of Sichuan University, Chengdu, 610041, People’s Republic of China
Correspondence: Hong Tang, Email [email protected]
Introduction: Hepatocellular carcinoma (HCC) is a solid tumor with a rich blood supply, and anti-angiogenesis has important clinical significance. Hepatitis B Virus-Encoded MicroRNA 3 (HBV-miR-3) has recently been reported to be involved in HCC development. In this study, we aim to elucidate the role of HBV-miR-3 in promoting HBV-related HCC angiogenesis through Factor Inhibiting Hypoxia-inducible factor 1 (FIH-1).
Results: By analyzing HBV-related HCC tissue samples, we found that high expression of HBV-miR-3 was associated with poor overall survival and HBV-miR-3 expression was significantly correlated with VEGFR2 and FIH-1 expressions. In vitro, HBV-miR-3 agomir repressed FIH-1 expression and promoted HIF-1α/VEGFA signaling activation in HepG2 cells, resulting in increased HUVEC lumen formation in HepG2-HUVEC co-culture model. Conversely, HBV-miR-3 antagomir induced FIH-1 expression and inhibited HIF-1α/VEGFA signaling activation in HepG2.2.15 cells, resulting in decreased HUVEC lumen formation in HepG2.2.15-HUVEC co-culture model. The effect of HBV-miR-3 to HCC angiogenesis was also confirmed by a mouse tumor bearing model. We also confirmed that HBV-miR-3 repressed FIH-1 expression via targeting the 3’-UTR of FIH-1 mRNA by luciferase activity assay.
Conclusion: HBV-miR-3 was related to HCC patients’ overall survival and it promoted angiogenesis by repressing FIH-1 expression. HBV-miR-3 may be a new marker for predicting prognosis and a novel target for anti-angiogenic treatment of HBV-related HCC.
Keywords: HBV-miR-3, FIH-1, HBV-related HCC, angiogenesis
Graphical Abstract:

Introduction
Hepatocellular carcinoma (HCC) is a solid tumor with abundant blood supply, and angiogenesis is involved in the process of HCC development.1,2 Most HCCs in China are related to hepatitis B virus infection.3 In previous studies, hepatitis B virus (HBV) was confirmed to promote the progression of angiogenesis of HCC by the HBV X protein (HBx) and small protein of hepatitis B virus surface antigens (SHBs).4 HBx increases the protein level of HIF-1α by direct binding with HIF-1α, thus leading to the decrease of interaction with pVHL, resulting in inhibiting the ubiquitin-dependent degradation of HIF-1α.5 HBx could also enhance Histone deacetylase 1 (HDAC1) transcription, which leads to increased deacetylation of the oxygen-dependent degradation (ODD) domain of HIF-1α, inhibiting its degradation.6 Small protein of hepatitis B virus surface antigens (SHBs) could also promote HCC angiogenesis via endoplasmic reticulum stress signaling to upregulate the expression of VEGFA.7 However, whether HBV modulates HCC angiogenesis in other methods remains unknown.
HBV-encoded miRNAs (HBV-miRs) were first discovered by deep sequencing in 2016, which are a group of v-miRs generated from 3.5-kb, 2.4-kb, and 2.1-kb transcripts.8 Among all the HBV-miRs, HBV-miR-3 was mostly studied. HBV-miR-3 is located at nucleotides (nt) 373 to 393 of the HBV genome. HBV-miR-3 could be detected in the serum of chronic hepatitis B patients with high viral load.9 HBV-miR-3 could control viral replication8,10 and promote HCC cell proliferation by regulating post-transcription of Phosphatase and tensin homolog (PTEN), Protein phosphatase 1A (PPM1A) and DIX domain containing 1 (DIXDC1) of the hosts.11,12 Thus, the role of HBV-miR-3 in HCC development should be further revealed.
Hypoxia-inducible factor 1 (HIF-1α) is the most important regulator of hypoxia signaling pathway. HIF-1α stabilization requires the inhibition of molecules including von Hippel–Lindau protein (pVHL) ubiquitin E3 ligase complex, prolyl hydroxylases (PHDs), or Hypoxia Inducible Factor 1 Subunit Alpha Inhibitor (HIF1AN), which is also named Factor Inhibiting HIF-1 (FIH-1).13 FIH-1 was recently reported to be a tumor suppressor and target of many tumor promoters. For example, MicroRNA-31 contributes to colorectal cancer development by targeting FIH-1.14,15 MiR-H16 which was encoded by herpes simplex virus-1 (HSV-1) induced NOTCH activation in HSV-1 infected glioma cells by downregulating FIH-1 expression.16 Long noncoding RNA (lncRNA) Cancer Susceptibility 2 (CASC2) inhibits the growth, migration, and invasion of thyroid cancer cells through sponging miR-18a-5p/FIH1 axis.17 Exosomal miR-135b enhances angiogenesis by targeting FIH-1 in myeloma cells.18 Therefore, how FIH-1 participates in tumor progression was worth studying. Here, we report that HBV-miR-3 promotes angiogenesis via FIH-1 in HCC, which suggests that HBV-miR-3 may be a new marker and therapeutic target for HBV-related HCC.
Materials and Methods
Clinical Samples
A total 108 HCC tissues were obtained after obtaining informed consent from patients who underwent liver resection at Western China Hospital of Sichuan University from 2018 to 2019. All subjects gave their informed consent for inclusion before they participated in the study in which the tissue would be used for future study. The study was conducted in accordance with the Declaration of Helsinki, and the protocols were approved by the Ethics Committee of Western China Hospital of Sichuan University (2016–91).
Cell Lines and Reagents
The HCC cell line HepG2 was purchased from Procell Life Science & Technology (Wuhan, China). HepG2.2.15 and HUVECs were purchased from Cellcook (Guangzhou, China). HCC cells were maintained in Dulbecco’s modified Eagle medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with fetal bovine serum (FBS; Gibco) at 37°C, 5% CO2. HUVECs were maintained in an endothelial cell growth medium-2 (EGM-2 BulletKit; Lonza, Basel, Switzerland) at 37°C, 5% CO2.
MicroRNA and Plasmid Transfection
The HBV-miR-3 agomir, agomir_Mut, antagomir, and their negative controls were purchased from GenePharma (Shanghai, China). Cells were cultured in culture medium without FBS or antibiotics for 24 hr prior to transfection. The agomir and antagomir were transfected using Lipofectamine RNAiMAX (Invitrogen) in accordance with the manufacturer’s protocol. At 4–6 hr post-transfection, the cells were washed with PBS to completely remove the agomir or antagomir constructs in the medium and were then cultured in DMEM with 10% FBS. Transfected cells were harvested at 48 hr, total cellular RNA, protein, and supernatants were isolated for RT-qPCR, western blot, ELISA analyses, or lumen formation assay. All transfections were performed in triplicate.
Real-Time Quantitative PCR
Total RNA isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) was then subjected to cDNA synthesis using Prime ScriptTM RT reagent Kit (Takara, Japan). MiRNA was extracted and purified with miRNeasy® Mini kit (Hilden, Germany) according to the manufacturer’s instructions. ACTB, HIF-1α, VEGFA, FIH-1, and VEGFR2 quantitative PCR probes were synthesized from Tsingke Biotechnology (Beijing, China). The expression of selected genes was determined in triplicate using LightCycler®96 (Roche, Rotkreuz, Switzerland). HBV-miR-3 RT, U6 RT for cDNA reverse transcription and HBV-miR-3 Forward primer, U6 Forward primer, miRNA Reverse primer designed for stem-loop RT-PCR were synthesized from Tsingke Biotechnology (Beijing, China). The expression of selected genes was determined in triplicate using C1000TM Thermal Cycler (Bio-rad, USA). The relative fold changes were calculated by 2−ΔΔCT method.
Western Blotting
Cell lysates were extracted using RIPA buffer (Cell Signaling Technology), and the following primary antibodies were used for Western blotting: anti-VEGFA monoclonal, ab214424 (Abcam); anti-HIF-1α monoclonal, ab51608 (Abcam); Anti-HIF1AN/FIH-1 monoclonal, ab233141 (Abcam), and anti-β-actin monoclonal (Zsbio, Beijing, China). Immune complexes were visualized using enhanced chemiluminescence detection reagents (4A BIOTECH, Beijing, China).
ELISA for VEGFA Detection
At 4–6 hr post-transfection, the cells were washed with PBS to completely remove the agomir or antagomir constructs in the medium and were then cultured in DMEM with 0.5% FBS. At 48 hr the supernatants of HepG2 and HepG2.2.15 cells were harvested for ELISA or lumen formation assay. VEGFA using RayBio® Human VEGF-A ELISA Kit (RayBiotech, USA) was used to examine VEGFA expression in cell culture medium following the manufacturer’s protocol. Absorbance was determined in a microtiter plate reader (Bio-rad, USA) with dual-wavelength measurement (450 nm).
Lumen Formation Assay
The lumen formation assay was assessed in 96-well plates coated with a growth factor reduced Matrigel (Corning, NY, USA) as described previously.19 HUVECs (3 × 104 cells/well) were seeded on top of Matrigel (50 μL/well)-coated wells in EGM-2 medium. When capillary-like structures (lumens) are formed, the upper medium is replaced by different kinds of HepG2 or HepG2.2.15 cell culture supernatants. After 6 hr, lumens were taken photos by phase-contrast microscopy OLYMPUS CKX53 (Olympus, Japan). Junctions and lumen lengths of HUVEC were analyzed by Image J.
Dual-Luciferase® Reporter Assay
The segments of HBV-miR-3 binding sites WT or MUT in the 3’-UTR of FIH-1 were separately constructed into the luciferase pmirGLO reporter vector (Promega, USA) containing firefly luciferase (Luc2) as a reporter and Renilla luciferase (RLuc) as a control. The plasmids were transfected using Lipofectamine 3000 or co-transfected with HBV-miR-3 antagomir using Lipofectamine 2000. At 48 h post-transfection, cell lysates were collected and the luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol.
Animal Studies
NOD/ShiLtJGpt-Prkdcem26Cd52Il2rgem26Cd22/Gpt Coisogenic Genetically Engineered Immunodeficient mice (NCG mice) were purchased from GemPharmatech Co. Ltd (Nanjing, China). Mice were housed under specific pathogen-free conditions with a 12-h light/dark cycle and provided ad libitum access to tap water and food. HepG2 or HepG2.2.15 cells (106 cells) were resuspended in 200 μL of a 1:1 DMEM: Matrigel (Corning, BD Biosciences) mixture and subcutaneously injected into 4- to 6-week-old NCG mice. Once tumors had reached a measurable size, mice were randomly divided into two groups (each n = 4). Each HepG2 xenograft was injected with 5nmol HBV-miR-3 agomir or agomir NC dissolved in PBS for 12 days, and each HepG2.2.15 xenograft was injected with 15μg HBV-miR-3 antagomir or antagomir NC dissolved in PBS for 16 days. The size of subcutaneous tumors was recorded twice a week. The experimental protocol was approved by the Sichuan University Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences.
Statistical Analysis
Overall survival analysis, Student’s t-test, one-way ANOVA, and Pearson correlation analysis were performed using GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA). A p value of less than 0.05 was considered significant.
Results
High HBV-miR-3 Expression is Associated with Poor Prognosis of HBV-Related HCC Patients and HCC Angiogenesis
By exploring the levels of HBV-miR-3 in HBV-related HCC, we found that HBV-miR-3 can only be detected in HCC tissues of HBsAg(+)HBcAb(+) patients (n=108), not detected in HCC tissues of HBsAg(-)HBcAb(+) patients (n=23, Figure 1A). Next, we analyzed HBV-miR-3 expressions in these HBsAg(+)HBcAb(+) patients by RT-qPCR. Sequences of the primers and HBV-miR-3-related microRNAs used in this study are detailed in Table S1. The relative HBV-miR-3 expressions in HCC tissues were normalized to Patient 1 and listed in Table S2. We ranked HBV-miR-3 expressions in HCC tissues of the 108 patients and divided them equally into 2 groups (HBV-miR-3 High and HBV-miR-3 Low). Patients with high HCC tissue HBV-miR-3 (n=54) showed poorer overall survival than patients with low HCC tissue HBV-miR-3 (n=54, P = 0.0308, R=0.2815, Figure 1B). We also evaluated the clinicopathological characteristics of HBV-miR-3 at high and low HCC, and again, HBV-miR-3 high HCC was positively associated with microscopic portal vein invasion (MVI), advanced BCLC stages, and tumor size (Table 1).
 |
Table 1 Clinical Characteristics of HBV-miR-3 High and Low HCC Patients in Cohort |
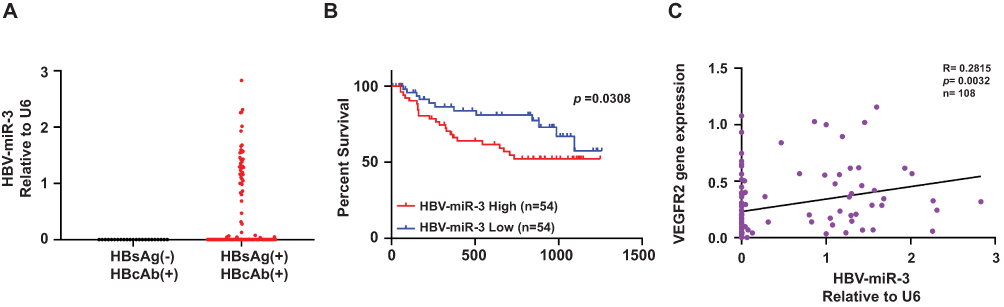 |
Figure 1 High HBV-miR-3 expression is associated with poor prognosis of HBV-related HCC patients and HCC angiogenesis. (A) HBV-miR-3 levels in HBsAg(-)HBcAb(+) patients (n=23) and HBsAg(+)HBcAb(+) patients (n=108). (B) Kaplan–Meier survival curves according to HBV-miR-3 expression in the tissue of HBV-related HCC patients (classified as low or high using the median value of all patients normalized to patient 1). Survival curves of two groups were compared using the Log rank test. (C) Pearson Correlation analysis of HBV-miR-3 and VEGFR2 gene expression. |
To further explore the involvement of HBV-miR-3 in HBV-related HCC process, we next analyzed the correlation between HBV-miR-3 and the specific angiogenetic marker vascular endothelial growth factor receptor 2 (VEGFR2) by RT-qPCR. The relative VEGFR2 expression levels of HCC tissues were also normalized to Patient 1 and listed in Table S2. We found that HBV-miR-3 was correlated with VEGFR2 (P = 0.0032, Figure 1C). These data indicate that high HBV-miR-3 in tumor tissues is related to dismal overall survival and involved in angiogenesis process of HBV-related HCC, so it could be regarded as a prognostic biomarker of HBV-related HCC.
HBV-miR-3 Can Be Detected in HBV Infected HCC Cell Line and Negatively Correlated with FIH-1
We next investigate the role of HBV-miR-3 in angiogenic signaling in HCC. First, we analyzed HBV-miR-3 level in HepG2 and HepG2.2.15 cells. HepG2.2.15 cell line is derived from HCC cell line HepG2 with stable transfection HBV expression.20 We found that HBV-miR-3 can only be detected in HepG2.2.15 not in HepG2 (Figure 2A). We also explored the protein expression of HIF-1α, VEGFA, and FIH-1, which are in classical hypoxia signaling. Interestingly, we found that HIF-1α and VEGFA expression were higher in HepG2.2.15 cells than HepG2 cells but on the contrary FIH-1 expression was lower in HepG2.2.15 cells than HepG2 cells (Figure 2B). This indicated that HBV was involved in hypoxia signaling of HCC. To clarify whether HBV-miR-3 regulates hypoxia signaling, we searched the potential HBV-miR-3 binding sites in the 3ʹ-UTR of a lot of proteins’ mRNA in TargetScan (https://www.targetscan.org/vert_80/). We found that FIH-1 was among the candidates. Next, we checked the 3ʹ-UTR sequence of FIH-1 by NCBI Gene (https://www.ncbi.nlm.nih.gov/gene/) and found two possible HBV-miR-3 binding sites. We found that the two possible binding sites 5’-ACATCC-3’ were located in 3319–3324 and 4735–4740 in 3ʹ-UTR FIH-1 (Figure 2C). We also verified that there are no possible binding sites in mRNAs of pVHL, PHD1-3, HIF-1α, and VEGFA. To confirm the correlation between HBV-miR-3 and FIH-1, we first investigated FIH-1 expression by Western blot in those 108 patients (Figure S1A). The relative FIH-1 expression levels were normalized to Patient 1 and listed in Table S2.We found that FIH-1 expressions were negatively correlated with HBV-miR-3 levels (Figure 2D). Also, we explored whether HBx affects FIH-1 and the results showed that HBx could not affect FIH-1 expression (Figure S1B). These data indicate that HBV-miR-3 may regulate FIH-1 protein expression in HCC by interacting with the 3ʹ-UTR in mRNA of FIH-1.
 |
Figure 2 HBV-miR-3 can be detected in HBV infected HCC cell line and negatively correlated with FIH-1. (A) HBV-miR-3 levels in HepG2 and HepG2.2.15 cells. (B) HIF-1α, VEGFA and FIH-1 protein expressions in HepG2 and HepG2.2.15 cells. (C) Predicted HBV-miR-3 binding sites in the 3’-UTR of FIH-1 mRNA. (D) Pearson Correlation analysis of HBV-miR-3 and FIH-1 protein expression in HBV-related HCC patients. The error bars represent the SD from at least three independent biological replicates. |
HBV-miR-3 Promotes VEGFA Secretion from HCC Cells by Regulating FIH-1/HIF-1α/VEGFA Signaling
Hypoxia signaling pathway plays a crucial role in tumor development. We then investigated the mechanism by which HBV-miR-3 regulating hypoxia signaling in HCC. HepG2 cells were transfected with different doses of HBV-miR-3 agomir. The qPCR results showed that the mRNAs of FIH-1and HIF-1α were not changed but VEGFA mRNA levels were increased in a dose-dependent manner (Figure 3A, B, and C). Next, we explored the protein expressions of hypoxia signaling. Western blot results showed that the protein expression of FIH-1 was reduced, while HIF-1α and VEGFA were induced in a dose-dependent manner following the transfection of HBV-miR-3 agomir (Figure 3D). In addition, we confirmed that HBV-miR-3 did not affect PHDs in HepG2 cells (Figure S2A). The ELISA results also showed that VEGFA levels in the supernatant of HepG2 cells were increased in a dose-dependent manner (Figure 3E). To examine the angiogenetic effect of HBV-miR-3, we tested the lumen formation of human umbilical vein endothelial cell (HUVEC) by transferring HBV-miR-3 agomir-transfected HepG2 cell culture medium to HUVECs (Figure 3F). In the lumen formation assay, the conditioned medium of HBV-miR-3 agomir-transfected HepG2 cells induced junctions and lumen lengths of HUVECs in an agomir-dose-dependent manner (Figure 3G-I). These results suggest that HBV-miR-3 inhibits FIH-1 expression and promotes VEGFA secretion from HCC. It is involved in angiogenesis of HBV-related HCC by up-regulating HIF-1α/VEGFA expression.
 |
Figure 3 HBV-miR-3 promotes VEGFA secretion from HCC cells by regulating FIH-1/HIF-1α/VEGFA signaling. (A–C) Gene expression of FIH-1, HIF-1α and VEGFA in HepG2 cells after been transfected with different concentration of HBV-miR-3 agomir and negative control. (D) Western blot analysis of FIH-1, HIF-1α and VEGFA protein expression of HepG2 cells after been transfected with different concentration of HBV-miR-3 agomir. (E) ELISA analysis of VEGFA levels of supernatant of HepG2 cells transfected with different concentration of HBV-miR-3 agomir and negative control. (F) Schematic of lumen formation assay. Culture supernatants were collected from HBV-miR-3 agomir transfected HepG2 cells at 48 h. HUVEC were treated with the supernatants from HepG2 for 6h. (G) Representative pictures of lumen formation assay. Scale bar=200 μm. (H) Junction numbers of HUVEC of lumen formation assay. (n=3). (I) Total lumen lengths of HUVEC for lumen formation assay. (n=3). The error bars represent the SD from at least three independent biological replicates. ANOVA was used to calculate p values, represented as * p< 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001; n.s, not significant. |
To confirm whether HBV-miR-3 directly affects lumen formation of HUVECs, we transfected HUVECs with HBV-miR-3 and lumen formation assay was performed. Western blot results showed that tip cell marker VEGFR2 and DLL4 were inhibited by HBV-miR-3, while stalk cell marker Jagged1 was increased but Notch-1 was not changed (Figure S3A). The lumen formation assay showed no differences among HUVECs transfected with different concentrations of HBV-miR-3 agomir (Figure S3B). These data indicate that HBV-miR-3 does not have a direct angiogenetic effect on HUVECs.
Inhibition of HBV-miR-3 Decreases VEGFA Secretion from HCC Cells by Regulating FIH-1/HIF-1α/VEGFA Signaling
We further confirm the effect of HBV-miR-3 in regulating angiogenesis. HepG2.2.15 cells were transfected with different doses of HBV-miR-3 antagomir for silencing the effect of HBV-miR-3. The qPCR results showed that the mRNAs of FIH-1 and HIF-1α were not altered but VEGFA mRNA levels were decreased in a dose-dependent manner (Figure 4A-C). Western blot results showed that the protein expression of FIH-1 was induced, while HIF-1α and VEGFA were reduced in a dose-dependent manner following the transfection of HBV-miR-3 antagomir (Figure 4D). We also confirmed that inhibition of HBV-miR-3 did not affect PHDs in HepG2.2.15 cells (Figure S2B). Also, ELISA result indicated that VEGFA levels in the supernatants of HepG2.2.15 cells were decreased in a dose-dependent manner (Figure 4E). In the lumen formation assay, the conditioned medium of HBV-miR-3 antagomir-transfected HepG2.2.15 cells reduced junctions and lumen lengths of HUVECs in an antagomir-dose-dependent manner (Figure 4F-I). These results suggest that silencing HBV-miR-3 promotes FIH-1 expression, resulting in inhibition of angiogenesis in HCC.
 |
Figure 4 Downregulating HBV-miR-3 induces FIH-1 protein expression. (A–C) Gene expression of FIH-1, HIF-1α and VEGFA in HepG2.2.15 cells after been transfected with different concentration of HBV-miR-3 antagomir and negative control. (D) Western blot analysis of FIH-1, HIF-1α and VEGFA protein expression of HepG2.2.15 cells after been treated with different concentration of HBV-miR-3 antagomir. (E) ELISA analysis of VEGFA levels of supernatant of HepG2 cells treated with different concentration of HBV-miR-3 antagomir and negative control. (F) Schematic of the lumen formation assay. Culture supernatants were collected from HBV-miR-3 antagomir transfected HepG2.2.15 cells at 48 h. HUVEC were treated with the supernatants from HepG2.2.15 for 6h. (G) Representative pictures of lumen formation assay. Scale bar=200 μm. (H) Junction numbers of HUVEC of lumen formation assay. (n=3). (I) Total lumen lengths of HUVEC of lumen formation assay. (n=3). The error bars represent the SD from at least three independent biological replicates. ANOVA was used to calculate p values, represented as ** p < 0.01; *** p < 0.001; **** p < 0.0001; n.s, not significant. |
Next, we confirmed whether HBV-miR-3 antagomir can affect HIF-1α/VEGFA signaling in non-HBV-related HCC cells such as Huh7. We transfected Huh7 cells with HBV-miR-3 antagomir and Western blot analysis revealed that HBV-miR-3 antagomir does not have biological effect on HIF-1α/VEGFA signaling (Figure S4). The inhibition of HIF-1α/VEGFA signaling in HBV-miR-3 antagomir-transfected HepG2.2.15 cells may be due to the suppression of HBV-miR-3.
HBV-miR-3 Promotes Angiogenesis by Repression FIH-1 and Inducing HIF-1α/VEGFA Signaling in vivo
Based on all the in vitro findings, we thus assessed the angiogenic effect of HBV-miR-3 on HCC xenografts. Compared with agomir NC, HBV-miR-3 agomir significantly promoted the growth of tumors derived from HepG2 cells (Figure 5A-C) and HBV-miR-3 antagomir significantly inhibited the growth of tumors derived from HepG2.2.15 cells (Figure 5D-F). In addition, HBV-miR-3 reduced FIH-1 and induced HIF-1α, VEGFA, and VEGFR2 expression of tumors derived from HepG2 cells (Figure 5G-I). While inhibiting HBV-miR-3-induced FIH-1 and reduced HIF-1α, VEGFA, and VEGFR2 expression of tumors derived from HepG2.2.15 cells (Figure 5J-L), suggesting that HBV-miR-3 is an angiogenetic factor of HBV-related HCC and it could be a promising therapeutic target option for HCC patients.
 |
Figure 5 HBV-miR-3 induces HCC angiogenesis and inhibiting HBV-miR-3 reduces HCC angiogenesis in vivo. (A–C) Effect of HBV-miR-3 on the growth of HepG2 xenografts. Mice were sacrificed after being injected with 5nmol HBV-miR-3 agomir or agomir NC twice a week for 12 days. (D, E and F) Effect of inhibiting HBV-miR-3 on the growth of HepG2.2.15 xenografts. Mice were sacrificed after being injected with 15μg HBV-miR-3 antagomir or antagomir NC twice a week for 16 days. (G and H) Western blot analysis of FIH-1, HIF-1α, VEGFA and VEGFR2 expression in xenograft tumors derived from HepG2 cells. β-Actin was used as the reference for quantifying protein expression. (I) Representative IHC staining images of FIH-1, HIF-1α and VEGFR2 expression in HepG2 cell xenografts. Scare bars=100μm. (J and K) Western blot analysis of FIH-1, HIF-1α, VEGFA and VEGFR2 expression in xenograft tumors derived from HepG2.2.15 cells. β-Actin was used as the reference for quantifying protein expression. (L) Representative IHC staining images of FIH-1, HIF-1α and VEGFR2 expression in HepG2 cell xenografts. Scare bars=100μm. The error bars represent the SD from at least three independent biological replicates. Student’s t-test was used to calculate p values, represented as * p < 0.05; ** p < 0.01, *** p < 0.001; **** p < 0.0001. |
HBV-miR-3 Represses the Expression of FIH-1 by Targeting the 3’-UTR of FIH-1 mRNA
To further confirm the mechanism of how HBV-miR-3 regulates angiogenesis in HCC, we constructed the mutation of HBV-miR-3 by replacing the conservative binding sites in HBV-miR-3 agomir (Figure 6A). Next, HepG2 cells were transfected with 10nM HBV-miR-3 agomir, HBV-miR-3_Mut agomir, or agomir control. Western blot results showed that compared to control, the protein expression of FIH-1 was reduced in HBV-miR-3 agomir transfected cells but not in HBV-miR-3_Mut agomir transfected cells, while HIF-1α and VEGFA were induced in HBV-miR-3 agomir transfected HpG2 cells but not in HBV-miR-3_Mut agomir transfected HepG2 cells (Figure 6B). ELISA results indicated that VEGFA levels in the supernatants of HepG2 cells transfected with 10nM HBV-miR-3 agomir were decreased but not changed in the supernatants of HepG2 cells transfected with 10nM HBV-miR-3_Mut agomir (Figure 6C). Moreover, in lumen formation assay, the junctions and lumen lengths of HUVECs were induced by supernatants of HBV-miR-3 agomir transfected HpG2 cells but not by supernatants of HBV-miR-3_Mut agomir transfected HepG2 cells (Figure 6D-F). These data verified the functional sequences of HBV-miR-3.
 |
Figure 6 HBV-miR-3 inhibits FIH-1 protein expression by targeting 3’-UTR of FIH-1 mRNA. (A) Mutant sequences of HBV-miR-3. (B) Western blot analysis of FIH-1, HIF-1α and VEGFA protein expression of HepG2 cells after been transfected with HBV-miR-3 agomir control, 10nM agomir and mutant agomir. (C) ELISA analysis of VEGFA levels of supernatant of HepG2 cells transfected with HBV-miR-3 agomir, HBV-miR-3 agomir_Mut and negative control. (D) Representative pictures of lumen formation assay. Scale bar=200 μm. (E) Junction numbers of HUVEC of lumen formation assay. (n=3). (F) Total lumen lengths of HUVEC for lumen formation assay. (n=3). (G) Wild-type and mutant fragments of the 3ʹ-UTR of FIH-1 mRNA. (H and I) Luciferase activity of HepG2 cells transfected with HBV-miR-3 agomir and the wild-type or mutant fragment of the 3ʹ-UTR of FIH-1 mRNA. (J) Luciferase activity of HepG2.2.15 cells co-transfected with HBV-miR-3 antagomir and the wild-type of the 3ʹ-UTR of FIH-1. The error bars represent the SD from at least three independent biological replicates. Student’s t-test was used to calculate p values, represented as * p< 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. The error bars represent the SD from at least three independent biological replicates. ANOVA was used to calculate p values, represented as * p< 0.05; ** p < 0.01; n.s, not significant. |
We also constructed luciferase reporter vectors containing the mutations of 3’-UTR the wild-type (WT) or mutant21 forms of the HBV-miR-3-binding sequence on the FIH-1 3’-UTR. To control for specificity in this assay, 6 nt mutations were introduced into the candidate binding sites at the 3ʹ-UTR of FIH-1 (Figure 6G). HepG2.2.15 cells were transfected with luciferase reporter plasmids carrying each FIH-1-WT or FIH-1-MUT 3ʹ-UTR segments. The luciferase activities of the MUT groups were higher than those of the WT groups (Figure 6H and I). Next, HepG2.2.15 cells were co-transfected with HBV-miR-3 antagomir/NC together with luciferase reporter vectors carrying 2 FIH-1-WT segments (3’UTR-1 and 3’UTR-2). The luciferase activity of antagomir co-transfected groups was higher than that of NC groups (Figure 6J). These data indicate that the predicted candidate binding sites are specific and critical for HBV-miR-3 to interact with mRNA of FIH-1.
Discussion
HBV is the most common pathogen for HCC with 55% of worldwide HCC cases reported in China.22 The mechanisms of HBV inducing liver cancer include integration of HBV genome to host genome, accumulation of HBV genome mutations, expression of HBx protein, expression of the truncated mutant HBsAg and inflammatory response.23–25 Although anti-HBV therapy with nucleoside analogues (NAs) can significantly reduce the risk of liver cancer and the recurrence rate of liver cancer after surgery, the occurrence and recurrence of liver cancer cannot be completely avoided. An investigation of the mechanisms of HBV regulating the occurrence and development of liver cancer has clinical significance.
Viral microRNAs (v-miR) regulate multiple transcripts of a virus or host.26,27 It was found that many viruses encoded v-miRs, suggesting that viruses also exploit this basic model of gene regulation. For example, Epstein–Barr virus (EBV) encoded 24 kinds of v-miRs, which can modulate various biological processes including viral replication, latency maintenance, immune escape, cell proliferation, apoptosis, metabolism, and tumor metastasis;28 Kaposi sarcoma herpesvirus (KSHV) encoded miRNAs could affect the differentiation status of infected cells, and thereby contribute to KSHV-induced oncogenesis;29 Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) encoded v-miRs subsequently targeted functional genes of host to cause illnesses.30 HBV-encoded miRNAs (HBV-miRs) were first discovered by deep sequencing in 2016,8 which are a group of v-miRs generated from 3.5-kb, 2.4-kb, and 2.1-kb transcripts. Till now, HBV-miRs including HBV-miR-6, -7, and −8 were discovered.31 Among all the HBV-miRs, HBV-miR-3 was mostly studied. It was reported HBV-miR-3 was involved in regulating viral replication and host genes to induce HCC cell proliferation. HBV-miR-3 could be detected in the serum of chronic hepatitis B patients with high viral load, which is correlated with the HBV-DNA and HBV pre-genomic RNA (pgRNA) loads in the serum. Oral NAs cannot inhibit the serum HBV-miR-3 load, but the use of long-acting interferon therapy can effectively reduce its load in the serum.9 HBV-miR-3 can inhibit HBV replication by binding to 3.5-kb mRNA, inhibiting the production of core protein (HBc) and pgRNA.8 HBV-miR-3 can also activate the JAK/STAT signaling pathway by inhibiting the expression of Suppressor Of Cytokine Signaling 5 (SOCS5) in host hepatocytes, promoting the M1 polarization of macrophages to secrete more interleukin-6 (IL-6) and to enhance interferon-related immune esponse, leading to inhibition of HBV replication.10 It is worth noting that in studies related to liver cancer, HBV-miR-3 promoted the occurrence and development of liver cancer by inhibiting the expression of known tumor suppressors such as PPM1A, DIXDC1, and PTEN.11,12 The role of HBV-miR-3 in HCC angiogenesis is lack of studying, therefore we aimed to investigate the functional roles of HBV-miR-3 in the activation of hypoxia signaling in HCC. Recently, new HBV-miRs HBV-miR-6, -7 and −8 were discovered.31
Hepatocellular carcinoma is a solid tumor with the characteristics of tissue hypoxia and active angiogenesis.1 VEGFA is the most important cell factor in angiogenesis and regulated directly by HIF-1. The heterodimer HIF-1 is composed of a continuously synthesized and degraded HIF-1α subunit and a constitutively expressed HIF-1β subunit. HIF-1α is mediated by ubiquitination and proteasomal degradation, whose stabilization requires the inhibition of pVHL, PHDs, and FIH-1. HIF-1α keeps stabilization, transferring into the nucleus and forms the HIF-1α/HIF-1β/CBP-p300 complex. The complex interacts with hypoxia response element32 and triggers the transcription of hypoxia-inducible genes (Figure 7A). Under normoxia condition, FIH-1 hydroxylates Asn803 of HIF-1α thereby blocking the interaction of HIF-1α with CBP-p300. Asn803-hydroxylated HIF-1α was subsequently hydroxylated in Pro402 and Pro564 by PHD1 and PHD2. HIF-1α with three hydroxylated residues then binds with pVHL, leading to ubiquitination and proteasomal degradation (Figure 7B).33 Inhibition of PHDs, FIH-1, or pVHL blocks degradation of HIF-1α thus elevates the expression of VEGFA. Viruses could enhance HIF-1α levels by modulating its transcription, translation, or stabilization through viral proteins.34 Human papillomavirus (HPV) derived E6 could mediate p53 degradation to enhance HIF-1α stabilization and E7 could bind to HDACs to enhance HIF transcription complex formation.35,36 EBV derived EBNA-3 and EBNA-5 binded to PHD-1 and PHD-2 to inhibit the breakdown of HIF-1α.37 KSHV derived vIRF-3 could bind directly to HIF-1α, leading to an increase in nuclear translocation and activity.38 In previous studies, HBV was confirmed to promote the progression of angiogenesis of HCC by HBx and small protein of SHBs.4 HBx increased the protein level of HIF-1α by the direct interacting with HIF-1α, thus repressing the ubiquitin-dependent degradation of HIF-1α by inhibition the interaction with pVHL.5 HBx could also enhance HDAC1 transcription, leading to increased deacetylation of the ODD of HIF-1α, inhibiting its degradation.6 HBx could also enhance HDAC1 transcription, leading to increased deacetylation of the ODD of HIF-1α, inhibiting its degradation.6 Despite HIF-1α, HBx protein was also reported promoting expression of angiopoietin-2 (Ang-2) in liver tissue via the activation of MAPK39 and to promote expression of VEGFA through mTOR signaling.40 In this study, we newly found that HBx does not affect the expression of FIH-1. In addition to HBx, small protein of hepatitis B virus surface antigens (SHBs) could promote HCC angiogenesis via endoplasmic reticulum stress signaling to upregulate the expression of VEGFA.7 Also, Pre-S mutants could elevate VEGFA expression in ground glass hepatocytes (GGH) in HBV infection.41 Whether any other mechanism of HBV regulates angiogenesis still need to be clarified. HBV-miRs were discovered in recent years and studied far from enough.
 |
Figure 7 HBV-miR-3 promotes HBV-related HCC by targeting 3’UTR of FIH-1 mRNA. (A) Under hypoxia condition, HIF-1α keeps stabilization and transferring into nucleus, forming HIF-1α/HIF-1β/CBP-p300 complex, interacting with HRE and triggering hypoxia inducible genes. (B) Under normoxia condition, HIF-1α was hydroxylated by FIH-1, PHD1 and PHD2, then binding with pVHL and degraded by proteasome. (C) HBV-miR-3 is generated from 3.5-kb, 2.4-kb, and 2.1-kb transcripts of cccDNA. HBV-miR-3 inhibits FIH-1 expression, leading to the up-regulation of HIF-1α and VEGFA to promote angiogenesis in HCC. |
In this study, we first demonstrated that HBV-miR-3 in HBsAg(+)HBcAb(+) HCC tissues rather than HBsAg(-)HBcAb(+), and high HBV-miR-3 levels in HCC tissues are associated with poorer prognosis. Next, we demonstrated the correction between HBV-miR-3 and angiogenesis marker VEGFR2 in HCC. These findings implied that HBV-miR-3 is involved in angiogenesis process of HBV-related HCC. Based on these findings, we confirmed that HBV-miR-3 reduced FIH-1 but induced HIF-1α/VEGFA expression and VEGFA secretion from HCC cells, which resulted in the activation of HUVEC lumen formation. Vise versa, inhibition of HBV-miR-3-induced FIH-1 but reduced HIF-1α/VEGFA expression and VEGFA secretion from HCC cells, which resulted in the suppression of HUVEC lumen formation. We also confirm the angiogenic effect of HBV-miR-3 by HCC xenograft models in vivo. In addition, we demonstrated the mechanism that HBV-miR-3 suppressed FIH-1 expression by targeting the sequence 5’-ACATCC-3’ in 3319–3324 and 4735–4740 in 3’-UTR of FIH-1 mRNA. These results indicate that HBV-miR-3 inhibiting FIH-1 expression to increase the stabilization of HIF-1α, thus stimulating VEGFA expression and promoting angiogenesis in HCC. Therefore, HBV-miR-3 and FIH-1 may be a potential therapeutic target for HCC.42 It is also interesting that HBV-miR-3 did not affect HUVECs. We think this phenomenon may be due to HUVEC lumen formation being dominantly modulated by VEGFA, and the key regulator NOTCH-1 of tip-stalk cell transition was not regulated by HBV-miR-3. But whether HBV-miR-3 could regulate other HCC-related biomarkers such as Stanniocalcin 2, APEX1 or affect HCC tumor microenvironment remodeling is still worth further investigation.43–45
Our study first showed the crucial mechanism of HBV-miR-3 by which HBV affects HCC angiogenesis: inhibiting FIH-1 thus inducing HIF-1α/VEGFA (Figure 7C). We discovered another mechanism for how HBV regulates angiogenesis in HCC besides viral proteins. In fact, HBV-miR-3 can also target many other sites of 3’-UTR of multiple proteins affecting different cell functions. Using TargetScan and miRBD, we found other targets of HBV-miR-3 including tumor suppressors AIM1 (absent in melanoma 1), ESR1 (estrogen receptor 1), and USP31 (ubiquitin-specific peptidase 31), as well as tumor promoter CTNND1 (catenin delta 1), which means HBV-miR-3 is an important host gene regulator in chronic hepatitis B pa or HCC patients and needs to be studied in more depth.
Conclusion
Our study revealed that HBV-miR-3 targets 3’-UTR of FIH-1 mRNA to inhibit FIH-1 expression, thus activating HIF-1α/VEGFA to promote angiogenesis in HCC. Further studies on HBV-miR-3 may clarify more mechanisms of how HBV affects HCC.
Abbreviations
3’-UTR, Three prime untranslated region; AIM1, Absent in melanoma 1; Ang-2, Angiopoietin-2; APEX1, Apyrimidinic endodeoxyribonuclease 1; BCLC stages, Barcelona clinic liver cancer stages; CASC2, Cancer Susceptibility 2; CBP-p300, CREB-binding protein and p300; CTNND1, Catenin delta 1; DIXDC1, DIX domain containing 1; DLL4, delta-like canonical Notch ligand 4; EBNAs, Epstein–Barr virus nuclear antigens; EBV, Epstein–Barr virus; ELISA, enzyme-linked immunosorbent assay; ESR1: Estrogen receptor 1; FIH-1, Factor Inhibiting Hypoxia-inducible factor 1; GGH, Ground glass hepatocytes; HBc, Hepatitis B core protein; HBcAb, Hepatitis B core antibody; HBsAg, Hepatitis B surface antigen; HBV, Hepatitis B virus; HBV-miRs, HBV-encoded miRNAs; HBV-miR-3, Hepatitis B Virus-Encoded MicroRNA; HBx, HBV X protein; HCC, Hepatocellular carcinoma; m HDAC1, Histone deacetylase 1; HUVEC, Human umbilical vein endothelial cell; HIF-1α, Hypoxia-inducible factor 1-alpha; HIF-1β, Hypoxia-inducible factor 1-beta; HIF1AN, Hypoxia Inducible Factor 1 Subunit Alpha Inhibitor; HPV, Human papillomavirus; HSV-1, herpes simplex virus-1; IL-6, Interleukin-6; KSHV, Kaposi sarcoma herpesvirus; lncRNA, Long noncoding RNA; MVI, microscopic portal vein invasion; NAs, nucleoside analogues; NCBI, National Center for Biotechnology Information; ODD, oxygen-dependent degradation; pgRNA, pre-genomic RNA; PHDs, prolyl hydroxylases; PPM1A, Protein phosphatase 1A; PTEN, Phosphatase and tensin homolog; pVHL, Von Hippel-Lindau protein; SARS-CoV-2, Severe acute respiratory syndrome coronavirus 2; SHBs, Small protein of hepatitis B virus surface antigen; SOCS5, Suppressor Of Cytokine Signaling 5; USP31, Ubiquitin specific peptidase 31; VEGFA, Vascular Endothelial Growth Factor A; VEGFR2, Vascular endothelial growth factor receptor 2.
Data Sharing Statement
The datasets used and materials used in this study are available upon reasonable request.
Ethics Approval and Consent to Participate
Clinical Samples
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the ethics committee of West China Hospital of Sichuan University (No. 2016-91).
Animal Study
The animal study was approved by the Ethics Committee of West China Hospital, Sichuan University (No.20230303014), and conformed to the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences.
Consent for Publication
All involved parties consent to publication.
Acknowledgments
We thank Yitian Wang and Jiayi Wang for their excellent technical assistance.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Key Research and Development Program of China (NO.2022YFC2304800) and 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYGD20009).
Disclosure
The authors declare that they have no competing interests.
References
1. Morse MA, Sun W, Kim R., et al. The role of angiogenesis in hepatocellular carcinoma. Clin Cancer Res. 2019;25(3):912–920. doi:10.1158/1078-0432.CCR-18-1254
2. Li H. Angiogenesis in the progression from liver fibrosis to cirrhosis and hepatocelluar carcinoma. Expert Rev Gastroenterol Hepatol. 2021;15(3):217–233. doi:10.1080/17474124.2021.1842732
3. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. 2018;68(6):394–424. doi:10.3322/caac.21492
4. Reyes A, Duarte LF, Farias MA, et al. Impact of hypoxia over human viral infections and key cellular processes. Int J Mol Sci. 2021;22:15. doi:10.3390/ijms22157954
5. Moon EJ, Jeong CH, Jeong JW, et al. Hepatitis B virus X protein induces angiogenesis by stabilizing hypoxia-inducible factor-1alpha. FASEB J. 2004;18(2):382–384. doi:10.1096/fj.03-0153fje
6. Yoo YG, Na TY, Seo HW, et al. Hepatitis B virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene. 2008;27(24):3405–3413. doi:10.1038/sj.onc.1211000
7. Wu SX, Ye SS, Hong YX, et al. Hepatitis B virus small envelope protein promotes hepatocellular carcinoma angiogenesis via endoplasmic reticulum stress signaling to upregulate the expression of vascular endothelial growth factor A. J Virol. 2022;96(4): e0197521. doi:10.1128/jvi.01975-21
8. Yang X, Li H, Sun H, et al. Hepatitis B virus-encoded microRNA controls viral replication. J Virol. 2017;91(10). doi:10.1128/JVI.01919-16
9. Gan W, Chen X, Wu Z, et al. The relationship between serum exosome HBV-miR-3 and current virological markers and its dynamics in chronic hepatitis B patients on antiviral treatment. Ann Translat Med. 2022;10(10):536. doi:10.21037/atm-22-2119
10. Zhao X, Sun L, Mu T, et al. An HBV-encoded miRNA activates innate immunity to restrict HBV replication. J Mol Cell Bio. 2020;12(4):263–276. doi:10.1093/jmcb/mjz104
11. Chavalit T, Nimsamer P, Sirivassanametha K, et al. Hepatitis B virus-encoded microRNA (HBV-miR-3) regulates host gene ppm1a related to hepatocellular carcinoma. MicroRNA. 2020;9(3):232–239. doi:10.2174/2211536608666191104105334
12. Tang J, Xiao X, Jiang Y, et al. miR-3 encoded by hepatitis b virus downregulates PTEN protein expression and promotes cell proliferation. J Hepatocell Carcinoma. 2020;7:257–269. doi:10.2147/JHC.S271091
13. Volkova YL, Pickel C, Jucht AE, Wenger RH, Scholz CC. The asparagine hydroxylase fih: a unique oxygen sensor. Antioxid Redox Signaling. 2022;37(13–15):913–935. doi:10.1089/ars.2022.0003
14. Chen T, Ren Z, Ye LC, et al. Factor inhibiting HIF1alpha (FIH-1) functions as a tumor suppressor in human colorectal cancer by repressing HIF1alpha pathway. Cancer Biol Ther. 2015;16(2):244–252. doi:10.1080/15384047.2014.1002346
15. Chen T, Yao LQ, Shi Q, et al. MicroRNA-31 contributes to colorectal cancer development by targeting factor inhibiting HIF-1alpha (FIH-1). Cancer Biol Ther. 2014;15(5):516–523. doi:10.4161/cbt.28017
16. Otani Y, Yoo JY, Chao S, et al. Oncolytic HSV-infected glioma cells activate notch in adjacent tumor cells sensitizing tumors to gamma secretase inhibition. Clin Cancer Res. 2020;26(10):2381–2392. doi:10.1158/1078-0432.CCR-19-3420
17. Liu QY, Gao LY, Xu L, et al. CASC2 inhibits the growth, migration, and invasion of thyroid cancer cells through sponging miR-18a-5p/FIH1 axis. Kaohsiung J Med Sci. 2021;37(4):268–275. doi:10.1002/kjm2.12331
18. Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood. 2014;124(25):3748–3757. doi:10.1182/blood-2014-05-576116
19. Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5(4):628–635. doi:10.1038/nprot.2010.6
20. Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci USA. 1987;84(4):1005–1009. doi:10.1073/pnas.84.4.1005
21. Shirabe K, Mano Y, Muto J, et al. Role of tumor-associated macrophages in the progression of hepatocellular carcinoma. Surg Today. 2012;42(1):1–7. doi:10.1007/s00595-011-0058-8
22. Zhu RX, Seto WK, Lai CL, Yuen MF. Epidemiology of hepatocellular carcinoma in the Asia-pacific region. Gut Liver. 2016;10(3):332–339. doi:10.5009/gnl15257
23. Larsson SB, Tripodi G, Raimondo G, et al. Integration of hepatitis B virus DNA in chronically infected patients assessed by Alu-PCR. J med virol. 2018;90(10):1568–1575. doi:10.1002/jmv.25227
24. Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. 2016;64(1 Suppl): S84–S101. doi:10.1016/j.jhep.2016.02.021
25. Wang ML, Wu DB, Tao YC, et al. The truncated mutant HBsAg expression increases the tumorigenesis of hepatitis B virus by regulating TGF-beta/smad signaling pathway. Virol J. 2018;15(1):61. doi:10.1186/s12985-018-0972-0
26. O’Brien J, Hayder H, Zayed Y, Peng C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol. 2018;9:402. doi:10.3389/fendo.2018.00402
27. Grundhoff A, Sullivan CS. Virus-encoded microRNAs. Virology. 2011;411(2):325–343. doi:10.1016/j.virol.2011.01.002
28. Dong M, Chen JN, Huang JT, Gong LP, Shao CK. The roles of EBV-encoded microRNAs in EBV-associated tumors. Crit Rev Oncol/Hematol. 2019;135:30–38. doi:10.1016/j.critrevonc.2019.01.014
29. Hansen A, Henderson S, Lagos D, et al. KSHV-encoded miRNAs target MAF to induce endothelial cell reprogramming. Genes Dev. 2010;24(2):195–205. doi:10.1101/gad.553410
30. Zhang S, Amahong K, Sun X, et al. The miRNA: a small but powerful RNA for COVID-19. Briefings Bioinf. 2021;22(2):1137–1149. doi:10.1093/bib/bbab062
31. Loukachov V, van Dort KA, Jansen L, Reesink HW, Kootstra NA. Identification of a novel HBV encoded miRNA using next generation sequencing. Viruses. 2022;14:6. doi:10.3390/v14061223
32. Moawad AW, Szklaruk J, Lall C, et al. Angiogenesis in hepatocellular carcinoma; pathophysiology, targeted therapy, and role of imaging. J Hepatocell Carcinoma. 2020;7:77–89. doi:10.2147/JHC.S224471
33. Peet D, Linke S. Regulation of HIF: asparaginyl hydroxylation. Novartis Found Symp. 2006;272:37–49.
34. Cuninghame S, Jackson R, Zehbe I. Hypoxia-inducible factor 1 and its role in viral carcinogenesis. Virology. 2014;456:370–83.
35. Ravi R, Mookerjee B, Bhujwalla ZM, et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14(1):34–44. doi:10.1101/gad.14.1.34
36. Bodily JM, Mehta KP, Laimins LA. Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. 2011;71(3):1187–1195. doi:10.1158/0008-5472.CAN-10-2626
37. Darekar S, Georgiou K, Yurchenko M, et al. Epstein-Barr virus immortalization of human B-cells leads to stabilization of hypoxia-induced factor 1 alpha, congruent with the Warburg effect. PLoS One. 2012;7(7): e42072. doi:10.1371/journal.pone.0042072
38. Shin YC, Joo CH, Gack MU, Lee HR, Jung JU. Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1 alpha to induce vascular endothelial growth factor expression. Cancer Res. 2008;68(6):1751–1759. doi:10.1158/0008-5472.CAN-07-2766
39. Sanz-Cameno P, Martin-Vilchez S, Lara-Pezzi E, et al. Hepatitis B virus promotes angiopoietin-2 expression in liver tissue: role of HBV x protein. Am J Pathol. 2006;169(4):1215–1222. doi:10.2353/ajpath.2006.051246
40. Yen CJ, Lin YJ, Yen CS, et al. Hepatitis B virus X protein upregulates mTOR signaling through IKKbeta to increase cell proliferation and VEGF production in hepatocellular carcinoma. PLoS One. 2012;7(7): e41931. doi:10.1371/journal.pone.0041931
41. Yang JC, Teng CF, Wu HC, et al. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology. 2009;49(6):1962–1971. doi:10.1002/hep.22889
42. Rani S, Roy S, Singh M, Kaithwas G. Regulation of transactivation at C-TAD domain of HIF-1alpha by Factor-Inhibiting HIF-1alpha (FIH-1): a potential target for therapeutic intervention in cancer. Oxid Med Cell Longev. 2022;2022:2407223. doi:10.1155/2022/2407223
43. Wu Z, Cheng H, Liu J, et al. The oncogenic and diagnostic potential of stanniocalcin 2 in hepatocellular carcinoma. J Hepatocell Carcinoma. 2022;9:141–155. doi:10.2147/JHC.S351882
44. Cao L, Cheng H, Jiang Q, Li H, Wu Z. APEX1 is a novel diagnostic and prognostic biomarker for hepatocellular carcinoma. Aging. 2020;12(5):4573–4591. doi:10.18632/aging.102913
45. Cheng H, Fan X, Ye E, et al. Dual tumor microenvironment remodeling by glucose-contained radical copolymer for MRI-guided photoimmunotherapy. Adv Mater. 2022;34(25): e2107674. doi:10.1002/adma.202107674
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.