")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Health-related quality of life and its correlates in Japanese patients with myotonic dystrophy type 1
Authors Endo M, Odaira K, Ono R, Kurauchi G, Koseki A, Goto M, Sato Y, Kon S, Watanabe N, Sugawara N, Takada H , Kimura E
Received 14 September 2018
Accepted for publication 4 December 2018
Published 14 January 2019 Volume 2019:15 Pages 219—226
DOI https://doi.org/10.2147/NDT.S187607
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Taro Kishi
Makiko Endo,1 Kaori Odaira,2 Ryohei Ono,3 Go Kurauchi,4 Atsushi Koseki,5 Momoko Goto,3 Yumi Sato,4 Seiko Kon,6 Norio Watanabe,7 Norio Sugawara,8 Hiroto Takada,6 En Kimura9
1Clinical Research Unit, National Center Hospital, National Center of Neurology and Psychiatry, Kodaira, Tokyo 187-8551, Japan; 2Regional Medical Liaison Office, National Hospital Organization, Aomori Hospital, Namioka, Aomori 038-1331, Japan; 3Section for Development and Disability Training, National Hospital Organization, Aomori Hospital, Namioka, Aomori 038-1331, Japan; 4Department of Rehabilitation, National Hospital Organization, Aomori Hospital, Namioka, Aomori 038-1331, Japan; 5Section for Development and Disability Training, National Hospital Organization, Hanamaki Hospital, Hanamaki, Iwate 025-0033, Japan; 6Department of Neurology, National Hospital Organization, Aomori Hospital, Namioka, Aomori 038-1331, Japan; 7School of Public Health, Graduate School of Medicine, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan; 8Department of Clinical Epidemiology, Translational Medical Center, National Center of Neurology and Psychiatry, Kodaira, Tokyo 187-8551, Japan; 9Department of Clinical Research Support, Translational Medical Center, National Center of Neurology and Psychiatry, Kodaira, Tokyo 187-8551, Japan
Purpose: Myotonic dystrophy type 1 (DM1) is a common form of muscular dystrophy that presents with a variety of symptoms that can affect patients’ quality of life (QoL). Despite the importance of clarifying patients’ subjective experience in both physical and psychosocial aspects for improved symptom management, there is lack of evidence concerning QoL of patients with DM1 in Japan.
Patients and methods: A cross-sectional study was performed with 51 DM1 patients who completed questionnaires that measured health-related QoL (HRQoL), depression, and daytime sleepiness. Activities of daily living, body mass index (BMI), and genetic information were also collected, together with general demographic information. Correlation analyses using these variables were performed. Furthermore, regression analysis was utilized to assess the relationship that HRQoL, depression, and daytime sleepiness scores have with other variables.
Results: Physical component summary (PCS) score was affected by the disease more than the mental component summary (MCS) score among study participants. Moderate correlation was observed between PCS and depression, PCS and Barthel index, and depression and daytime sleepiness. Regression analysis revealed that age, sex, cytosine–thymine–guanine repeats, and BMI did not predict the aforementioned dependent variables.
Conclusion: DM1 symptoms influenced physical component scores more than mental component scores, although the state of physical wellness seemed to affect patients’ mood. Explaining the QoL of these patients only using biologic and genetic characteristics was not sufficient. We conclude that social and psychological aspects of these patients’ lives and the nature of adjustments made by patients due to DM1 to require further examination in order to improve the standard of care.
Keywords: depression, excessive daytime sleepiness, chronic illness, psychosocial perspective
Introduction
Myotonic dystrophy type 1 (DM1) is the most common form of muscular dystrophy and is caused by CTG (cytosine–thymine–guanine) triplet repeat expansion in the 3′ untranslated region of the myotonic dystrophy protein kinase gene.1 It is an autosomal dominant disorder that presents with multisystemic symptoms such as myotonia; disturbance of cardiac, digestive, and metabolic function; cataracts; fatigue; and daytime sleepiness.2 The correlation between the size of the CTG triplet repeat expansion and symptom severity is not strong, although associations exist between small expansions of 50–90 and mild symptoms, and between expansions of over 1,000 and congenital DM1.3,4 Because of this symptom variety, a multidisciplinary approach that consists of medical, rehabilitative, and psychosocial teams for the treatment and care of patients is essential to tackle this currently incurable disease. Due to the chronic nature of DM1, it is crucial to examine patients’ perceptions of life with the disease and whether issues in question are related to one’s biologic, psychological, or social aspects. A number of previous studies on patients with neuromuscular disease utilized measures for health-related quality of life (HRQoL), a concept that embodies respondent’s perception of biologic, psychological, social, economic, and spiritual well-being.5 Existing studies on HRQoL of patients with muscular disease, including those with DM1, have used both generic and disease-specific instruments in assessments.6 Most studies examined the relationship between QoL and biologic, neurologic, genetic, and psychosocial factors.7–9 Although the results differ slightly between studies, all have concluded that DM1 has a significant influence on patients’ QoL. Regarding the QoL of Japanese DM1 patients, a study by Fujino et al presented insightful findings on the association between QoL and cognitive functions. While QoL in this study was measured using the Muscular Dystrophy Quality of Life Scale (MDQoL-60), which was developed for Japanese muscular dystrophy patients,10 some previous DM1 QoL-related research in other countries have used generic measures that target a range of diseases or the general population,7,9,11 such as the Short-Form 36 Health Survey (SF-36).12,13 Disease- and culture-specific measures such as the MDQoL-60 are relevant in investigating the frequency and severity of particular conditions and are useful in researching outcomes, whereas generic measures prove useful in comparisons across diseases or with the general population.14 In this study, we aimed to explore the perception of QoL of Japanese DM1 patients by adopting widely employed generic questionnaires that have been used frequently in previous studies on DM1 and other diseases that have similar symptoms to DM1, in order to gain a perspective on the standing of Japanese DM1 patients regarding the spectrum of QoL and other major symptoms such as depression and daytime sleepiness. Moreover, we investigated factors that may have influenced the QoL of study participants.
Methods
Inclusion and exclusion criteria
The study included adult DM1 patients (age, 18–69) whose diagnoses were genetically confirmed and were autonomously mobile with or without use of assistive orthosis or wheelchair. Autonomous mobility was included in the criteria to control for participants’ level of physical activity. Participants were recruited through outpatient visits at Aomori Hospital of Namioka city, Aomori, and the National Center of Neurology and Psychiatry (NCNP) in Tokyo and also through publications in patient organization media. Patients were excluded if inclusion criteria were not met. The ethics review committees of Aomori Hospital and NCNP approved this study (approval numbers: 29–19 and A2016-050, respectively), and patients were required to provide written informed consent prior to their participation. The study was conducted in accordance with the Declaration of Helsinki. A gift voucher worth 1,000 Japanese yen was offered to participants in return.
Study procedure and measures
Participants’ demographic data were collected from their medical records if they were patients of Aomori Hospital or NCNP. If not, the data were collected through direct inquiry upon visiting for the study. Activity of daily living data were obtained using the Barthel index questionnaire,15 and height and weight were measured at the study sites for body mass index (BMI) calculation. Participants then completed a set of questionnaires, which consisted of Japanese versions of the Short-Form 36 Health Survey, version 2 (SF-36v2); Center for Epidemiologic Studies Depression Scale (CES-D); and Epworth Sleepiness Scale (ESS).12,13,16–18 These data were collected from the participants by study evaluators who went through all questionnaires in face-to-face interviews.
The first questionnaire, SF-36v2, is a widely used self-administered questionnaire that measures respondent’s HRQoL and can be applied to generic target populations. It consists of 36 items that are divided into 8 domains: physical function, role limitations due to physical function, bodily pain, general health, vitality, social function, role limitations due to emotional function, and mental health. Moreover, physical and mental component summary scores (PCS and MCS, respectively) can be derived by combining relevant domain scores. Standardized norm-based scoring allows results to be compared to normative data from the Japanese population (mean =50, SD =10).19 Second, CES-D is a 20-item self-reported scale to assess depression in the general population. Scores range from 0 to 60 and a score of 16 or above implies the possibility of depression. The final measure, ESS, evaluates excessive daytime sleepiness. It consists of eight questions to which respondent self-reports, and the score ranges from 0 to 24. Higher score implies higher daytime sleepiness. All questionnaires were translated into Japanese, and had confirmed validity and reliability.12,13,17,18
Statistical analyses
Descriptive statistics were calculated for demographic and clinical data, SF-36v2 domain and summary scores, CES-D score, and ESS score (Table 1). Pearson’s product–moment correlation analysis was performed to examine the strength of relationships between demographic, clinical, QoL, depression, and sleepiness data (Table 2). Furthermore, multiple linear regression was conducted to analyze the effect of demographic and clinical variables on SF-36v2, CES-D, and ESS scores (Table 3). Statistical significance was considered at a value of P<0.05. These analyses were performed using R software version 3.4.1 for Windows.
 | Table 1 Participant characteristics |
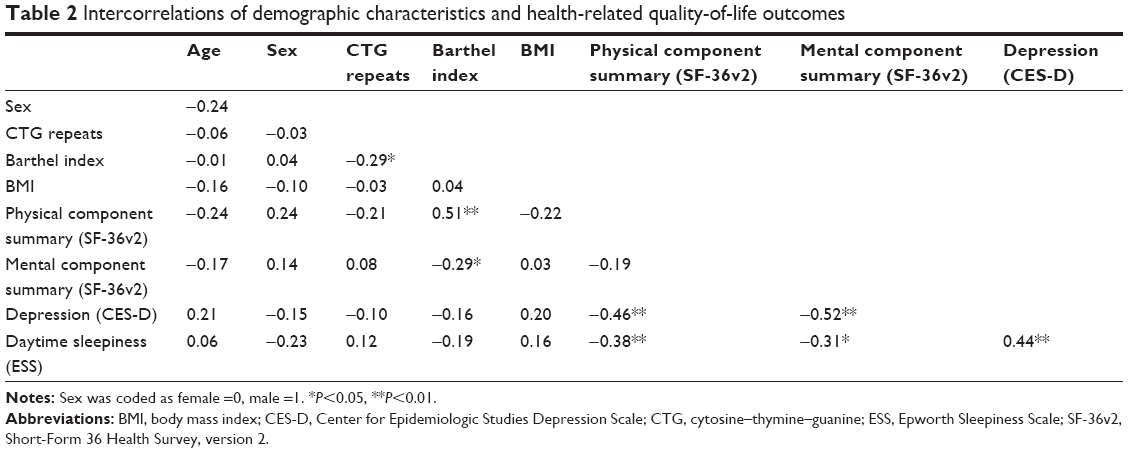 | Table 2 Intercorrelations of demographic characteristics and health-related quality-of-life outcomes |
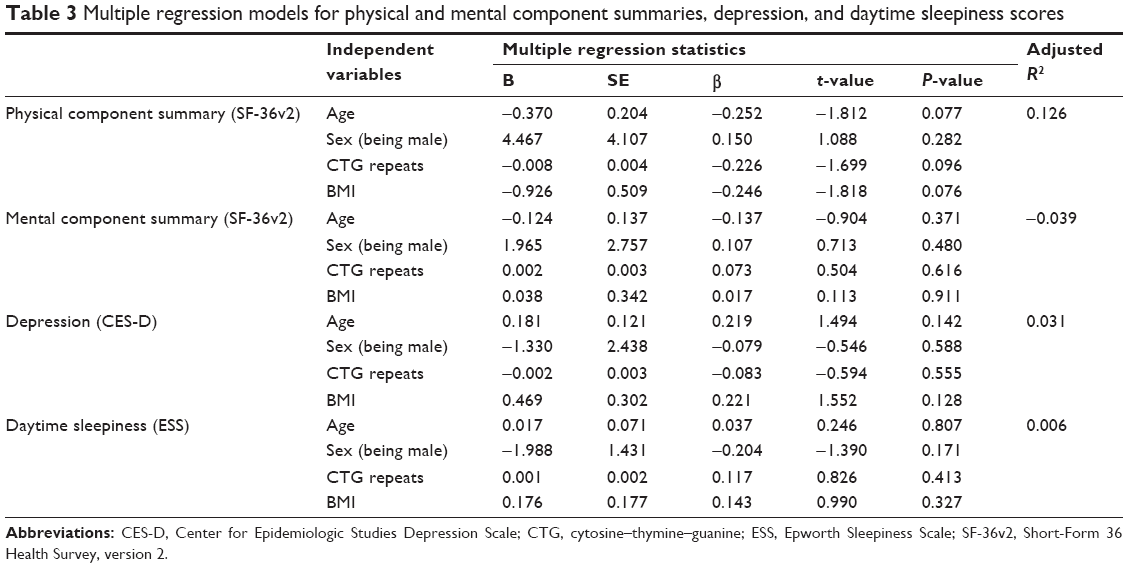 | Table 3 Multiple regression models for physical and mental component summaries, depression, and daytime sleepiness scores |
Ethics approval and consent to participate
Ethics Committees of the National Hospital Organization, Aomori Hospital, and National Center of Neurology and Psychiatry approved the study protocol. Participants read the information sheet and were provided an oral explanation by HT and EK. All participants signed the consent form.
Results
During October 2016 and November 2017, 51 adult DM1 patients (25 men, 26 women; mean age ± SD: 44.7±10.3 years) participated in the study. A flowchart of participant inclusion is shown in Figure 1. Out of the 51 patients, 48 patients were recruited through regular outpatient visits at Aomori Hospital; the remaining 3, who reside in Tokyo and its suburbs, joined through a recruiting notice that we publicized in newsletters and on websites of patient organizations.
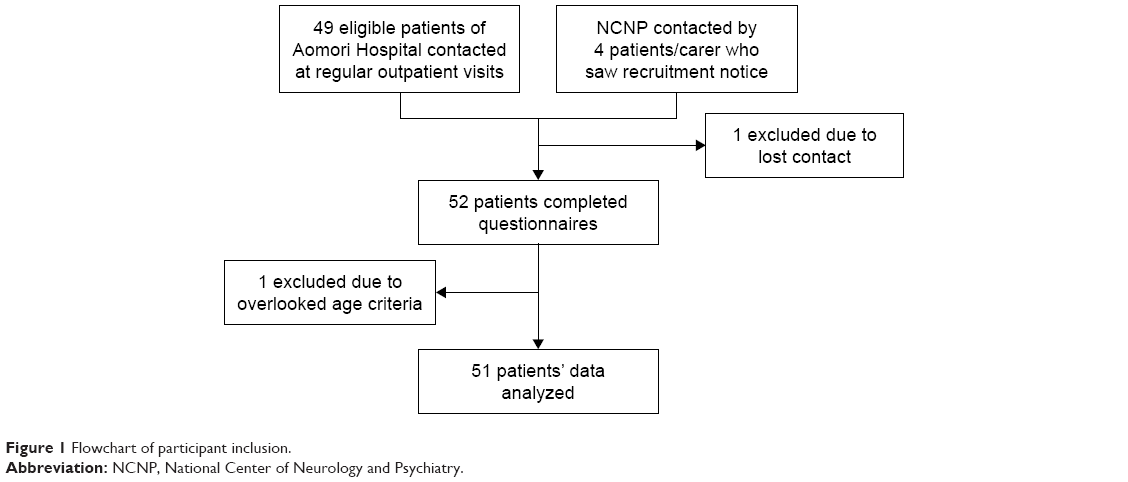 | Figure 1 Flowchart of participant inclusion. |
Table 1 shows demographic and clinical characteristics of the participants, and their scores for SF-36v2, CES-D, and ESS in mean ± SD. SF-36v2 norm-based mean scores for PCS and MCS of the participants in this study were 29.0 and 52.3, respectively. The mean for CES-D was 14.3, thereby not reaching the cutoff score of 16. Nevertheless, 19 patients (37.3% of the total) scored 16 or above. Additionally, the status of HRQoL of DM1 compared to the scores of Japanese general population is illustrated in Figure 2.20 It shows that mental health and social function domains of DM1 are close to the scores of general population, whereas domains related to physical health have wider gaps between DM1 and the population. In Table 2, intercorrelations of demographic and clinical characteristics, SF-36v2 summary component scores, CES-D scores, and ESS scores are presented. Moderate correlation was observed between PCS and depression (r=−0.46, P<0.01), PCS and Barthel index (r=0.51, P<0.01), and depression and daytime sleepiness (r=0.44, P<0.01). Table 3 shows the results of the multiple linear regression analysis performed for PCS, MCS, CES-D, and ESS scores. The analysis revealed that age, sex, CTG repeats, and BMI did not predict aforementioned dependent variables (P>0.07).
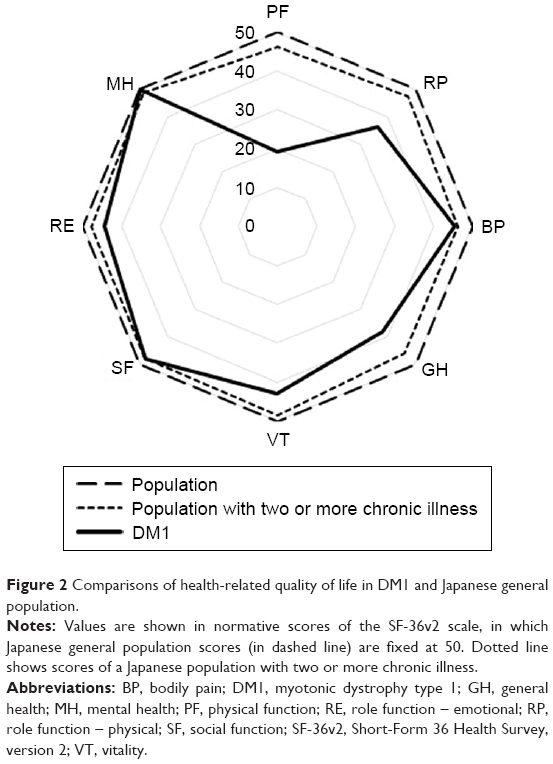 | Figure 2 Comparisons of health-related quality of life in DM1 and Japanese general population. |
Discussion
DM1 symptoms influenced physical component scores more than mental component scores in the SF-36v2 among the study participants. Compared to the mean of 2007 normative score for Japanese individuals with two or more chronic illnesses,19 the participants’ mean PCS score was lower by 16.9. On the other hand, the mean MCS score of this study was higher, even than the 2007 normative score from a Japanese population without chronic illness19 by 1.3. Although results among previous studies vary,6,7,21 a study by Laberge et al showed a tendency similar to that in our study.9 They revealed that SF-36 scores for physical domains in the study participants were lower than in the general Canadian population, while the MCS scores did not differ. A Dutch study by Kalkman et al also showed SF-36 results that were close to those of our study participants, where scores of the domains related to physical health were lower than emotional and social domain scores.22 As one of the reasons for higher MCS score in DM1 population, a response shift,23 a phenomenon suggested by past studies to explain higher QoL scores of DM1 patients, may also apply to the participants of this study.9,11 Response shift is a process in which patients accommodates their own illness through changes in internal standards, values, and concepts.23 Due to the chronic nature of the illness, this life adjustment phenomenon can be highly relevant for patients who live with muscular disease. Moreover, because these patients live with the disease over years, it may be difficult for them to feel or notice any changes related to their condition that may have occurred during the past month or year, the timeframe to which SF-36v2 questions refer.
It has also been argued in previous studies that cognitive impairment affects DM1 patients’ perception of QoL.7–9 In a study on DM1 patients’ unawareness of their disease, Baldanzi et al showed that despite limitations in social function, such as inability to work or withdrawal from relationships, patients appeared unconcerned according to their caregivers’ accounts.24 They further argued that DM1 patients’ unawareness of their disease was related specifically to lower scores in visuospatial memory, cognitive flexibility, comprehension, and conceptualization. Emotional QoL that is higher than the national average among the participants in this study may be explained by issues concerning disease awareness in DM1. In a systematic review and meta-analysis of the cognitive profile of DM1 patients by Okkersen et al, studies included in the analysis showed significantly worse cognitive performance in patients than in control participants. Large effect sizes were observed for global cognition, intelligence, visual memory, visuospatial perception, visuoconstruction, psychomotor speed, and social cognition,25 domains in which findings by Baldanzi et al overlap to an extent. In terms of social perception, our study found a similar tendency of the SF-36v2 social function domain score (48.0±11.5) being close to the Japanese normative score. We were unable to assess patients’ cognitive function in this study, rendering direct comparison with other results impossible. However, the mean CTG repeat size of our study participants was higher than that of participants in studies reviewed by Okkersen et al so that we can assume that our participants may have similar cognitive symptoms. The higher CTG repeat size of 894.6 in our study may be due to the fact that the Aomori Hospital study site is one of the few medical institutions that are specialized in neuromuscular diseases in the region, thereby attracting patients with severe symptoms. Despite this fact, we view that the study participants’ clinical states were not severe, as the high genetic variability of disease condition and prognosis is known to exist in DM1.26 To summarize, better emotional and social QoL despite limitations in physical function in our participants may have influenced the cognitive symptoms of DM1, leading to disease unawareness among this population.
The mean score for depression in study participants measured using the CES-D was 14.3±8.5, not reaching the cutoff of 16, which is defined as a likely presence of depression. In a study on the development of the Japanese version of CES-D,17 the mean score of the healthy control population was 8.9±7.1, a score that suggests a higher prevalence of depressive symptoms in our study participants. Prior studies on depression in myotonic dystrophy have had mixed results, with some concluding a higher presence and severity of symptoms, while others found no significant depression in this population.27–30 In fact, researchers have not yet reached an agreement on the cause of DM1 depression.29,30 Some studies indicated that the expression of apathy characterized by low motivation and diminished affect and concerns may be partially attributable to the prevalence of depression,29,31 while another emphasized the overlap of certain physical symptoms of depression with major DM1 symptoms such as fatigue, muscle weakness, and sleep disorder.30 Our study did not investigate participant apathy; thus, no comparison with these results can be made, although a recent study on Japanese DM1 patients showed an apathy prevalence that exceeds the cutoff score of the scale being used.8,32 In terms of fatigue, a question is included in the vitality domain of SF-36v2, in which our participants scored 43.0±11.3, a score that is lower than the national average of 50. As both restricted physical function and daytime sleepiness were also observed in our participants, our result can be explained on the grounds of somatic symptom overlap between depression and DM1.
In regard to daytime sleepiness, it was present in our study participants although the mean score did not exceed 10, a score that is typically interpreted as a presence of excessive daytime sleepiness.33 Our result appears to be in line with previous studies that utilized ESS.8,34,35 Daytime sleepiness is known to be one of the most common symptoms to affect DM1 patients,2,36–38 and together with the presence of fatigue they negatively influence HRQoL.35 However, it is commonly acknowledged that there is a gap between patients’ subjectively reported sleepiness and objective data.38,39 Our mean score, which did not reach pathologic levels of the symptom, may also be explained by this characteristic of perceiving sleepiness in the patients.
The mental state of study participants as derived from SF-36v2 and CES-D did not show a significant adverse condition, but our results also indicated that depressive mood measured using CES-D had a moderate correlation with PCS in SF-36v2 and daytime sleepiness assessed using ESS. These results suggest that despite higher mean MCS scores measured by the SF-36v2 than the national normative score, the physical condition of participants does seem to affect their mental condition. The association between daytime sleepiness and mental health of DM1 has already been reported in a previous study.9 This relationship can be understood by viewing sleep dysregulation as one of the main symptoms in mental illness. Regarding the association observed in this study between depression and physical function, our result is congruent with previous data that explain this finding as a natural progression of disease that worsens with time.7 However, there also exists contradictory data that indicate an association between depression and earlier phases of the disease.30 It is well understood that DM prognosis varies widely with factors such as age of onset and type of symptoms that are present in a patient; therefore, discussions related to disease severity and symptoms need to be considered carefully and should be based on further studies.
While correlation was observed between PCS and CES-D scores, a significant correlation was not detected between PCS and MCS. The reason for this difference in results can be partly explained by the timeframe of the questions for each measure. SF-36v2 collects information on the respondent’s health over the past month, whereas the CES-D questions respondent’s mood and behavior over the past week. DM1 patients may find it difficult to feel changes in their condition during a longer period of time due to the chronic nature of the disease. Short-term mood fluctuations may be easier to recall as memories of recent events, to which a particular mood may be attached, and might be more vividly remembered by patients. It was revealed in previous studies where DM1 patients were followed longitudinally over years that their QoL improved while muscular weakness worsened when measuring using the common HRQoL questionnaires, SF-36 and Individualized Neuromuscular Quality of Life questionnaire.11,40 Several potential explanations such as the speed of progression of particular symptoms, a response shift that occurred in patients during years of living with the disease, and other cognitive and behavioral effects of DM1 have been suggested.11,40 For use in clinical trials that aim for therapeutic developments that cure the disease, a questionnaire’s validity, reliability, and responsiveness are crucial in measuring improvements in disease states, such that it is undoubtedly appropriate to attach caution in utilizing these questionnaires.11,40,41 Nevertheless, it is also a reality that the patients need to live with chronic diseases until a cure is developed. Adjustment to particular symptoms and a life with illness becomes important in coping with the disease in terms of emotional wellbeing and functional preservation, and these results that show positive QoL despite the significant effect of illness shed light on one of the important aspects of DM1. To deepen our understanding further on the cause of high mental QoL or improving QoL amidst worsening muscle function, more studies on DM1 patients’ behavior, cognition, emotion, and interpersonal dynamics are necessary to clarify what exactly is a good or poor adjustment for DM1.42
Finally, the multiple linear regression analysis for PCS, MCS, CES-D, and ESS scores showed that these variables had no significant association with a group of independent variables comprising age, sex, CTG repeats, and BMI for our study participants. With this result, we can assume that the biologic and/or genetic characteristics of participants cannot fully explain their state of QoL, but various aspects of life such as employment or other social participation status, relationship satisfaction, and living environment need also to be taken into account.6,7,9 As factors that influence the QoL of patients with myotonic dystrophy, previous studies that are covered in Graham et al have reported variables such as pain, disease severity, fatigue, employment status, mood, and sex.6 Adding to these elements, Laberge et al revealed in their study that DM1 health-related QoL was affected significantly by factors such as daytime sleepiness, psychological distress, and social participation dissatisfaction, as well as other commonly mentioned factors such as fatigue and unemployment.9 These findings show that it is most important to examine various aspects of these patients’ lives in order to improve their standard of care, and that treatment and care should be provided not only via a medical approach but also include psychosocial aspects. In this respect, the results of the OPTIMISTIC study that provided psychological intervention in the form of cognitive behavioral therapy to DM1 patients and caregivers as a method of maintaining and improving DM1 patients’ function have definitely added significant value to care improvement research.43
This study is not without its limitations. First, the sample population consisted mostly of DM1 patients who reside in Aomori prefecture, an area in northern Japan; thus, the results of this study cannot be taken as representing all Japanese patients as region-specific elements are presumed to be influencing the outcome to an extent. Second, a limited number of samples that we were able to access restricted us when performing some statistical analyses, such as determining the number of variables and controlling for clinical factors in the regression analysis. Increased sample size would have enabled us to further clarify factors related to QoL. Finally, and in relation to the second point, the data of some major variables that are frequently utilized in DM1 research, such as disease severity, disease duration, cognitive function, and employment status, were not collected in this study due to resource limitation. Future research can further contribute to DM1 knowledge by collecting information concerning these factors from a wider regional source.
Conclusion
Despite the aforementioned limitations, this study adds valuable insights to the already existing research as it is the first study to investigate HRQoL of Japanese DM1 patients with the use of SF-36v2. Our results suggest that for some patients, improvement of QoL may not necessarily be equivalent to treated disease. In this regard, care based on psychosocial perspectives should be an effective addition to the existing standard, and future research on the development of a supporting method or program tailored to the needs of DM1 patients is crucial.
Availability of data and material
The datasets used in this study are available from the corresponding author upon reasonable request.
Acknowledgments
The authors would like to thank the participant patients/caregivers and patient advocate groups for their understanding and support for this study. This work is funded by JSPS Kakenhi grant number JP16K09735.
Disclosure
The authors report no conflicts of interest in this work.
References
Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68(4):799–808. | ||
Harper P. Myotonic Dystrophy. 2nd ed. Oxford: Oxford University Press; 2009. | ||
Barceló JM, Mahadevan MS, Tsilfidis C, MacKenzie AE, Korneluk RG. Intergenerational stability of the myotonic dystrophy protomutation. Hum Mol Genet. 1993;2(6):705–709. | ||
Tsilfidis C, MacKenzie AE, Mettler G, Barceló J, Korneluk RG. Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nat Genet. 1992;1(3):192–195. | ||
Doi Y. Introduction – conceptual issues on quality of life (QOL) and importance of QOL research. J Natl Instit Public Health. 2004;53(3):176–180. | ||
Graham CD, Rose MR, Grunfeld EA, Kyle SD, Weinman J. A systematic review of quality of life in adults with muscle disease. J Neurol. 2011;258(9):1581–1592. | ||
Antonini G, Soscia F, Giubilei F, et al. Health-related quality of life in myotonic dystrophy type 1 and its relationship with cognitive and emotional functioning. J Rehabil Med. 2006;38(3):181–185. | ||
Fujino H, Shingaki H, Suwazono S, et al. Cognitive impairment and quality of life in patients with myotonic dystrophy type 1. Muscle Nerve. 2018;57(5):742–748. | ||
Laberge L, Mathieu J, Auclair J, Gagnon É, Noreau L, Gagnon C. Clinical, psychosocial, and central correlates of quality of life in myotonic dystrophy type 1 patients. Eur Neurol. 2013;70(5–6):308–315. | ||
Kawai M, Ono M, Yatabe K, Ohya Y, Saito T, Sugiyama H, et al. Development of MDQoL-60 for Patients with Muscular Dystrophy [Kainyuu no koukahantei no tameno kinnjisutorofi-QOL hyouka shakudo MDQoL-60 no kaihatsu]. Hasuda, Saitama: Higashisaitama National Hospital; 2005. | ||
Peric S, Vujnic M, Dobricic V, et al. Five-year study of quality of life in myotonic dystrophy. Acta Neurol Scand. 2016;134(5):346–351. | ||
Fukuhara S, Bito S, Green J, Hsiao A, Kurokawa K. Translation, adaptation, and validation of the SF-36 Health Survey for use in Japan. J Clin Epidemiol. 1998;51(11):1037–1044. | ||
Fukuhara S, Ware JE Jr, Kosinski M, Wada S, Gandek B. Psychometric and clinical tests of validity of the Japanese SF-36 Health Survey. J Clin Epidemiol. 1998;51(11):1045–1053. | ||
Ware JE, Gandek B, Guyer R, Deng N. Standardizing disease-specific quality of life measures across multiple chronic conditions: development and initial evaluation of the QOL Disease Impact Scale (QDIS®). Health Qual Life Outcomes. 2016;14(1):84. | ||
Mahoney FI, Barthel DW. Functional evaluation: the Barthel index. Md State Med J. 1965;14:61–65. | ||
Radloff L. The CES-D Scale: a self-report depression scale for research in the general population. Appl Psychol Meas. 1977;1(3):385–401. | ||
Shima S, Shikano T, Kitamura T. Atarashii yokuutsusei no jikohyouka shakudo ni tsuite [New self-reporting scale on depression]. Seishin Igaku. 1985;27:717–723. | ||
Takegami M, Suzukamo Y, Wakita T, et al. Development of a Japanese version of the Epworth Sleepiness Scale (JESS) based on item response theory. Sleep Med. 2009;10(5):556–565. | ||
Fukuhara S, Suzukamo Y. Manual of SF-36v2 Japanese version. 3rd ed. Vol. 2004. Kyoto: iHope International Inc.; 2015. | ||
Tieleman AA, Jenks KM, Kalkman JS, Borm G, van Engelen BGM. High disease impact of myotonic dystrophy type 2 on physical and mental functioning. J Neurol. 2011;258(10):1820–1826. | ||
Peric S, Stojanovic VR, Basta I, et al. Influence of multisystemic affection on health-related quality of life in patients with myotonic dystrophy type 1. Clin Neurol Neurosurg. 2013;115(3):270–275. | ||
Kalkman JS, Schillings ML, van der Werf SP. Experienced fatigue in facioscapulohumeral dystrophy, myotonic dystrophy, and HMSN-I. J Neurol Neurosurg Psychiatr. 2005;76(10):1406–1409. | ||
Sprangers MA, Schwartz CE. Integrating response shift into health-related quality of life research: a theoretical model. Soc Sci Med. 1999;48(11):1507–1515. | ||
Baldanzi S, Bevilacqua F, Lorio R, et al. Disease awareness in myotonic dystrophy type 1: an observational cross-sectional study. Orphanet J Rare Dis. 2016;11:34. | ||
Okkersen K, Buskes M, Groenewoud J, et al. The cognitive profile of myotonic dystrophy type 1: a systematic review and meta-analysis. Cortex. 2017;95:143–155. | ||
De Antonio M, Dogan C, Hamroun D, et al. Unravelling the myotonic dystrophy type 1 clinical spectrum: a systematic registry-based study with implications for disease classification. Rev Neurol. 2016;172(10):572–580. | ||
Brumback RA, Carlson KM. The depression of myotonic dystrophy: response to imipramine. J Neurol Neurosurg Psychiatry. 1983;46(6):587–588. | ||
Duveneck MJ, Portwood MM, Wicks JJ, Lieberman JS. Depression in myotonic muscular dystrophy. Arch Phys Med Rehabil. 1986;67(12):875–877. | ||
Gallais B, Montreuil M, Gargiulo M, Eymard B, Gagnon C, Laberge L. Prevalence and correlates of apathy in myotonic dystrophy type 1. BMC Neurol. 2015;15:148. | ||
Winblad S, Jensen C, Månsson JE, Samuelsson L, Lindberg C. Depression in myotonic dystrophy type 1: clinical and neuronal correlates. Behav Brain Funct. 2010;6(1):25. | ||
Bungener C, Jouvent R, Delaporte C. Psychopathological and emotional deficits in myotonic dystrophy. J Neurol Neurosurg Psychiatry. 1998;65(3):353–356. | ||
Starkstein SE, Mayberg HS, Preziosi TJ, Andrezejewski P, Leiguarda R, Robinson RG. Reliability, validity, and clinical correlates of apathy in Parkinson’s disease. J Neuropsychiatry Clin Neurosci. 1992;4(2):134–139. | ||
Johns M. The official website of the Epworth Sleepiness Scale (ESS) and the Epworth Sleepiness Scale for Children and Adolescents (ESS-CHAD). Available from: http://epworthsleepinessscale.com/about-the-ess/. Accessed December 20, 2018. | ||
Bonanni E, Carnicelli L, Crapanzano D, et al. Disruption of sleep-wake continuum in myotonic dystrophy type 1: beyond conventional sleep staging. Neuromuscul Disord. 2018;28(5):414–421. | ||
Laberge L, Dauvilliers Y, Bégin P, Richer L, Jean S, Mathieu J. Fatigue and daytime sleepiness in patients with myotonic dystrophy type 1: to lump or split? Neuromuscul Disord. 2009;19(6):397–402. | ||
Heatwole C, Bode R, Johnson N, et al. Patient-reported impact of symptoms in myotonic dystrophy type 1 (PRISM-1). Neurology. 2012;79(4):348–357. | ||
Kierkegaard M, Harms-Ringdahl K, Widén Holmqvist L, Tollbäck A. Perceived functioning and disability in adults with myotonic dystrophy type 1: a survey according to the International Classification of Functioning, Disability and Health. J Rehabil Med. 2009;41(7):512–520. | ||
Laberge L, Gagnon C, Dauvilliers Y. Daytime sleepiness and myotonic dystrophy. Curr Neurol Neurosci Rep. 2013;13(4):340. | ||
van Hilten JJ, Kerkhof GA, van Dijk JG, Dunnewold R, Wintzen AR. Disruption of sleep-wake rhythmicity and daytime sleepiness in myotonic dystrophy. J Neurol Sci. 1993;114(1):68–75. | ||
Peric S, Heatwole C, Durovic E, et al. Prospective measurement of quality of life in myotonic dystrophy type 1. Acta Neurol Scand. 2017;136(6):694–697. | ||
Heatwole C, Johnson N, Dekdebrun J, et al. Myotonic dystrophy patient preferences in patient-reported outcome measures. Muscle Nerve. Epub 2018 Jan 12. | ||
Moss-Morris R. Adjusting to chronic illness: time for a unified theory. Br J Health Psychol. 2013;18(4):681–686. | ||
Okkersen K, Jimenez-Moreno C, Wenninger S, et al. Cognitive behavioural therapy with optional graded exercise therapy in patients with severe fatigue with myotonic dystrophy type 1: a multicentre, single-blind, randomised trial. Lancet Neurol. 2018;17(8):671–680. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.