Back to Journals » OncoTargets and Therapy » Volume 13
HDAC1/2 Inhibitor Romidepsin Suppresses DEN-Induced Hepatocellular Carcinogenesis in Mice
Authors Afaloniati H ![]() , Angelopoulou K
, Angelopoulou K ![]() , Giakoustidis A, Hardas A
, Giakoustidis A, Hardas A ![]() , Pseftogas A
, Pseftogas A ![]() , Makedou K
, Makedou K ![]() , Gargavanis A
, Gargavanis A ![]() , Goulopoulos T, Iliadis S, Papadopoulos V, Papalois A, Mosialos G, Poutahidis T
, Goulopoulos T, Iliadis S, Papadopoulos V, Papalois A, Mosialos G, Poutahidis T ![]() , Giakoustidis D
, Giakoustidis D
Received 17 February 2020
Accepted for publication 27 May 2020
Published 15 June 2020 Volume 2020:13 Pages 5575—5588
DOI https://doi.org/10.2147/OTT.S250233
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Hara Afaloniati,1 Katerina Angelopoulou,1 Alexander Giakoustidis,2 Alexandros Hardas,3 Athanasios Pseftogas,4 Kali Makedou,5 Athanasios Gargavanis,2 Thomas Goulopoulos,2 Stavros Iliadis,5 Vasileios Papadopoulos,2 Apostolos Papalois,6 George Mosialos,4 Theofilos Poutahidis,3 Dimitrios Giakoustidis2
1Laboratory of Biochemistry and Toxicology, School of Veterinary Medicine, Faculty of Health Sciences, Aristotle University of Thessaloniki, Thessaloniki, Greece; 2First Department of Surgery, Medical School, Aristotle University of Thessaloniki, General Hospital Papageorgiou, Thessaloniki, Greece; 3Laboratory of Pathology, School of Veterinary Medicine, Faculty of Health Sciences, Aristotle University of Thessaloniki, Thessaloniki, Greece; 4School of Biology, Faculty of Sciences, Aristotle University of Thessaloniki, Thessaloniki, Greece; 5Department of Biological Chemistry, Medical School, Faculty of Health Sciences, Aristotle University of Thessaloniki, Thessaloniki, Greece; 6Experimental, Educational and Research Center, ELPEN, Pikermi, Attica, Greece
Correspondence: Dimitrios Giakoustidis
First Department of Surgery, Medical School, Aristotle University of Thessaloniki, General Hospital Papageorgiou, Thessaloniki, Greece
Tel +30 6932306133
Email [email protected]
Background: Hepatocellular carcinoma (HCC) is a frequently diagnosed cancer and a leading cause of cancer-related death worldwide. Its rapid progression, combined with the limited treatment options at late stages, imposes the need for early detection and aggressive intervention. Based on the knowledge that hepatocarcinogenesis is significantly influenced by histone acetylation, we directed our search for novel HCC therapeutics among histone deacetylation inhibitors (HDACi). The aim of the present study was to investigate the effect of HDAC1/2 inhibitor Romidepsin in the well-established mouse model of diethylnitrosamine (DEN)-induced HCC.
Materials and Methods: C56BL/6 mice were treated with Romidepsin at the critical point of 10 months after DEN challenge and their livers were examined 2 months later using histopathology and morphometry. Protein levels were assessed in serum using ELISA and in liver tissues using Western blot and immunohistochemistry (in-situ detection). Gene expression was quantified using real-time PCR.
Results: Romidepsin suppressed cancer progression. This effect was associated with decreased proliferation and increased apoptosis of cancer cells. The cell cycle regulator CK2a, the anti-inflammatory molecule PPAR-γ, and the tumor suppressors PTEN and CYLD were upregulated in treated HCC. By contrast, the expression of PI3K, NF-κB p65 and c-Jun was reduced. In line with this result, the levels of two major apoptosis regulators, ie, BAD and the multifunctional protein c-Met, were lower in the blood serum of treated mice compared to the untreated mice with HCC.
Conclusion: These findings suggest that Romidepsin, a drug currently used in the treatment of lymphoma, could also be considered in the management of early-stage HCC.
Keywords: diethylnitrosamine, DEN, histone deacetylases, HDAC, HDAC inhibitors, HDACi, hepatocellular carcinoma, HCC, Romidepsin
Introduction
Liver cancer is estimated to be the sixth most frequently diagnosed cancer and the fourth cancer-related cause of death worldwide.1 The incidence of the disease is high in the developing countries and lower but constantly increasing in industrialized ones.2 The most common type of liver cancer is hepatocellular carcinoma (HCC). HCC is an aggressive malignant tumor with a multistep development, arising from low-grade dysplastic nodules, proceeding to high-grade dysplastic nodules and eventually to malignant tumors.3,4
HCC treatment approaches as well as patient prognosis vary according to the disease stage. For early-stage HCC, resection, local ablation or liver transplantation is followed and the median survival is five years. For intermediate stages, transarterial chemoembolization is recommended, whereas for advanced stages systematic therapy with sorafenib is the only choice with a median survival of 2.5 years and 10 months, respectively.5,6 Moreover, due to the high risk of recurrence – up to 70% of patients with early-stage HCC – and the presence of unresectable tumors in a proportion of patients, there is a major need for new therapeutic strategies.7 Although HCC is by large chemo-resistant, systemic anticancer treatment in the early stages of the disease still constitutes a promising approach.8,9
Hepatocarcinogenesis is underlined by multiple biological processes comprising both genetic modifications, such as mutations and copy number variations, as well as epigenetic alterations like methylation and histone modifications.4,10-13 Histone acetylation, a widely studied histone modification, is controlled by the opposing activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs add an acetyl group to a histone lysine, which causes chromatin decondensation and thus transcriptional activation. On the other hand, deacetylation of histones by HDACs leads to chromatin condensation and gene silencing. Moreover, HDACs remove the acetyl group of a variety of protein targets, such as transcription factors and cellular proteins implicated in cell growth, differentiation and apoptosis.14,15 HDAC inhibitors (HDACi) restrain the activity of HDAC, leading to re-expression of silenced genes and re-activation of proteins. Recently, HDACi attracted a great interest, as they have been shown to have anti-tumor effects in various types of cancers, including HCC.6,16,17 In HCC, HDACi exert their antitumor effect via activation of both classical and alternative cell death molecular cascades, thus expanding the overall survival. Moreover, combination of HDACi with targeted therapies was shown to eliminate cell proliferation by increasing autophagy and apoptosis.17–19
Romidepsin, an HDACi, is a cyclic peptide that inhibits mainly HDAC1 and HDAC2 enzyme activity.20 It is a systemic drug (also known as FR901228, FK228, or depsipeptide) approved by the Food and Drug Administration (FDA) for the treatment of patients with relapsed or refractory cutaneous T-cell lymphoma (CTCL) and peripheral T-cell lymphoma (PTCL).21 However, its possible effectiveness in the treatment of HCC remains unknown. To our knowledge, there are only two reports that demonstrate that Romidepsin inhibits cell cycle and induces apoptosis in HCC cell lines and mouse xenograft models.22,23
The aim of the present study was to investigate the effect of Romidepsin in the widely used and well-characterized mouse model of diethylnitrosamine (DEN)-induced HCC. The evolution of this spontaneous, chemically-induced HCC parallels many aspects of human liver neoplasmatogenesis; it mimics both organism response and cancer progression, as well as tumor-related gene expression patterns and pathways.24,25 In this study, we demonstrate that Romidepsin suppressed HCC in mice in a manner that is consistent with relevant alterations in the expression of selected cancer-associated molecules.
Materials and Methods
Animals and Experimental Design
C56BL/6 male mice weighing 25–27 gr were purchased from the Hellenic Pasteur Institute. Mice were kept in stainless cages at constant temperature (22 to 24°C) and allowed free access to food and water during the 24-h day/night cycle. All experimental procedures were performed according to the Guide for the Care and Use of Laboratory Animals.26 Ethical approval and licensing (License reference no 4956/09-08-2012) were provided by the competent National Veterinary Administration Authorities according to Greek legislative (Decree no. 2015/92, 160/91) and European Communities Council directive (no. 86/609/EEC).
A total of 23 male mice were used. Of these, 17 were treated with the carcinogen DEN, and 6 remained untreated. DEN (5 mg/kg of body weight) was given with a single i.p. injection at the age of 14 days for the induction of HCC. At the age of 10 months, 8 of the 17 mice were further i.p. injected with 0.03 mg of the HDAC1/2 inhibitor Romidepsin (Abcam, Cambridge, UK; 0.03 mg/per mouse) twice a week for three consecutive weeks. Romidepsin was dissolved in dimethyl sulfoxide (DMSO) to a final concentration of 10 mM and further diluted in 500 μL saline prior to i.p. administration. An equal amount of DMSO was diluted in saline and injected i.p. in the remaining 9 mice. Mice were killed with an overdose of ketamine and xylazine at twelve months of age.
Histopathology, Immunohistochemistry, and Morphometry
Formalin-fixed livers were embedded in paraffin, cut at 5 μm, and stained with hematoxylin and eosin or immunohistochemistry (IHC). The extent of HCC lesions was scored in whole liver sections of mice on a 0 to 4 scale according to the following scheme. 0, no HCC; 1, HCC <25%; 2, HCC=25–50%; 3, HCC=50–75%; HCC >75% of the liver section area. Primary antibodies for IHC included rabbit antibodies against Ki-67, DKK1 (Abcam, Cambridge, UK), cleaved Caspase-3, NF-κB, c-Jun (Cell Signaling, Beverly, MA), β-catenin, Cyclin D1, PTEN, PPAR-γ, PI3K3 (ThermoFisher Scientific/Lab Vision, Fremont, CA). Heat-induced antigen retrieval was performed with citrate buffer, pH 6, for c-Jun, cleaved caspase-3, NF-κB, β-catenin, Cyclin D1, PPAR-γ and PI3K or with EDTA buffer, pH 8, for Ki-67 and DKK1. Rabbit primary antibody binding was detected with goat anti-rabbit polymer HRP (ZytoChem Plus, Berlin, Germany). Color was developed with Diaminobenzidine substrate-chromogen (Biogenex, Fremont, CA) and tissues were counterstained with hematoxylin.
For quantitative histomorphometry, IHC-positive cells were counted in hepatocellular cancer images of x40 representative high power fields and results were recorded as number of cells or pixels per image as previously described.27 The ImageJ image processing and analysis program (NIH, Bethesda, MD) was used for all histomorphometrical assessments.
Enzyme-Linked Immunosorbent Assay
Hepatocyte growth factor receptor (c-Met/HGFR) and Bcl2-associated agonist of cell death (BAD) serum levels were determined using a quantitative sandwich enzyme immunoassay technique from Cusabio Biotech Co., Ltd. (Wuhan, China). Standards and serum samples were diluted 1:2 in Sample Diluent and assayed in duplicate in 96-well microplates precoated with antibodies specific for c-Met/HGFR or BAD, respectively. A 2h incubation at 37°C was followed by the addition of the appropriate antibody, a 1-h incubation at 37°C, washing, the addition of avidin conjugated horseradish peroxidase (HRP), a 1-h incubation at 37°C, washing and the addition of the appropriate substrate. Finally, the reactions were stopped and within 5 min the optical density at 450 nm was determined in a microplate reader. A standard curve was created and the concentration of the samples was calculated, taking into account the initial dilution of the samples. The inter-assay and intra-assay precision for both assays were <10% and <8%, respectively. The detection range for c-Met/HGFR was 0.078–5 ng/mL and for BAD 31.2–2000 pg/mL.
Western Blot
Cells were washed twice with ice-cold phosphate-buffered solution (PBS). Cells lysis was achieved using SDS lysis buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 10% Glycerol and 3% β-mercaptoethanol), followed by heating at 95°C for 5 min. The samples were analyzed by SDS-PAGE and proteins were electrophoretically transferred to nitrocellulose membrane for Western blot analysis. For immunoblotting, the following primary antibodies were used: anti-CYLD mouse monoclonal (Santa Cruz Biotechnology Inc), anti-PPAR-γ mouse monoclonal (Invitrogen), anti-p38 rabbit polyclonal (Invitrogen) and anti-PTEN rabbit polyclonal (Invitrogen). Membrane-bound antibodies were detected by an enhanced chemiluminescence detection kit (Pierce) using a Typhoon FLA 7000 imaging system (GE Healthcare Life Sciences). Bands were quantified using ImageJ processing and analysis program (NIH, Bethesda, MD).
Quantitative Gene Expression Analysis
Real-time PCR based on the SYBR Green chemistry was used to quantitatively determine gene expression, as previously described.28 Primer sequences, their positions within the corresponding genes, and amplicon sizes of the 7 genes examined, as well as the corresponding housekeeping gene Gapdh, are presented in Table 1.
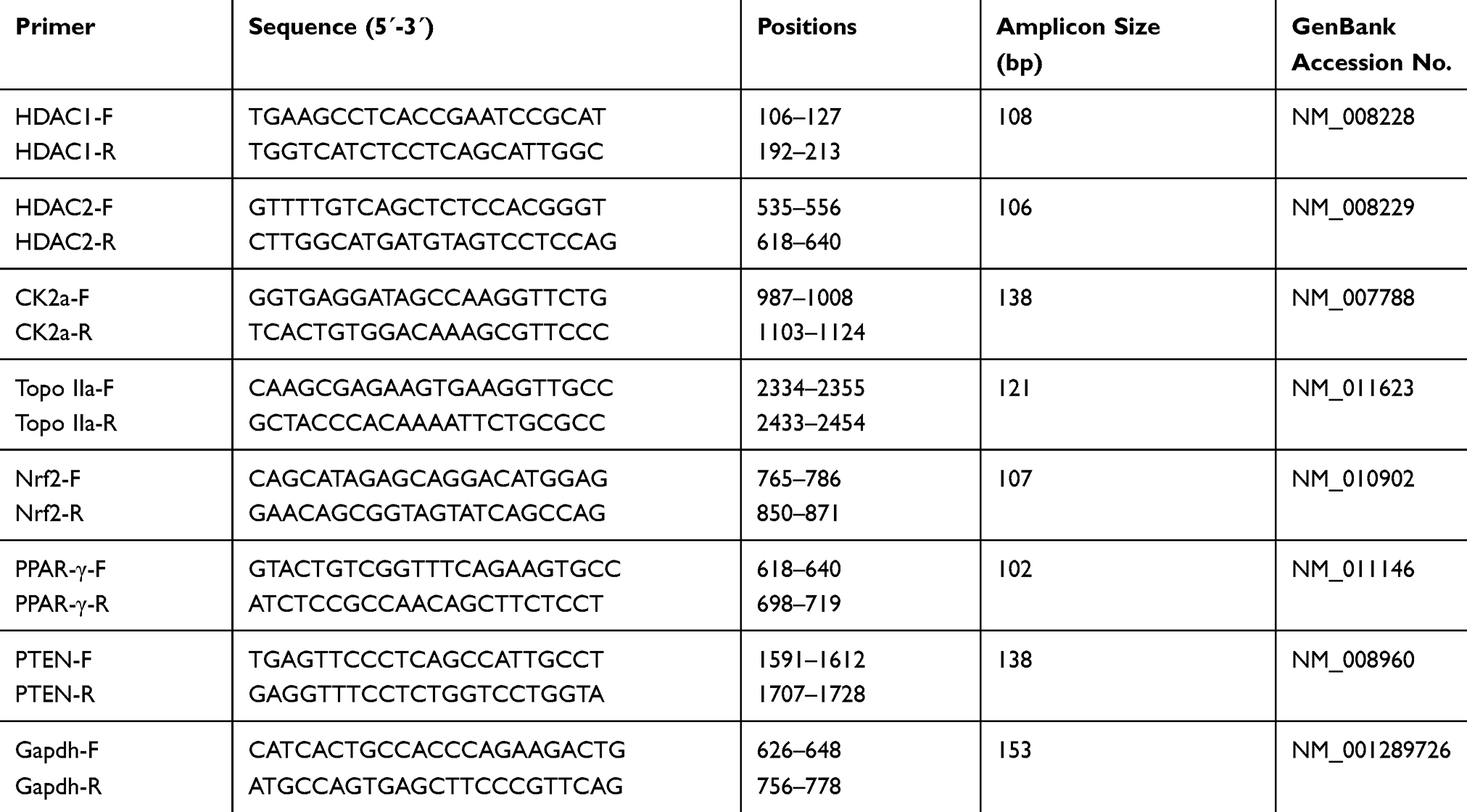 |
Table 1 Primers Used for Gene Expression Analysis |
Statistical Analyses
Histomorphometry, relative gene expression, Western blot and serum protein measurements data were compared between groups using Mann Whitney U analysis. Statistical significance was set at P<0.05. All analyses were performed with the GraphPad Prism version 5.0 for windows (GraphPad Software, San Diego, CA, USA). Data representation was done with bar graphs depicting the mean and standard error of the parameter assessed for each experimental group.
Results
Inhibition of HDAC1/2 Reduces DEN-Induced Hepatocellular Cancer
In our previous research, we used a DEN mouse model of HCC, which at 10 months after carcinogen challenge, consistently presented with multiple hepatocellular adenomas.29,30 The same mouse model was used here to evaluate the effect of HDAC blockade on HCC evolution and growth. For that, we treated mice with the HDAC1/2 inhibitor Romidepsin at the critical point of 10 months after DEN challenge and examined their livers 2 months later.
At the end of the experiment (12 months), the livers of the control mice receiving no carcinogen were normal (n = 6). At the same time, the livers of all carcinogen-treated mice that remained further untreated (n = 9) showed the whole spectrum of DEN-induced carcinogenesis lesions, including hepatocellular adenomas, early hepatocellular carcinomas and large, highly infiltrative hepatocellular carcinomas that were obliterating extensive areas of normal liver. Most carcinomas appeared to arise in large hepatocellular adenomas that were sharply demarcated from surrounding liver tissue. These carcinomas consistently had a trabecular formation. The larger expansive carcinomas lacking clear demarcation, however, in addition to trabecular growth patterns contained areas of solid and acinar histological architectures, and multifocal necrosis and hemorrhage (Figure 1).
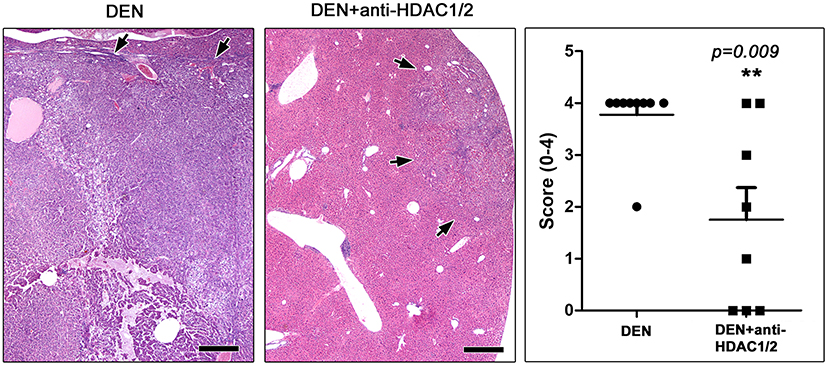 |
Figure 1 Anti-HDAC1/2 treatment counteracts HCC at 10 months after DEN administration. Large, expansive HCC in a DEN-treated mouse that received no further treatment. A small well-circumscribed hepatic cell tumor from a mouse that was further treated with Romidepsin is shown on the side for comparison. Arrows depict tumor tissue margins. Histopathological scores reflecting the extent of neoplastic invasion of liver tissue suggest that Romidepsin treatment suppressed liver tumors at statistically significant levels. Hematoxylin and Eosin. Scale bars: 500 μm. Numbers on the y-axis of bar graph correspond to the mean±SEM of histological score; ** p<0.001. Abbreviations: DEN, diethylnitrosamine; HDAC, histone deacetylase. |
Interestingly, in the experimental group of DEN-challenged mice that were treated with HDAC1/2 inhibitors (n = 8), 3 of the 8 mice had no detectable neoplastic lesions upon histological examination of the liver. The neoplastic lesions in the remaining mice of this group were qualitatively comparable with, but smaller than those of mice that did not receive therapy (Figure 1). To assess the effect of HDAC1/2 blockade more precisely, the extent of HCC lesions was scored semi-quantitatively in whole liver sections. The statistical analysis of histopathological scores suggested that HDAC1/2 inhibition counteracted the evolution and growth of DEN-induced HCC in the liver of mice at a statistically significant level (Figure 1).
By using ELISA, we assessed met proto-oncogene (c-Met) and BAD protein levels in mice serum. c-Met, a tyrosine protein kinase receptor, is found increased in liver cancer and correlates with poor prognosis in human patients.31 BAD is an important apoptosis and cell replication regulator of hepatocytes that has been shown to inhibit liver carcinogenesis.32 We found that both c-Met and BAD were significantly higher in the serum of mice treated with Romidepsin by comparison with the untreated controls (Figure S1).
HDAC1/2 Inhibition Affects Apoptosis and Proliferation of HCC Cells
HDAC1/2 inhibitors have been shown to induce apoptosis and cell cycle arrest.33 To examine whether the tumor-suppressing effect of Romidepsin observed in this study is associated with these cellular events, we used IHC to label apoptotic and proliferating cells in liver sections. With Caspase-3-specific IHC we first found that apoptotic cells in HCC lesions are particularly scarce, which matches our previous observations in DEN-induced early hepatocellular carcinogenesis.29,30 Notably, morphometric counts of Caspase-3-positive cancer cells suggested that HDAC1/2 inhibition upregulates apoptosis in HCC at statistically significant levels (Figure 2). Likewise, using the proliferation marker Ki-67, we found that HCC tumors of Romidepsin-treated mice contained significantly less Ki-67-positive cancer cells compared to their untreated counterparts (Figure 2A).
 |
Figure 2 HDAC1/2 inhibition affects cell cycle and apoptosis in HCC. (A) Morphometric counts of immunohistochemically stained liver sections show that Romidepsin treatment increases apoptosis (Caspase-3) and decreases proliferation (Ki-67) in neoplastic hepatocytes. This effect co-existed with significant differences in the expression of cell cycle and apoptosis regulators such as CK2a (B) and HDAC2 (C). IHC; Diaminobenzidine chromogen, Hematoxylin counterstain. Scale bars: 25 μm. Numbers on the y-axis of bar graphs correspond to the mean±SEM of the parameters assessed. *p<0.05, ** p<0.001. Abbreviations: CK2a, casein kinase 2, alpha 1 polypeptide; DEN, diethylnitrosamine; HDAC2, histone deacetylase 2; Ki-67, antigen identified by monoclonal antibody Ki 67; R, Romidepsin. |
HDAC1/2 Inhibition Alters the Expression of Selected Cell Cycle and Apoptosis Regulators
Cyclin D1 is a major regulator of cell cycle that binds to HDACs and has been shown to be downregulated in HCC after HDAC inhibition.34 For that, we next examined Cyclin D1 expression in mouse liver tumors with IHC. Cyclin D1 expression in HCC was multifocally diffuse with predominance in expanding fronts of tumors, multi-sized intratumoral nests of large atypical karyomegalic cancer cells and areas of solid growth pattern. The distribution of Cyclin D1-positive cells as well as their numbers appeared comparable in tumors from both experimental groups of mice. Morphometrical counts of Cyclin D1-expressing cells confirmed this observation (Figure S2A). By examining other selected pleiotropic major cell cycle and growth regulators, that also affect apoptosis, at the gene or protein expression levels, we found that suppression of HCC growth by Romidepsin correlated significantly with the upregulation of casein kinase 2, alpha 1 polypeptide (CK2a) (Figure 2B) and (Figure 2C), but not of HDAC1 (Figure S2B) and topoisomerase (DNA) II alpha (Topo IIa) (Figure S2B).
HDAC1/2 Blockade Modifies the Expression of Critical Inflammation and Cellular Stress-Associated Molecules
NF-κB p65, nuclear factor of kappa light chain enhancer of activated B cells, p65 (NF-κB p65) and jun proto-oncogene (c-Jun) are critical inflammation and cellular stress response pleiotropic molecules with major roles in cellular processes including proliferation and apoptosis, and in cancer.35 By IHC and morphometrical analysis, we found that tumor cells with nuclear c-Jun expression were relatively few in both experimental groups, but significantly, less in the livers of mice treated with Romidepsin compared to the untreated controls (Figure 3A). IHC for NF-κB p65 showed a consistently positive signal in intratumoral nonparenchymal liver and inflammatory cells. Neoplastic hepatocytes, however, had a multifocally diffuse positivity with cytoplasmic but not nuclear NF-κB p65 presence, which appeared less extensive in the tumors of treated mice. Indeed, using a morphometric approach based on counts of positively IHC-stained image pixels, we found that HCC of Romidepsin-treated mice had significantly lower levels of NF-κB p65 expression compared to HCC of untreated control mice (Figure 3A).
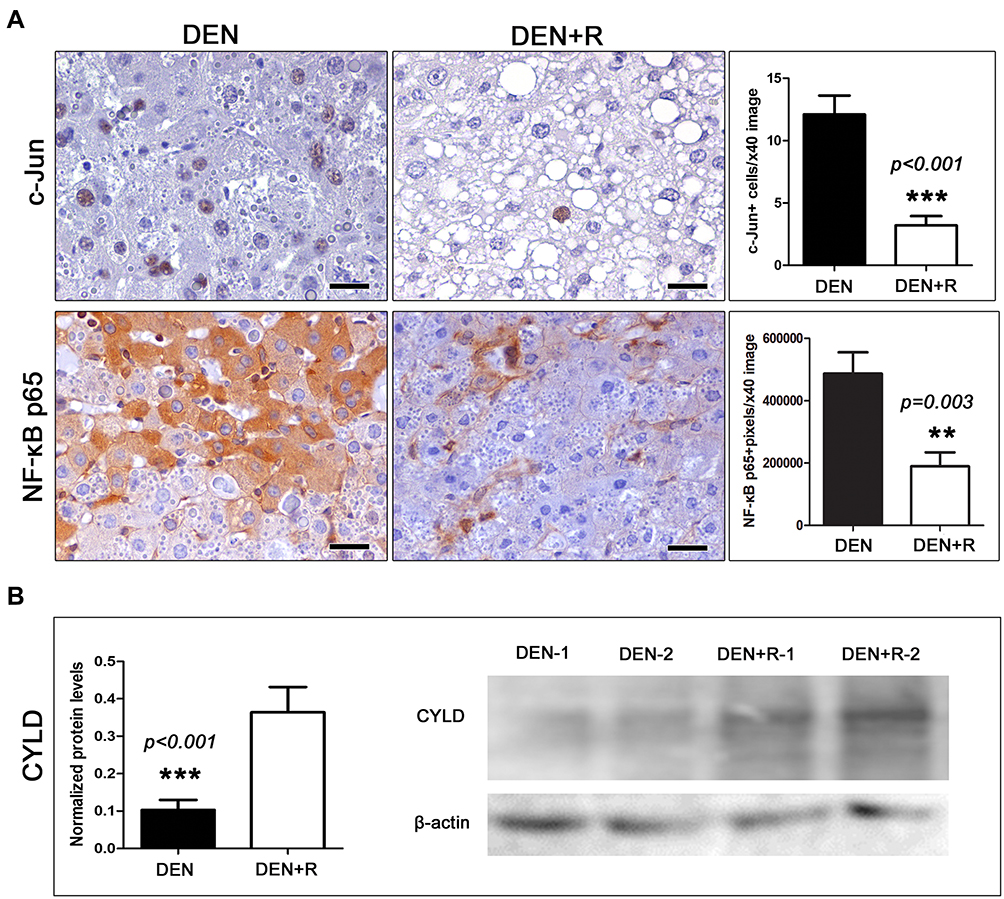 |
Figure 3 Effects of HDAC1/2 depletion on the expression of selected cell-stress-associated proteins. (A) HCC cells show significantly decreased cytoplasmic NF-κB and nuclear c-Jun expression after treatment with Romidepsin. (B) The treatment also caused an increase of the expression of the NF-κB suppressor protein CYLD, as assessed by Western Blot analysis. Shown are bands from liver tissue of two selected representative mice per group. IHC; Diaminobenzidine chromogen, Hematoxylin counterstain. Scale bars: 25 μm. Numbers on the y-axis of bar graphs correspond to the mean±SEM of the parameters assessed. **p<0.01, ***p<0.001. Abbreviations: c-Jun, jun proto-oncogene; CYLD, CYLD lysine 63 deubiquitinase; DEN, diethylnitrosamine; NF-κB p65, nuclear factor of kappa light chain enhancer of activated B cells, p65; R, Romidepsin. |
We next examined livers for the expression of the deubiquitinase CYLD, which counteracts NF-κB and JNK activation.36–38 Western blot analysis indicated that Romidepsin significantly increased CYLD expression (Figure 3B), which is consistent with previous findings in HCC cell lines.39
Another key transcription factor that activates anti-oxidative stress responses and cross-talks with the NF-κB pathway is nuclear factor, erythroid derived 2, like 2 (Nrf2).40 Quantitative gene expression analysis, however, showed that Nrf2 levels were comparable between Romidepsin-treated and untreated mice with HCC (Figure S2B). Likewise, mitogen-activated protein kinase 14 (p38 MAPK), which crosstalks with both JNK/c-Jun and NF-κB pathways in hepatocarcinogenesis,41 was found to be in comparable levels in the two experimental groups examined (Figure S2B).
Peroxisome proliferator activated receptor gamma (PPAR-γ) has been reported to have important roles in cell metabolism, immune response, inflammation and cancer, including HCC.42–44 For that, we next examined PPAR-γ levels in liver samples. By immunohistochemistry and morphometrical analysis, we found that liver tumors of mice treated with Romidepsin had significantly increased PPAR-γ-positive HCC cells, compared to the untreated HCC mice (Figure 4). Quantitative gene expression and Western blot analysis of liver samples were in line with this observation, showing significantly increased PPAR-γ expression, both at gene and protein levels, after HDAC1/2 blockade (Figure 4).
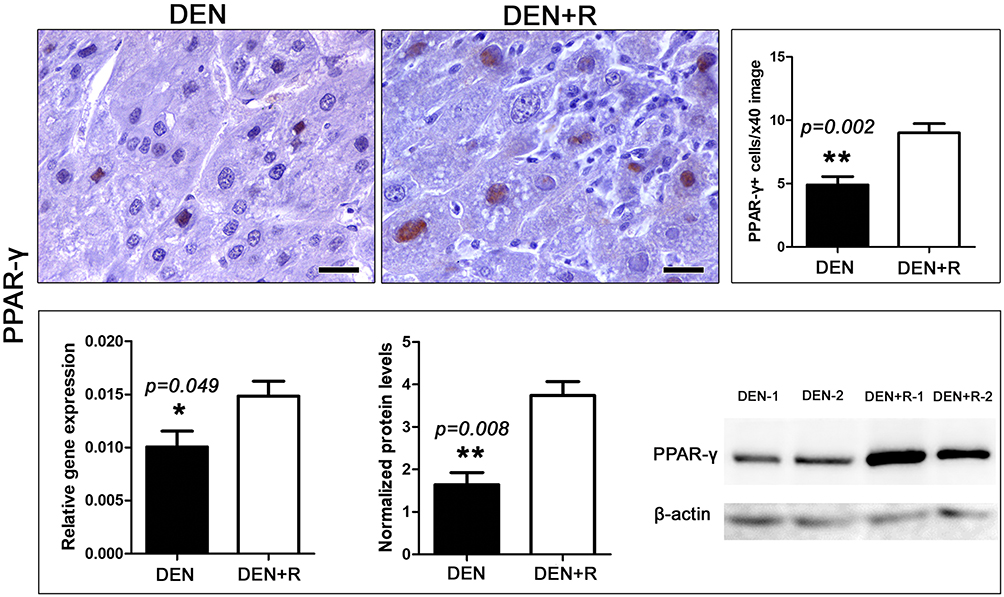 |
Figure 4 HDAC1/2 blockade affects PPAR-γ expression. The anti-tumor treatment significantly increased the number of HCC cells with positive nuclear PPAR-γexpression immunohistochemical signal. This result is line with PPAR-γgene and protein expression analysis as assessed by Real-time PCR and Western Blot analysis, respectively. Western Blot from two selected liver samples per experimental group, also used in Figure 3, is shown. IHC; Diaminobenzidine chromogen, Hematoxylin counterstain. Scale bars: 25 μm. Numbers on the y-axis of bar graphs correspond to the mean±SEM of the parameters assessed. *p<0.05, ** p<0.001. Abbreviations: DEN, diethylnitrosamine; PPAR-γ, peroxisome-proliferator-activated receptor gamma; R, Romidepsin. |
The Inhibition of HDAC1/2 Affects PI3K/Akt but Not Wnt/β-Catenin Signaling in HCC
Romidepsin has been reported to inhibit not only HDAC1/2 but phosphoinositide-3-kinase regulatory subunit 1 (PI3K) as well. PI3K is a key activator protein of the PI3K/Akt pathway that promotes cancer.45 To examine whether Romidepsin treatment alters PI3K expression in livers with HCC we used PI3K-specific IHC. We found that in the non-tumoral liver tissue, PI3K is aptly expressed and has a centrilobular distribution (acinar zone 3 with often expansion into zone 2). PI3K presence was similar in the two treatment groups (Figure S3). However, in neoplastic hepatocytes, PI3K was frequently reduced or even absent. In the non-treated mice, several HCC tumors showed inconsistently a positive PI3K-positive signal of multifocal distribution and variable density. Interestingly, HCC tumors of Romidepsin-treated mice consistently showed absence of PI3K positivity (Figure 5).
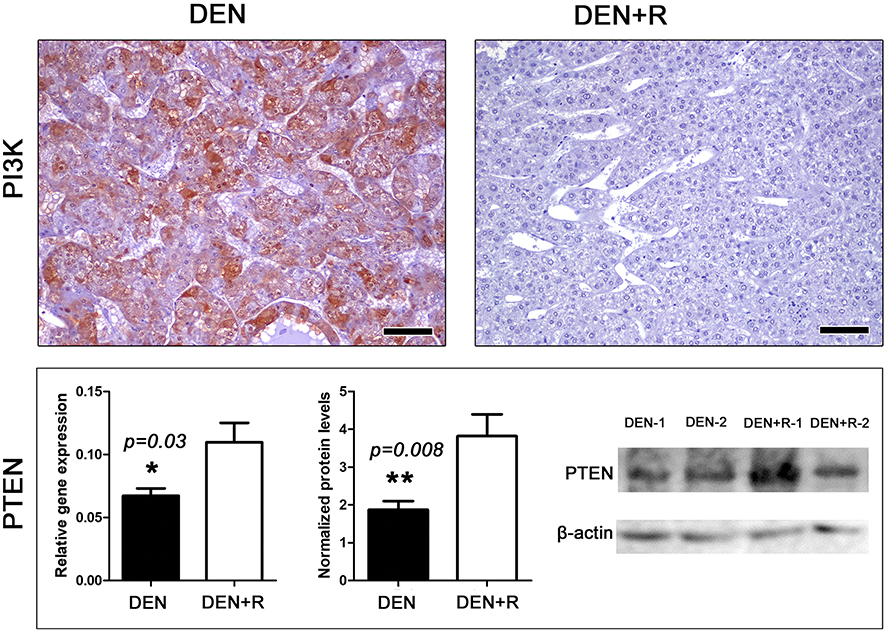 |
Figure 5 Romidepsin treatment ablates PI3K and upregulates PTEN expression in HCC. HCC from non-treated mouse shows able cytoplasmic PI3K expression. By contrast, PI3K immunohistochemical signal in HCC from Romidepsin-treated mouse is practically non-detectable. The treatment up-regulated PTEN expression as determined by both Real-time PCR and Western blot analyses. Western Blot from two selected liver samples per experimental group, also used in Figures 3 and 4, is presented. IHC; Diaminobenzidine chromogen, Hematoxylin counterstain. Scale bars: 25 μm. Numbers on the y-axis of bar graphs correspond to the mean±SEM of the parameters assessed. *p<0.05, ** p<0.001. Abbreviations: DEN, diethylnitrosamine; PI3K, phosphoinositide-3-kinase regulatory subunit 1; PTEN, phosphatase and tensin homolog; R, Romidepsin. |
The tumor suppressor phosphatase and tensin homolog (PTEN) is an upstream negative regulator of PI3K/Akt pathway, which is often involved in HCC evolution.46 Having found that PI3K is practically abolished from neoplastic hepatocytes after Romidepsin treatment, we next examined PTEN. Gene expression analysis revealed that the HDAC1/2 blocker upregulated PTEN expression at statistically significant levels (Figure 5), an observation that was further confirmed by the results of the Western Blot analysis for PTEN protein (Figure 5).
For testing the effects of Romidepsin on the Wnt/β-Catenin signaling pathway, which is activated in liver cancer,47,48 we selectively examined catenin (cadherin associated protein), beta 1 (β-catenin) and the dickkopf WNT signaling pathway inhibitor 1 (DKK1). As we have previously described,30 with β-catenin-specific IHC, DEN-induced hepatocellular adenomas are clearly discernible from adjacent non-tumoral liver tissue due to a denser membrane staining. In the present study, this staining pattern was seen not only in adenomas, but in early small-sized HCC lesions as well. In larger, more progressed tumors, however, β-catenin staining of neoplastic hepatocytes was significantly reduced or even completely abolished. These less differentiated tumors multifocally had areas showing HCC cells with prominent membrane and cytoplasmic (but extremely scarce nuclear) stabilization of β-catenin. At the same time, other areas of the tumors had minimal or absent β-catenin positivity. Β-catenin was mostly expressed in areas of the trabecular or acinar types, whereas areas of the solid type rarely showed foci with β-catenin expression (Figure S3). The qualitative comparison of Romidepsin-treated and not-treated livers suggested that HDAC1/2 blockade did not affect the extent and distribution patterns of β-catenin expression in HCC.
Similarly, both experimental groups were comparable in terms of DKK1 IHC-staining patterns (Figure S3). Irrespectively of Romidepsin treatment, small-sized tumors showed a rather diffuse DKK1 positivity. On the other hand, in larger tumors, DKK1-negative areas predominated, with DKK1-positivity restricting mostly at the boundaries and invasive fronts of tumors (Figure S3).
Discussion
HCC is a rapidly progressive malignancy with bad prognosis if diagnosed at late stages. Early diagnosis and treatment are, therefore, critical for an effective therapy. For the early stages, surgical approach remains the sole option, despite the high recurrence rate observed (70% within 5 years).5,6 Moreover, in HCC, neoadjuvant/adjuvant systemic chemotherapy combined with loco-regional operations is still controversial. Several agents, including sorafenib – the only FDA approved drug for first-line treatment of advanced stage HCC – have been evaluated for preventing and/or treating tumor recurrence, but they failed to demonstrate a clear positive effect.7,49
Given the limited options for HCC treatment and based on the knowledge that hepatocarcinogenesis is significantly influenced by histone modifications, we decided to search for novel HCC therapeutics among histone deacetylation inhibitors. Romidepsin seemed to be a promising option, since it constitutes an already licensed and established anti-cancer drug for CTCL and PTCL that could be easily processed to clinical trials in HCC. Moreover, other than virus reactivation in hepatitis B patients, Romidepsin has not been associated with adverse effects in the liver.50 Although Romidepsin has been shown to suppress growth of HCC cells in culture and in tumor transplant mouse models,22,23 its effectiveness in animal models of spontaneous HCC has not been tested. In these models, the early stages of the disease, where treatment may be more effective, can be better probed. In the present study, we used the well-established DEN-induced HCC mouse model with high similarity to human HCC, in which carcinomas arise from premalignant hepatocellular lesions and adenomas.24,25 In this model, Romidepsin treatment led to a significant suppression of tumorigenesis. Interestingly, at 12 months of age, where DEN-challenged mice had lesions representing the whole spectrum of HCC evolution including large, advanced-stage HCC tumors, in the Romidepsin-treated group almost one-third of mice had no detectable tumors whereas in the remaining ones a significant reduction in tumor size was observed. Romidepsin directly inhibits the action of histone deacetylases HDAC1 and HDAC2.33 We observed that in DEN-treated animals, Romidepsin administration led to increased HDAC2, but not HDAC1, gene expression. It is thus possible that exogenous inhibition of HDAC2 enzyme triggers a counteracting mechanism of compensatory expression at the gene level.
HDACi were shown to play a crucial role in cancer progression through regulation of cell proliferation and apoptosis.33 Although apoptosis has been reported to be a rare event in early HCC lesions in mouse,29,30 in our study despite the low number of apoptotic cells, morphometric analysis revealed a significant increase of Caspase-3-positive (apoptotic) neoplastic cells in Romidepsin-treated HCC mice, as compared to their non-treated counterparts, which supports a role of Romidepsin in promoting apoptosis.23 This was further strengthened by the significant elevation of BAD, a major apoptosis regulator, detected in serum of Romidepsin-treated mice, as compared to the HCC untreated controls. BAD was shown to delay cell proliferation and suppress carcinogenesis in the liver.32 We also examined the expression of Ki-67, a well-known marker of cell proliferation and poor prognosis. It was identified that HDAC1/2 inhibition was accompanied by a reduction in cell proliferation indicated by lower numbers of Ki-67-positive (cycling) cells. This finding is in accordance with a recent study that demonstrated lower Ki-67 expression in the hepatocytes of HDAC1/2 knockout mice during liver regeneration.51
HDACi were shown to have a role in cell cycle regulation in HCC.33 In our study, Romidepsin did not affect Cyclin D1 expression, although HDAC1 blockade has been previously shown to suppress Cyclin D1.34 We also investigated another important cell cycle regulator, the ubiquitously expressed multifunctional protein serine/threonine kinase CK2a. CK2a is overexpressed in HCC and it has been associated with bad prognosis in HCC.52 HDACi have been shown to promote CK2a transcriptional activation in HCC,53 which was also evident in the present study. Through this mechanism, HDACi suppress the tumorigenic effects of Topo IIa, since CK2a has been demonstrated to phosphorylate Topo IIa and lead to its subsequent degradation.53 Topo IIa is associated with more aggressive tumor phenotype, early recurrence and cancer-related death.54–56 Our results revealed comparable levels of Topo IIa mRNA among Romidepsin treated and non-treated groups. In the former group, however, Topo IIa protein may undergo a higher degree of degradation through CK2a phosphorylation, due to the increased levels of CK2a identified.
The majority of HCC arise from chronic liver disease that often associates with chronic inflammation, which predisposes the liver to dysplasia and the subsequent malignant transformation. One of the major factors involved in this inflammation-fibrosis-cancer process is NF-κB.57 NF-κB has been reported to possess oncogenic effects when activated in immune cells and nonparenchymal liver tissue, and a bi-directional role in liver parenchyma.58 Moreover, there is a controversy as to whether HDACi activate or inhibit NF-κB transcription.59,60 Interestingly, in our DEN-induced mouse carcinogenesis, liver tumors of the Romidepsin-treated mice had a significantly lower expression of NF-κB p65. A key negative regulator of NF-κB is the deubiquitinase CYLD, which was shown to be upregulated in liver cancer cells after HDACi treatment.39 Our results support this observation, since Romidepsin-associated HCC-suppression correlated with a significant elevation of liver CYLD protein levels, as compared to the HCC untreated controls. Thus, Romidepsin NF-κB p65 downregulation may be mediated, at least in part, by CYLD overexpression. Other molecules that contribute to NF-κB pleiotropic action in inflammation, oxidative stress and apoptosis, such as c-Jun, Nrf2 and p38 MAPK, have all been associated with more aggressive hepatocellular carcinogenesis.35,41,61 Among them, only c-Jun protein levels were found to be lower in Romidepsin-treated HCC mice. This finding is in accordance with the effect of HDACi on c-Jun expression in other types of cancer.62
The role of Romidepsin in regulating inflammatory response was further supported by the observation that PPAR-γ expression was significantly elevated in Romidepsin-treated HCC mice, as compared to the HCC untreated ones. PPAR-γ overexpression has been associated alongside with metabolism improvement, with inflammation inhibition, immune modulation and antimetastatic effects.43,44,63,64 Moreover, Thiazolidinedione (TZD), a synthetic ligand of PPAR-γ used in the treatment of type 2 diabetes, was shown to decrease the risk of HCC development in diabetes mellitus patients.65 For that, further studies are needed to investigate whether a Romidepsin/TZD combination therapy may further improve the tumor-suppressive outcome, especially in the early stages of HCC.
The Wnt/β-catenin and the PI3K/Akt are considered to be major HCC-related signaling pathways. Their activation is associated with more aggressive tumors, poor prognosis, and earlier recurrence in HCC.48,66,67 For that, we examined the expression of selected key molecules of these pathways. In Wnt/β-catenin pathway, after binding of the Wnt ligands to Frizzled receptors, β-catenin escapes degradation and translocates to the nucleus. This ligand-receptor binding leads to a larger complex with LRP5/6, the formation of which is inhibited by DKK1.48 In our experimental conditions, Romidepsin did not affect the expression levels of either β-catenin or DKK1 in mice with HCC. In PI3K/Akt pathway, the activation of the key molecule PI3K by tyrosine kinase receptors, G protein–coupled receptors, integrins, B and T cell receptors or cytokine receptors induces the intracellular signaling cascade. The major inhibitor of this pathway is PTEN, which directly inhibits PI3K activation.67 Romidepsin has been shown to inhibit PI3K in colorectal and prostate cancer cells.45 Here, the anti-oncogenic effect of Romidepsin in HCC correlated significantly with robust PI3K downregulation and PTEN tumor suppressor upregulation. These findings suggest that Romidepsin may at least in part exert its antitumor activity through inhibition of PI3K/Akt pathway.
Finally, the role of c-Met protein that regulates many cellular processes and signaling pathways was also investigated. In HCC, c-Met activation has been associated with tumor promotion, aggressiveness and poor prognosis.68,69 On the other hand, loss of c-Met has been shown to promote DEN-induced hepatocellular carcinogenesis through regulation of cell proliferation and oxidative stress.70,71 Our results support a tumor-suppressive role of c-Met, since a significant overexpression of c-Met was identified in serum of Romidepsin-treated HCC mice. c-Met seems to have distinct roles in different stages of HCC and this should be considered when designing novel treatments.68,70,71
In conclusion, this is the first study demonstrating that the HDAC1/2 inhibitor Romidepsin suppresses early-stage HCC. A possible mechanism associated with the identified tumor-suppressing effect of Romidepsin in DEN-induced HCC, which involves deregulations of selected cancer-related molecules, is diagrammatically presented in Figure 6. As stated, the therapeutic options for this rapidly progressive malignancy are still limited with tumor recurrence rate remaining high. Thus, histone acetylation, which has a crucial role in liver cancer progression, seems to be a promising novel approach for the management of early-stage HCC. In order to proceed to clinical trials; however, further studies are necessary to clarify the mechanism through which the licensed for lymphoma treatment Romidepsin exerts its antitumor effects in early liver carcinoma.
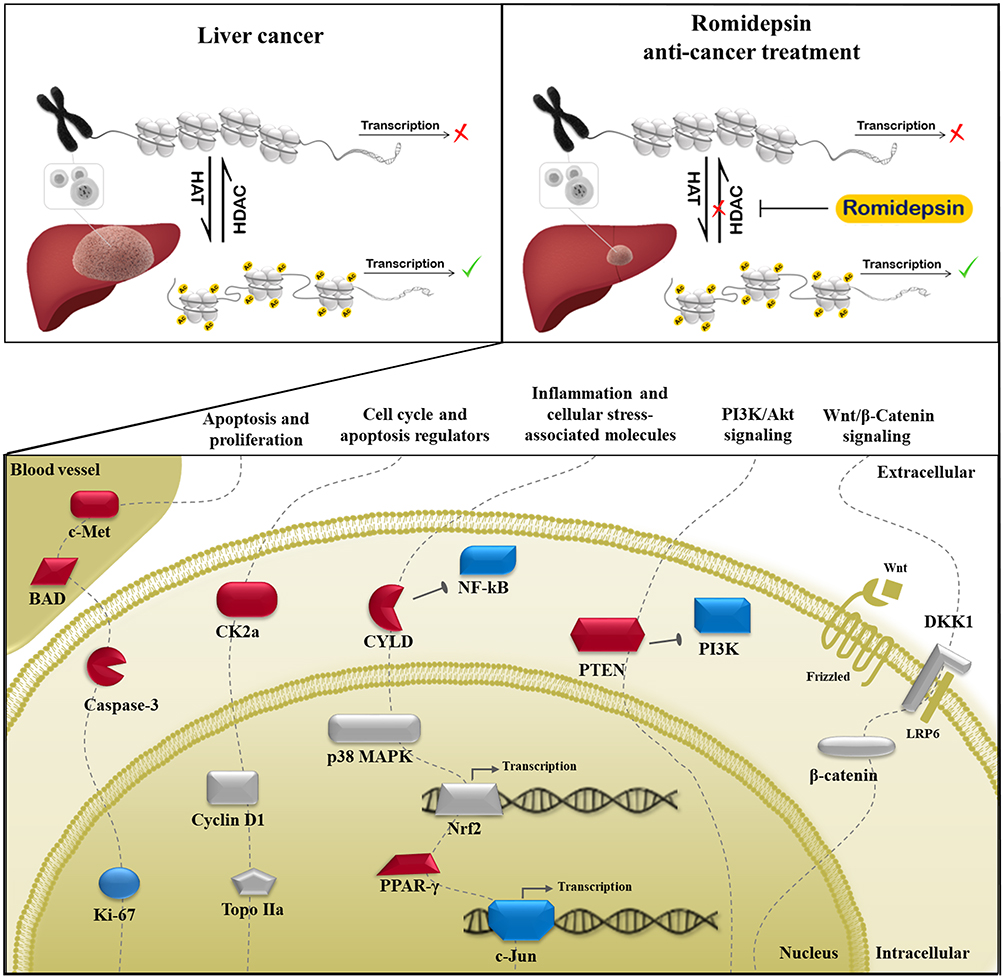 |
Figure 6 A possible mechanism associated with the tumor-suppressing effect of Romidepsin in DEN-induced HCC mouse model. Romidepsin suppresses liver tumorigenesis, at least in part, by mediating the deregulated expression of extra- and intra-cellular molecules involved in cell proliferation, apoptosis, cell cycle regulation, and inflammation processes, as well as in cancer-related signaling pathways (dashed lines). Molecules whose expression levels are upregulated, downregulated or non-affected by Romidepsin treatment are depicted in red, blue, and grey color, respectively. Abbreviations: BAD, Bcl2-associated agonist of cell death; CK2a, casein kinase 2, alpha 1 polypeptide; c-Met, met proto-oncogene; CYLD, CYLD lysine 63 deubiquitinase; DKK1, dickkopf WNT signaling pathway inhibitor 1; Frizzled, frizzled class receptor 9; HAT, histone acetyltransferases; HDAC, histone deacetylase; Ki-67, antigen identified by monoclonal antibody Ki 67; LRP6, low-density lipoprotein receptor-related protein 6; NF-κB p65, nuclear factor of kappa light chain enhancer of activated B cells, p65; c-Jun, jun proto-oncogene; Nrf2, nuclear factor, erythroid derived 2, like 2; p38 MAPK, mitogen-activated protein kinase 14; PI3K, phosphoinositide-3-kinase regulatory subunit 1; PPAR-γ, peroxisome-proliferator-activated receptor gamma; PTEN, phosphatase and tensin homolog; β-catenin, catenin (cadherin associated protein) beta 1; Topo IIa, topoisomerase (DNA) II alpha; Wnt, wingless-type MMTV integration site family. |
Abbreviations
BAD, Bcl2-associated agonist of cell death; CK2a, casein kinase 2, alpha 1 polypeptide; c-Met, met proto-oncogene; CTCL, cutaneous T-cell lymphoma; CYLD, CYLD lysine 63 deubiquitinase; DEN, diethylnitrosamine; DKK1, dickkopf WNT signaling pathway inhibitor 1; FDA, Food and Drug Administration; HATs, histone acetyltransferases; HCC, hepatocellular carcinoma; HDACi, HDAC inhibitors; HDACs, histone deacetylases; NF-κB p65, nuclear factor of kappa light chain enhancer of activated B cells, p65; c-Jun, jun proto-oncogene; Nrf2, nuclear factor, erythroid derived 2, like 2; p38 MAPK, mitogen-activated protein kinase 14; PI3K, phosphoinositide-3-kinase regulatory subunit 1; PPAR-γ, peroxisome-proliferator-activated receptor gamma; PTCL, peripheral T-cell lymphoma; PTEN, phosphatase and tensin homolog; β-catenin, catenin (cadherin associated protein), beta 1; Topo IIa, topoisomerase (DNA) II alpha.
Acknowledgments
This work was funded as Scholarship by the Experimental, Educational and Research Center ELPEN Pharmaceuticals. The authors are pleased to acknowledge the contribution of the personnel of the Experimental, Educational and Research Center of ELPEN Pharmaceutical, namely: A. Zacharioudaki, M. Karamperi, K. Tsarea, E. Karampela, S. Gerakis, E. Gerakis, A. Karaiskos, and N. Psychalakis, in the performance of the experiments.
Alexandros Hardas is presently affiliated with the Department of Pathobiology & Population Sciences, The Royal Veterinary College, Hatfield, UK.
Disclosure
Dimitrios Giakoustidis reports grants from ELPEN during the conduct of the study. The authors report no other possible conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Wallace MC, Preen D, Jeffrey GP, Adams LA. The evolving epidemiology of hepatocellular carcinoma: a global perspective. Expert Rev Gastroenterol Hepatol. 2015;9(6):765–779. doi:10.1586/17474124.2015.1028363
3. Kudo M. Multistep human hepatocarcinogenesis: correlation of imaging with pathology. J Gastroenterol. 2009;44(S19):112–118. doi:10.1007/s00535-008-2274-6
4. Severi T, van Malenstein H, Verslype C, van Pelt JF. Tumor initiation and progression in hepatocellular carcinoma: risk factors, classification, and therapeutic targets. Acta Pharmacol Sin. 2010;31(11):1409–1420. doi:10.1038/aps.2010.142
5. Heimbach JK, Kulik LM, Finn RS, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatol. 2018;67(1):358–380. doi:10.1002/hep.29086/suppinfo
6. Vogel A, Cervantes A, Chau I, et al. Hepatocellular carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up † incidence and epidemiology. Ann Oncol. 2019. doi:10.1093/annonc/mdy308
7. Akateh C, Black SM, Conteh L, et al. Neoadjuvant and adjuvant treatment strategies for hepatocellular carcinoma. World J Gastroenterol. 2019;25(28):3704–3721. doi:10.3748/wjg.v25.i28.3704
8. Gong Q, Qin Z, Hou F. Improved treatment of early small hepatocellular carcinoma using sorafenib in combination with radiofrequency ablation. Oncol Lett. 2017;14(6):7045–7048. doi:10.3892/ol.2017.7174
9. Invernizzi F, Iavarone M, Donato MF, et al. Early treatment with sorafenib and mTOR inhibitor in recurrent hepatocellular carcinoma after liver transplantation: safety and survival. Dig Liver Dis. 2018;50(1):49. doi:10.1016/j.dld.2018.01.052
10. Ogunwobi OO, Harricharran T, Huaman J, et al. Mechanisms of hepatocellular carcinoma progression. World J Gastroenterol. 2019;25(19):2279–2293. doi:10.3748/wjg.v25.i19.2279
11. Wahid B, Ali A, Rafique S, Idrees M. New insights into the epigenetics of hepatocellular carcinoma. Biomed Res Int. 2017;2017:1609575. doi:10.1155/2017/1609575
12. Zimmer V, Lammert F. Genetics and epigenetics in the fibrogenic evolution of chronic liver diseases. Best Pract Res Clin Gastroenterol. 2011;25(2):269–280. doi:10.1016/j.bpg.2011.02.007
13. Marquardt JU, Andersen JB, Thorgeirsson SS. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat Rev Cancer. 2015;15(11):653–667. doi:10.1038/nrc4017
14. Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26(37):5420–5432. doi:10.1038/sj.onc.1210610
15. Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6(10):a026831. doi:10.1101/cshperspect.a026831
16. Chrun ES, Modolo F, Daniel FI. Histone modifications: a review about the presence of this epigenetic phenomenon in carcinogenesis. Pathol Res Pract. 2017;213(11):1329–1339. doi:10.1016/j.prp.2017.06.013
17. Neureiter D, Stintzing S, Kiesslich T, Ocker M. Hepatocellular carcinoma: therapeutic advances in signaling, epigenetic and immune targets. World J Gastroenterol. 2019;25(25):3136–3150. doi:10.3748/wjg.v25.i25.3136
18. Khan FS, Ali I, Afridi UK, Ishtiaq M, Mehmood R. Epigenetic mechanisms regulating the development of hepatocellular carcinoma and their promise for therapeutics. Hepatol Int. 2017;11(1):45–53. doi:10.1007/s12072-016-9743-4
19. Tsilimigras DI, Ntanasis-Stathopoulos I, Moris D, Spartalis E, Pawlik TM. Histone deacetylase inhibitors in hepatocellular carcinoma: a therapeutic perspective. Surg Oncol. 2018;27(4):611–618. doi:10.1016/j.suronc.2018.07.015
20. Banik D, Moufarrij S, Villagra A. Immunoepigenetics combination therapies: an overview of the role of HDACs in cancer immunotherapy. Int J Mol Sci. 2019;20(9):2241. doi:10.3390/ijms20092241
21. Iyer SP, Foss FF. Romidepsin for the treatment of peripheral T-cell lymphoma. Oncologist. 2015;20(9):1084–1091. doi:10.1634/theoncologist.2015-0043
22. Zhou H, Cai Y, Liu D, et al. Pharmacological or transcriptional inhibition of both HDAC1 and 2 leads to cell cycle blockage and apoptosis via p21 Waf1/Cip1 and p19 INK4d upregulation in hepatocellular carcinoma. Cell Prolif. 2018;51(3):e12447. doi:10.1111/cpr.12447
23. Sun W-J, Huang H, He B, et al. Romidepsin induces G2/M phase arrest via Erk/cdc25C/cdc2/cyclinB pathway and apoptosis induction through JNK/c-Jun/caspase3 pathway in hepatocellular carcinoma cells. Biochem Pharmacol. 2017;127:90–100. doi:10.1016/j.bcp.2016.12.008
24. Brown ZJ, Heinrich B, Greten TF. Mouse models of hepatocellular carcinoma: an overview and highlights for immunotherapy research. Nat Rev Gastroenterol Hepatol. 2018;15(9):536–554. doi:10.1038/s41575-018-0033-6
25. Lee J-S, Chu I-S, Mikaelyan A, et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet. 2004;36(12):1306–1311. doi:10.1038/ng1481
26. National Research Council. Guide for the Care and Use of Laboratory Animals.
27. Ouzounidis N, Giakoustidis A, Poutahidis T, et al. Interleukin 18 binding protein ameliorates ischemia/reperfusion-induced hepatic injury in mice. Liver Transpl. 2016;22(2):237–246. doi:10.1002/lt.24359
28. Karamanavi E, Angelopoulou K, Lavrentiadou S, et al. Urokinase-type plasminogen activator deficiency promotes neoplasmatogenesis in the colon of micejama. Transl Oncol. 2014;7(2):174–187.e5. doi:10.1016/j.tranon.2014.02.002
29. Sklavos A, Poutahidis T, Giakoustidis A, et al. Effects of wnt-1 blockade in DEN-induced hepatocellular adenomas of mice. Oncol Lett. 2018;15(1):1211–1219. doi:10.3892/ol.2017.7427
30. Gavriilidis P, Poutahidis T, Giakoustidis A, et al. Targeting hepatocarcinogenesis model in C56BL6 mice with pan-aurora kinase inhibitor danusertib. J Cancer. 2018;9(5):914–922. doi:10.7150/jca.22329
31. Bupathi M, Kaseb A, Meric-Bernstam F, Naing A. Hepatocellular carcinoma: where there is unmet need. Mol Oncol. 2015;9(8):1501–1509. doi:10.1016/j.molonc.2015.06.005
32. Pierce RH, Vail ME, Ralph L, Campbell JS, Fausto N. Bcl-2 expression inhibits liver carcinogenesis and delays the development of proliferating foci. Am J Pathol. 2002;160(5):1555–1560. doi:10.1016/S0002-9440(10)61101-7
33. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18(7):1414. doi:10.3390/ijms18071414
34. Xie HJ, Noh JH, Kim JK, et al. HDAC1 inactivation induces mitotic defect and caspase-independent autophagic cell death in liver cancer. PLoS One. 2012;7(4):e34265. doi:10.1371/journal.pone.0034265
35. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121(7):977–990. doi:10.1016/j.cell.2005.04.014
36. Trompouki E, Hatzivassillou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature. 2003;424(6950):793–796. doi:10.1038/nature01803
37. Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature. 2003;424(6950):801–805. doi:10.1038/nature01802
38. Brummelkamp TR, Nijman SMB, Dirac AMG, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature. 2003;424(6950):797–801. doi:10.1038/nature01811
39. Kotantaki P, Mosialos G. The expression of tumor suppressor gene Cyld is upregulated by histone deacetylase inhibitors in human hepatocellular carcinoma cell lines. Cell Biochem Funct. 2016;34(7):465–468. doi:10.1002/cbf.3212
40. Ahmed SMU, Luo L, Namani A, Wang XJ, Tang X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochim Biophys Acta Mol Basis Dis. 2017;1863(2):585–597. doi:10.1016/j.bbadis.2016.11.005
41. Nakagawa H, Maeda S. Inflammation- and stress-related signaling pathways in hepatocarcinogenesis. World J Gastroenterol. 2012;18(31):4071. doi:10.3748/wjg.v18.i31.4071
42. Liu Y, Colby JK, Zuo X, Jaoude J, Wei D, Shureiqi I. The role of PPAR-δ in metabolism, inflammation, and cancer: many characters of a critical transcription factor. Int J Mol Sci. 2018. doi:10.3390/ijms19113339
43. Clark RB. The role of PPARs in inflammation and immunity. J Leukoc Biol. 2002;71(3):388–400.
44. Martin H. Role of PPAR-gamma in inflammation. Prospects for therapeutic intervention by food components. Mutat Res Fundam Mol Mech Mutagen. 2010;690(1–2):57–63. doi:10.1016/j.mrfmmm.2009.09.009
45. Saijo K, Imamura J, Narita K, et al. Biochemical, biological and structural properties of romidepsin (FK228) and its analogs as novel HDAC/PI3K dual inhibitors. Cancer Sci. 2015;106(2):208–215. doi:10.1111/cas.12585
46. Xu W, Yang Z, Xie C, et al. PTEN lipid phosphatase inactivation links the hippo and PI3K/Akt pathways to induce gastric tumorigenesis. J Exp Clin Cancer Res. 2018;37(1). doi:10.1186/s13046-018-0795-2
47. Giakoustidis A, Giakoustidis D, Mudan S, Sklavos A, Williams R. Molecular signalling in hepatocellular carcinoma: role of and crosstalk among WNT/ß-catenin, sonic hedgehog, notch and dickkopf-1. Can J Gastroenterol Hepatol. 2015;29(4):209–217. doi:10.1155/2015/172356
48. Khalaf AM, Fuentes D, Morshid AI, et al. Role of Wnt/β-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J Hepatocell Carcinoma. 2018;5:61–73. doi:10.2147/JHC.S156701
49. Zeng J, Lv L, Mei ZC. Efficacy and safety of transarterial chemoembolization plus sorafenib for early or intermediate stage hepatocellular carcinoma: a systematic review and meta-analysis of randomized controlled trials. Clin Res Hepatol Gastroenterol. 2016;40(6):688–697. doi:10.1016/j.clinre.2016.04.006
50. Ritchie D, Piekarz RL, Blombery P, et al. Reactivation of DNA viruses in association with histone deacetylase inhibitor therapy: a case series report. Haematologica. 2009;94(11):1618–1622. doi:10.3324/haematol.2009.008607
51. Xia J, Zhou Y, Ji H, et al. Loss of histone deacetylases 1 and 2 in hepatocytes impairs murine liver regeneration through Ki67 depletion. Hepatology. 2013;58(6):2089–2098. doi:10.1002/hep.26542
52. Zhang HX, Jiang SS, Zhang XF, et al. Protein kinase CK2α catalytic subunit is overexpressed and serves as an unfavorable prognostic marker in primary hepatocellular carcinoma. Oncotarget. 2015;6(33):34800–34817. doi:10.18632/oncotarget.5470
53. Chen MC, Chen CH, Chuang HC, Kulp SK, Teng CM, Chen CS. Novel mechanism by which histone deacetylase inhibitors facilitate topoisomerase IIα degradation in hepatocellular carcinoma cells. Hepatology. 2011;53(1):148–159. doi:10.1002/hep.23964
54. Watanuki A, Ohwada S, Fukusato T, et al. Prognostic significance of DNA topoisomerase IIalpha expression in human hepatocellular carcinoma. Anticancer Res. 2002;22(2B):1113–1119.
55. Panvichian R, Tantiwetrueangdet A, Angkathunyakul N, Leelaudomlipi S. TOP2A amplification and overexpression in hepatocellular carcinoma tissues. Biomed Res Int. 2015;2015:1–8. doi:10.1155/2015/381602
56. Wong N, Yeo W, Wong W-L, et al. TOP2A overexpression in hepatocellular carcinoma correlates with early age onset, shorter patients survival and chemoresistance. Int J Cancer. 2009;124(3):644–652. doi:10.1002/ijc.23968
57. Czauderna C, Castven D, Mahn FL, Marquardt JU. Context-dependent role of NF-κB signaling in primary liver cancer—from tumor development to therapeutic implications. Cancers (Basel). 2019;11(8):1053. doi:10.3390/cancers11081053
58. Luedde T, Schwabe RF. NF-κB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8(2):108–118. doi:10.1038/nrgastro.2010.213
59. Adam E, Quivy V, Bex F, et al. Potentiation of tumor necrosis factor-induced NF-B activation by deacetylase inhibitors is associated with a delayed cytoplasmic reappearance of I B. Mol Cell Biol. 2003;23(17):6200–6209. doi:10.1128/MCB.23.17.6200-6209.2003
60. Imre G, Gekeler V, Leja A, Beckers T, Boehm M. Histone deacetylase inhibitors suppress the inducibility of nuclear factor-κB by tumor necrosis factor-α receptor-1 down-regulation. Cancer Res. 2006;66(10):5409–5418. doi:10.1158/0008-5472.CAN-05-4225
61. Raghunath A, Sundarraj K, Arfuso F, Sethi G, Perumal E. Dysregulation of Nrf2 in hepatocellular carcinoma: role in cancer progression and chemoresistance. Cancers (Basel). 2018;10(12):481. doi:10.3390/cancers10120481
62. He W, Wu Y, Tang X, et al. HDAC inhibitors suppress c-Jun/Fra-1-mediated proliferation through transcriptionally downregulating MKK7 and Raf1 in neuroblastoma cells. Oncotarget. 2016;7(6):6727–6747. doi:10.18632/oncotarget.6797
63. Chi C-W, Hsu H-T. Emerging role of the peroxisome proliferator-activated receptor-gamma in hepatocellular carcinoma. J Hepatocell Carcinoma. 2014;127. doi:10.2147/jhc.s48512:127
64. Shen B, Chu ESH, Zhao G, et al. PPARgamma inhibits hepatocellular carcinoma metastases in vitro and in mice. Br J Cancer. 2012;106(9):1486–1494. doi:10.1038/bjc.2012.130
65. Huang MY, Chung CH, Chang WK, et al. The role of thiazolidinediones in hepatocellular carcinoma risk reduction: a population-based cohort study in Taiwan. Am J Cancer Res. 2017;7(7):1606–1616.
66. Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014;60(4):855–865. doi:10.1016/j.jhep.2013.11.031
67. Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29(36):4989–5005. doi:10.1038/onc.2010.236
68. Bouattour M, Raymond E, Qin S, et al. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology. 2018;67(3):1132–1149. doi:10.1002/hep.29496
69. Kim JH, Kim HS, Kim BJ, Jang HJ, Lee J. Prognostic value of c-Met overexpression in hepatocellular carcinoma: a meta-analysis and review. Oncotarget. 2017;8(52):90351–90357. doi:10.18632/oncotarget.20087
70. Takami T, Kaposi-Novak P, Uchida K, et al. Loss of hepatocyte growth factor/c-Met signaling pathway accelerates early stages of N-nitrosodiethylamine-induced hepatocarcinogenesis. Cancer Res. 2007;67(20):9844–9851. doi:10.1158/0008-5472.CAN-07-1905
71. Marx-Stoelting P, Borowiak M, Knorpp T, Birchmeier C, Buchmann A, Schwarz M. Hepatocarcinogenesis in mice with a conditional knockout of the hepatocyte growth factor receptor c-Met. Int J Cancer. 2009;124(8):1767–1772. doi:10.1002/ijc.24167
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.