Back to Journals » The Application of Clinical Genetics » Volume 13
Haptoglobin Gene Polymorphism in Patients with Sickle Cell Anemia: Findings from a Nigerian Cohort Study
Authors Olatunya OS ![]() , Albuquerque DM
, Albuquerque DM ![]() , Santos MNN
, Santos MNN ![]() , Kayode TS, Adekile A, Costa FF
, Kayode TS, Adekile A, Costa FF ![]()
Received 20 January 2020
Accepted for publication 26 March 2020
Published 8 May 2020 Volume 2020:13 Pages 107—114
DOI https://doi.org/10.2147/TACG.S246607
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Oladele Simeon Olatunya,1,2 Dulcineia Martins Albuquerque,1 Magnun Nueldo Nunes Santos,3 Tolorunju Segun Kayode,4 Adekunle Adekile,5 Fernando Ferreira Costa1
1Hematology and Hemotherapy Center, University of Campinas, Campinas, São Paulo State, Brazil; 2Department of Paediatrics, College of Medicine, Ekiti State University, Ado Ekiti, Ekiti State, Nigeria; 3Department of Clinical Pathology, School of Medical Sciences, University of Campinas, Campinas, São Paulo State, Brazil; 4Department of Chemical Pathology, Ekiti State University Teaching Hospital, Ado Ekiti, Ekiti State, Nigeria; 5Department of Pediatrics, Faculty of Medicine, Kuwait University, Jabriya, Kuwait
Correspondence: Oladele Simeon Olatunya
Email [email protected]
Purpose: To determine the various haptoglobin genotypes and their influence on the clinico-laboratory manifestations among young Nigerian sickle cell anemia (SCA) patients.
Patients and Methods: A total of 101 SCA patients and 64 controls were studied. SCA was diagnosed by polymerase chain reaction (PCR). Haptoglobin genotype was determined by PCR followed by agarose gel electrophoresis. The patients’ laboratory and clinical parameters were differentiated by haptoglobin genotypes.
Results: The Hp1 and Hp2 alleles frequencies were 0.62 and 0.38 in the patients and 0.73 and 0.27 in the controls, respectively, and these did not differ significantly (p> 0.05). The haptoglobin genotype distribution among the patients and controls were Hp1-1, 43 (42.6%); Hp2-1, 40 (39.6%); Hp2-2, 18 (17.8%) and Hp1-1, 35 (54.7%); Hp2-1, 24 (37.5%); Hp2-2, 5 (7.8%), respectively, with no difference between the two groups (P> 0.05). No significant difference was found in the clinical events and laboratory parameters of the patients when partitioned according to the various haptoglobin genotypes (P> 0.05).
Conclusion: This study found that haptoglobin gene polymorphism does not have a significant influence on the clinico-laboratory manifestations among SCA patients.
Keywords: haptoglobin gene polymorphism, sickle cell disease, clinico-laboratory manifestations
Introduction
Sickle cell disease (SCD) is an inherited disorder characterized by the mutation (HBB; glu(E)6valA; GAG-GTG; rs334) which produces the sickle hemoglobin (HbS) and homozygosity for the HbS in an individual results in sickle cell anemia (SCA) and a very severe disease spectrum.1–3 SCA prevalence is high in the sub-Saharan Africa (SSA).1–3 Individuals with SCA exhibit clinical variability both within the individuals and across patients.1–3 Many hypotheses have been advanced as reasons for the observed phenotypic diversities in SCA, and environmental influences, as well as socio-demographic characteristics, are believed to play key roles in SSA.2–4
Also, some genetic modifiers like the presence of α-thalassemia, modulators of fetal hemoglobin production, β-globin gene haplotypes and other non-globin genetic factors like the uridine diphosphate glucuronosyl transferase 1A (UGT1A1) promoter polymorphisms and glucose −6-phosphatase dehydrogenase deficiency (G6PD) play key roles.1–3,5
Furthermore, both traditional markers of hemolysis and newer hemolysis marker like red blood cell microparticles have proven useful in prognosticating the clinical course of SCA.6,7 However, none of these could fully predict and explain the clinical diversities seen in SCA. Therefore, the search for more candidate markers of the disease condition continues.
Haptoglobin is found in both mammals and humans. In humans, its polymorphism results in two dominant alleles (Hp1 and Hp2) located on chromosome 16q22 leading to the three genotypes described so far, namely: HP1-1, HP2-1, and HP2-2.8,9 The HP1-1 genotype is very common in Africa and Latin America but rare in Southeast Asia. However, it has the highest efficiency and biological activities in respect of mopping up free plasma Hb (plasma hemoglobin) and suppression of inflammation8,9 while the HP2-2 variant has the least capacity to do these.8,9 The HP2-1 variant has intermediate biological and anti-inflammatory abilities compared to HP1-1 and HP2-2.8,9
Some antibacterial and antioxidant properties have been associated with haptoglobin, and it is the major scavenger of free plasma Hb, with which it combines to form a complex that is scavenged and internalized by the scavenger receptor CD163, located on the surfaces of circulating monocytes and liver macrophages.8,9 Individuals with certain haptoglobin genotypes are susceptible to oxidative stress, endothelial dysfunction and increased risk/evolution of various clinical conditions.10–14 Adams et al10 linked HP2-2 genotype with heart disease and diabetes risk among European Americans. Similarly, HP2-1 and HP2-2 genotypes were linked with more complications among Ghanaians with diabetes mellitus11 while both Boettger et al12 and Langlois and Delanghe13 found relationships between haptoglobin polymorphisms and blood cholesterol levels in addition to the modulation of human immunodeficiency virus disease outcome. Also, an in-vitro study by Chintagari and colleagues,14 on SCD mice reported that haptoglobin lessens the expression of heme oxygenase-1 (HO-1).
Although different studies have described the frequencies of the various genotypes and alleles of haptoglobin among different populations of SCD patients,8,9,15 there is a lack of data regarding how haptoglobin gene polymorphism impacts the clinical course of SCD in the SSA where the bulk of patients with SCD are found. However, some earlier studies from more developed countries have suggested a possibility that the various pathophysiological processes (oxidative stress, inflammation and hemolysis, etc.) and their associated downstream events in SCA could be perturbed by the different haptoglobin genotypes.13–16
Despite the huge burden of SCA in Nigeria, there is a lack of data on how haptoglobin genotypes affect the laboratory and clinical expressions among patients with SCA in the country. This study describes the haptoglobin genotypes among a Nigerian SCA population and their influence on the clinico-laboratory manifestations of the patients.
Patients and Methods
Study Design and Setting
This was a cross-sectional descriptive study conducted at the pediatrics hematology unit of the Ekiti State University Teaching Hospital (EKSUTH) Ado Ekiti, Ekiti State Nigeria. The study participants were 102 young SCA patients who were not on hydroxyurea therapy. They were aged between 2 and 21 years (median of 9 years) and were regular attendees at the Pediatric Hematology unit. The patients have been on follow up for a median of 4 years, range 1–14 years. They were all in steady state at the time of recruitment, ie free from any crisis and transfusion for at least 1 month and 100 days, respectively. In addition, 68 unrelated children (20 Hb AS, and 48 Hb AA) attending the EKSUTH were selected as the controls. Patients having any chronic disease(s) other than SCA were excluded. Those on hydroxyurea or chronic blood transfusion were also excluded. To diagnose SCA, the Hb genotype was determined in Nigeria using cellulose acetate paper in alkaline buffer electrophoresis and high-performance liquid chromatography (HPLC). In addition, SCA diagnosis was confirmed by DNA studies at the Hematology and Hemotherapy Centre, University of Campinas (UNICAMP), Sao Paulo, Brazil as previously described.17
Ethical Approval
The study was performed according to the Declaration of Helsinki on research involving human subjects. The Ethics and Research Committee of EKSUTH approved the study with reference no: A67/2016/03/003. Also, the University of Campinas Ethics Committee approved the study with reference no: CAAE 54031115.9.0000.5404. Furthermore, written informed consent of parents/caregivers was obtained. In addition, patients’ assents and consents were obtained as applicable and the purpose of the study was explained to the participants` parents in clear and plain language.
Data Collection
Clinical and Laboratory Data
Data on clinical evolution and steady-state laboratory parameters were retrieved and recorded for each patient using a pretested review chart form. The steady-state parameters were performed with standard techniques and an average of at least two results (conducted at least 3 months apart) for each parameter was used for each patient. These included the complete blood count performed with Sysmex KX21N hematology analyser manufactured by Sysmex Corporation, Kobe, Japan and serum lactate dehydrogenase (LDH), aspartate transaminase (AST), and bilirubin. High-performance liquid chromatography (HPLC) using (Bio-Rad Variant D10, Hercules, CA, USA) was done to measure the HbF, HbS and other Hb variants.
Other information obtained for each patient were the details of their disease clinical evolution as well as their biodata as retrieved from their hospital records. The diagnoses of clinical events were arrived at using both clinical skills and appropriate investigations following independent reviews by physicians and a pediatric hematology specialist. The clinical evolution record obtained included the occurrence of vaso-occlusive crisis (VOC) ie number of severe bone pain crises that disrupt daily activities, required hospital admission and use of opioids analgesics within the preceding 1 year prior to recruitment. Also, the occurrences of lifetime chronic complications of SCA were determined. These included the presence of gallstone as determined by serial abdominal ultrasound scans done when indicated, presence of overt stroke, priapism and leg ulcer as previously described.18
Genetic Studies
The DNAs extracted from the whole blood sample drawn from the participants in tubes containing EDTA by using (Qiagen QIAamp DNA Blood Mini Kit, Cat No. 51104 Germany) was used to confirm SCD diagnosis by polymerase chain reaction (PCR) and perform all haptoglobin studies. The DNA studies were carried out blinded to the clinical and laboratory parameters of the participants at the Centro de Hematologia e Hemoterapia (Hemocentro), UNICAMP, Campinas, São Paulo State, Brazil.
Haptoglobin Genotypes Determination
Haptoglobin genotypes of the participants were determined by standard protocols as previously described.8,15,19,20 These involved selective PCR amplification of the DNA segments of the various alleles ie (Hp1 and Hp2 alleles), followed by gel electrophoresis.8,15,19,20
The reaction was performed in a VeritiTM Thermal Cycler (Applied Biosystems, Foster City, CA, USA) in 25 μL final volume containing 150 ng of DNA sample; 1X enzyme Buffer; 2.5 mM of MgCl2; 0.3 mM of dNTP mix; 0.1 µM of each primer (Table 1) and 0.06U of Taq DNA Polymerase (Biotools, Madrid, Spain). Thermal cycle conditions were as follows: preheating at 94°C by 5 minutes, followed by 30 cycles of 94°C for 45 seconds, 58°C for 1 minute, and 72°C for 1 minute and a final extension at 72°C for 7 minutes was performed.
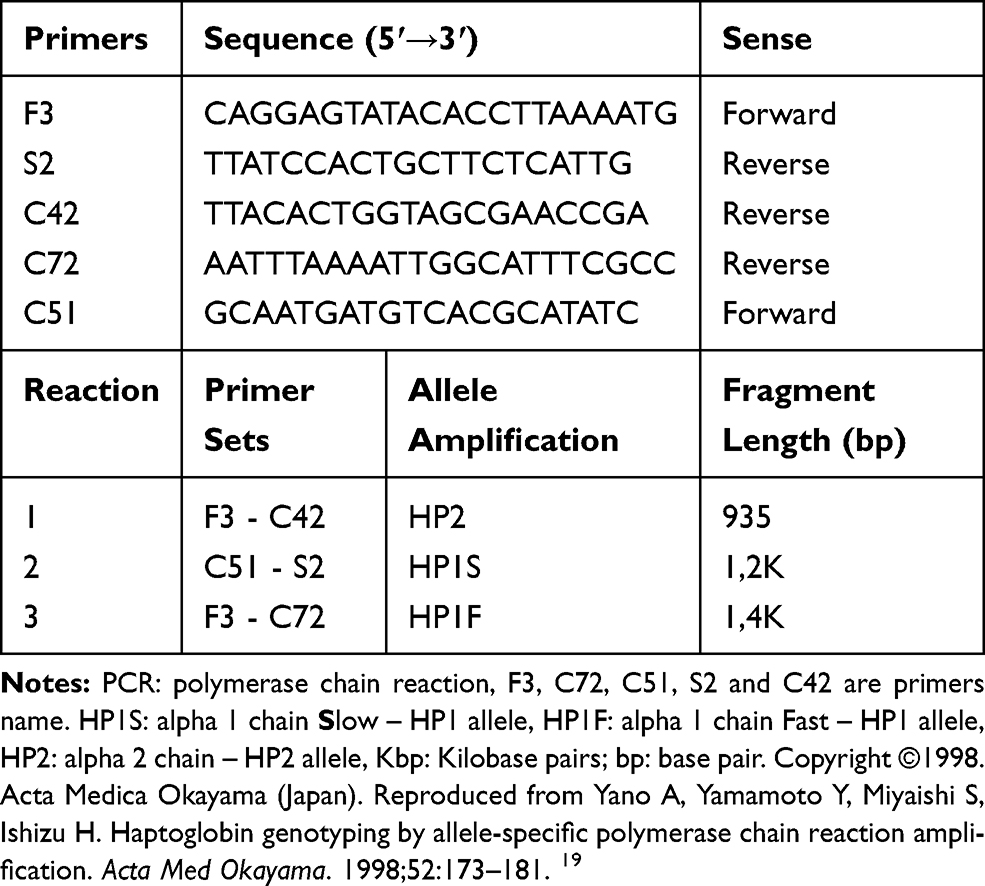 |
Table 1 Primer Sequences and Sets for Allele-Specific PCR Used to Amplify the Haptoglobin Alleles |
All samples were submitted to three reactions (1, 2 and 3 - Table 1) according to the strategy established by Yano et al19 (Figure 1), and the genotypes were defined after electrophoresis on 1.5% agarose gel and identification of the amplified fragment for each allele (Figure 2).
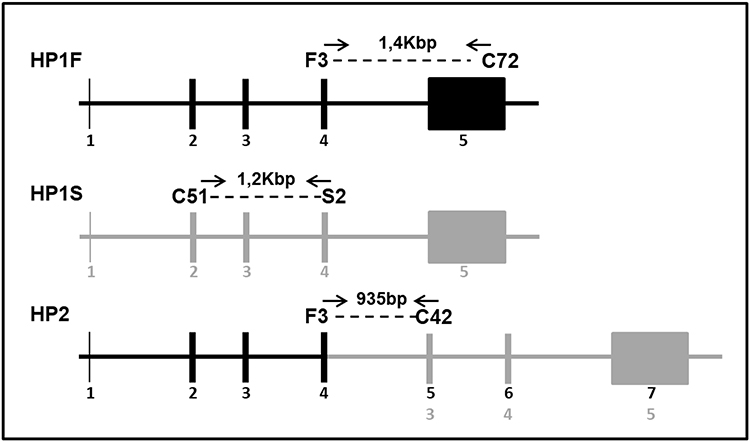 |
Figure 1 Strategy of selective amplification of the different Hp alleles by allele-specific PCR. Notes: F3, C72, C51, S2 and C42 are primers names (Table 1). HP1S: alpha 1 chain Slow – HP1 allele; HP1F: alpha 1 chain Fast – HP1 allele; HP2: alpha 2 chain – HP2 allele; Kbp: Kilobase pairs; bp: base pairs; Numbers 1 to 7: exon numbers. Copyright ©1998. Acta Medica Okayama (Japan). Adapted from Yano A, Yamamoto Y, Miyaishi S, Ishizu H. Haptoglobin genotyping by allele-specific polymerase chain reaction amplification. Acta Med Okayama. 1998;52:173–181.19 |
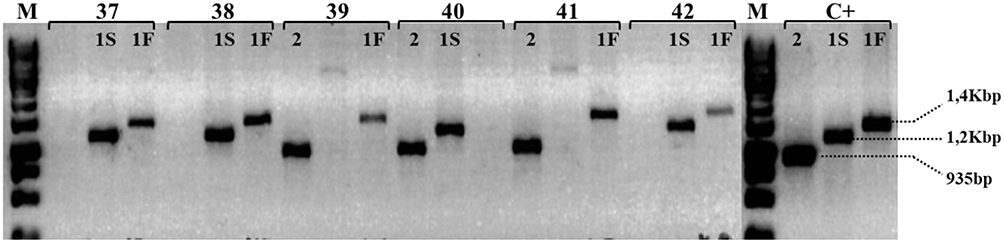 |
Figure 2 Haptoglobin genotyping by allele-specific PCR. Electrophoresis on 1.5% agarose gel demonstrating genotype identification using DNA from individuals (numbers 37 to 42) representing genotypes Hp 1S-1F, Hp 2-1F and Hp 2-1S. The allele 1F is represented by PCR product of 1,4Kbp (primers F3/C72); 1S by 1,2Kbp (primers C51/S2) and allele 2 by 935bp (primers F3/C42). M represents the size marker GeneRuller 1Kb (Thermo Scientific) and C+: are positive amplicons for each allele.Notes: F3, C72, C51, S2 and C42 are primers name (Table 1). 1S: alpha 1 chain Slow – HP1 allele; 1F: alpha 1 chain Fast – HP1 allele; 2: alpha 2 chain – HP2 allele; Kbp: Kilobase pairs; bp: base pairs. Copyright ©1998. Acta Medica Okayama (Japan). Reproduced from Yano A, Yamamoto Y, Miyaishi S, Ishizu H. Haptoglobin genotyping by allele-specific polymerase chain reaction amplification. Acta Med Okayama. 1998;52:173–181.19 |
Data Analysis
The GraphPad Prism Program, version 5 for Windows (San Diego, California, USA) was used to perform the statistical analysis. The normality of the quantitative variables was tested by the Shapiro–Wilk, Kolmogorov–Smirnov, and D`Agostino/Pearson Omnibus tests. The differences between quantitative variables among patients were assessed by using the Kruskal–Wallis analysis of variance (ANOVA) and Mann–Whitney test as appropriate, while the Chi-square or Fisher`s exact tests were used as applicable for the categorical variables. P values less than 0.05 were taken as statistically significant for all analyses.
Results
Haptoglobin genotypes determination was successful in 101 SCA patients and 64 controls (19 AS and 45 AA).
Biodata and Haptoglobin Genotypes of Participants
No sex or age distribution difference was observed between the patients and controls. Hp1-1 was the most common haptoglobin genotype and the distribution of the haptoglobin genotypes did not reveal any significant difference across the various groups of participants (Table 2). The frequencies of the haptoglobin genotypes among the patients were as follows: Hp1-1, 43 (42.6%); Hp2-1, 40 (39.6%); and Hp2-2, 18 (17.8%) and there was no significant difference between the patients and the controls (Table 2). The Hp1 and Hp2 alleles frequencies in the SCA patients were 0.62 and 0.38, respectively. The Hp1 and Hp2 alleles frequencies in the controls were 0.73 and 0.27, respectively. The distribution of both the haptoglobin genotypes and alleles did not reveal any significant difference p= 0.06 and 0.13, respectively (Table 2).
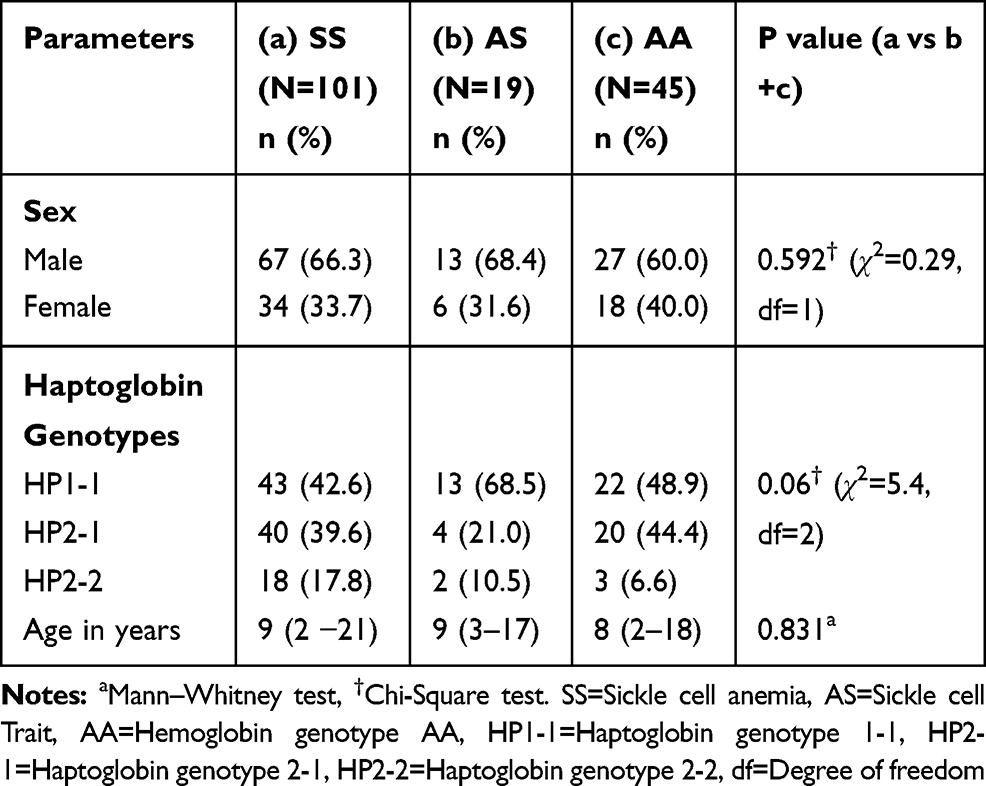 |
Table 2 Biodata and Haptoglobin Genotype Distribution Among Study Participants |
Laboratory Parameters in Patients and Controls
As expected, there were significant differences in the laboratory parameters of the patients and the controls (Supplement Table 1)
Clinical Events Among Patients
The number of VOC ie severe painful episodes (bone pain crisis) in the last 1 year prior to recruitment, ranged between 1 and 6 with a median of 2 (mean 2.5±1.3) and VOC occurred in 62 (61.4%) patients. The long-term sequalae of SCD found in the patients were: gallstone and leg ulcer each in six (5.9%) patients, respectively, while osteonecrosis, overt stroke, and priapism were found each in five (4.9%) patients, respectively.
Effect(s) of Haptoglobin Genotypes on Patients' Laboratory Parameters
The haptoglobin genotypes had no significant effect(s) on the laboratory parameters of the patients, including Hb and markers of hemolysis (Table 3).
 |
Table 3 Laboratory Parameters in SS Patients with Different Haptoglobin Genotypes |
Effects of Haptoglobin Genotypes on Patients' Clinical Events
As shown in Table 4, although patients with the HP2-2 genotype had more frequent VOC compared to the other genotypes, this did not attend statistical significance. Also, the partitioning of the haptoglobin genotypes did not differentiate the other clinical events amongst the patients.
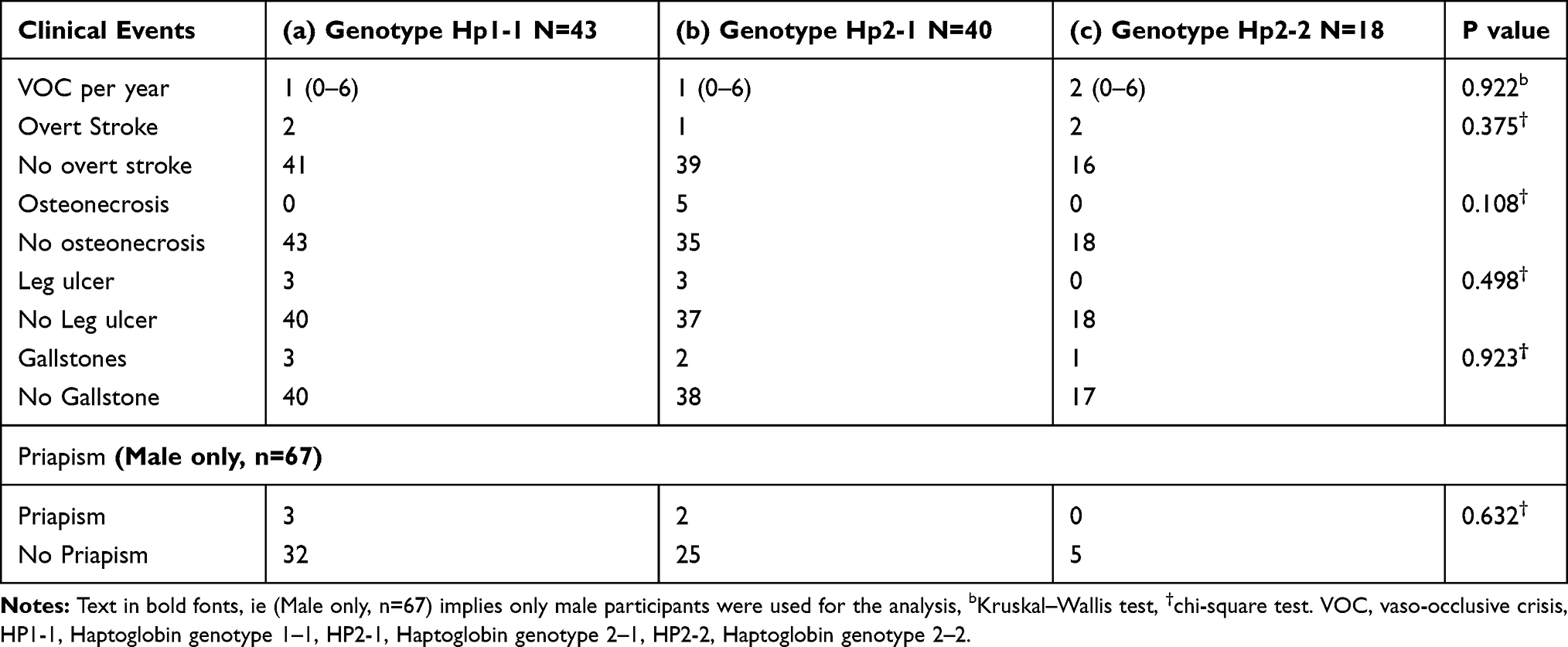 |
Table 4 Clinical Events in Patients with Different Haptoglobin Genotypes |
Discussion
The effects of some disease genetic markers could vary among different populations because of genetic variability and environmental factors. Sickle cell disease is a disorder of public health concern in the developing world1–4 and, there are very scanty literature data on the roles of genetic factors on SCA in these countries. This is especially so in Nigeria. These observations have created some gaps in understanding the effects of some genetic modifiers on the clinical phenotypes of SCA among Africans. This raises the need for more studies among many cohorts with diverse ethnic backgrounds in order to have more understanding of the impacts of genetic markers on SCA.
This study confirms that the Hp1 allele and its associated genotypes (Hp1-1 and Hp2-1) are common among the Nigerian SCA patients. Our finding conforms to the pattern observed in Northeast Brazil15 and an earlier study in Nigeria.8 On the contrary, the above pattern was scantly found in both southeast Brazil21 and Kuwait.8 While SCA patients from both southeast Brazil21 and Kuwait8 have a milder disease phenotype, their counterparts from Nigeria8 and Northeast Brazil15 have a more severe disease phenotype. Therefore, the differences in the genotype distributions across these studies8,21 and ours may reflect the genetic diversities of the patients involved with the studies as well as the roles of survival and environmental factors. The migration of people from Africa to the northern Brazil through the trans-Atlantic slave trade could account for the similarity between the haptoglobin genotypes in this study and that from northern Brazil hence, possible links between SCD in Africa and Brazil as earlier postulated.15
Though the number of patients with significant clinical events is quite low probably because the study participants were young patients who are yet to be fully exposed to some long-time sequalae/chronic complications of SCA; nonetheless, the lack of any effect of the haptoglobin genotypes on the clinical events among patients in this study contradicts the hypothesis of a possible link between haptoglobin genotypes and clinical events among SCD patients. Previous authors have hinted that haptoglobin genotypes could influence clinical and laboratory events in SCD through their ability to perturb hemolysis, oxidative and inflammatory injuries which are prominent aspects of SCA pathophysiological processes.15,16 Our findings also contradict the proposition that individuals with certain haptoglobin genotypes considered to have very weak biological activities (eg Hp2-2) are more prone to complications of certain health conditions.8–11,13,15,21,22
Several studies had postulated on the possible influence of haptoglobin genotypes on the clinical severity of medical conditions/diseases.8–11,13,15,21-23 Out of these, only the study by Adekile and Haider8 examined the relationship between haptoglobin genotypes and clinical events (occurrence of vaso-occlusive crises) among SCA patients and they found that haptoglobin genotype did not influence the occurrence of this clinical event among their Kuwaiti study cohort. However, the impact of haptoglobin genotype on the clinical events among Nigerians with SCA included in the same study8 was not examined thus making the current study the first to relate the phenotype of Nigerian SCA patients with haptoglobin genotype. Although patients with Hp2-2 genotype in this study tended to have more frequent VOC episodes, this did not attain statistical significance. Nevertheless, many other studies focused on other chronic health conditions, have associated the Hp2-2 genotype with different diseases and their complications which they linked probably, to the lower antioxidant and anti-inflammatory capacities of HP2-2 genotype.10–13,22
Similar to some earlier studies on SCD,21,24 there were no effects of the haptoglobin genotypes on the laboratory parameters of the studied patients. These observations contrast with findings by Guetta and colleagues,25 where some cytokines were associated with Hp-1 allele. Although we did not measure cytokines in the current study, nonetheless, as found in this study, the study by Pierrot-Gallo et al21 also observed that the haptoglobin gene polymorphism did not differentiate the hematological parameters of the SCA patients. However, both the current study and that by other authors21,24 did not examine plasma hemoglobin (a known marker of intravascular hemolysis)7 speculated to have possible links with haptoglobin genotypes.16 Nevertheless, in this study, we went further to examine how the haptoglobin genotype perturbs the serum lactate dehydrogenase, aspartate transaminase, and bilirubin levels (all known markers of intravascular hemolysis),7 and found no association. Our findings may suggest that other factors related to haptoglobin may probably influence the impact(s) of haptoglobin genotype on the phenotypic expression of SCA patients. Among such potential candidate factors are the CD163 scavenger receptors and heme oxygenase activities. The level and activities of CD163 scavenger receptors and heme oxygenase rather than the haptoglobin gene polymorphism may be more related to SCA manifestations and this could be explored in future studies.
Two major limitations of this study were the small sample size and the small number of various SCA phenotypes found which may probably, reflect the young age of the patients. In spite of the above limitations, the study demonstrated that HP1-1 genotype was the most common among the Nigerian SCA patients and the occurrence of haptoglobin gene polymorphism did not differ between the SCA patients and controls. In addition, it was the first study to relate the haptoglobin genotype to the clinical and laboratory manifestations among Nigerian SCA patients.
In conclusion, this study did not find a significant relationship between the haptoglobin gene polymorphism and the clinico-laboratory manifestations of young Nigerian SCA patients. This is in agreement with previous studies.8,21,24 Collectively, these findings suggest that haptoglobin gene polymorphism may not be a major genetic contributor and influencer of the clinical and laboratory expressions of SCA. We suggest more studies preferably, longitudinal and multicentred, to further expose and explore the relationship between haptoglobin genotypes and SCA manifestations.
Acknowledgments
We acknowledge with thanks, the laboratory supports provided by Prof A.G Falusi of the University of Ibadan, Southwest Nigeria. The authors also thank the participants and their parents for their supports during the study.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This study was supported by grants No 2014/00984-3 from the São Paulo Research Foundation, and grants No 2015/141693-0 from the Brazilian National Council for Scientific and Technological Development, Brazil.
Disclosure
The authors have no conflicts of interest to declare.
References
1. Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376:2018–2031. doi:10.1016/S0140-6736(10)61029-X
2. Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Eng J Med. 2017;376:1561–1573. doi:10.1056/NEJMra1510865
3. Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:e18010. doi:10.1038/nrdp/2018.10
4. Makani J, Ofori-acquah NO, Wonkman A, Ohene-frempong K. Sickle cell disease: new opportunities and challenges in Africa. Sci World J. 2013;:1–16. doi:10.1155/2013/193252
5. Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. Am J Hematol. 2012;87(8):795–803. doi:10.1002/ajh.23232
6. Nouraie M, Lee JS, Zhang Y, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica. 2013;98(3):464–472. doi:10.3324/haematol.2012.068965
7. Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127(3):750–760. doi:10.1172/JCI89741
8. Adekile AD, Haider MZ. Haptoglobin gene polymorphisms in sickle cell disease patients with different βS-globin gene haplotypes. Med Princ Pract. 2010;19:447–450. doi:10.1159/000320302
9. Wobeto VP, Zaccariotto TR, Sonati MF. Polymorphism of human haptoglobin and its clinical importance. Genet Mol Biol. 2008;31(3):602–620. doi:10.1590/S1415-47572008000400002
10. Adams JN, Cox AJ, Freedman BI, Langefeld CD, Carr JJ, Bowden DW. Genetic analysis of haptoglobin polymorphisms with cardiovascular disease and type 2 diabetes in the diabetes heart study. Cardiovasc Diabetol. 2013;12(1):31. doi:10.1186/1475-2840-12-31
11. Adinortey MB, Gyan BA, Adjimani JP, et al. Haptoglobin Polymorphism and association with complications in Ghanaian Type2 diabetes patients. Ind J Clin Biochem. 2011;26(4):366–372. doi:10.1007/s12291-011-0141-3
12. Boettger LM, Salem RM, Handsaker RE, et al. Recurring exon deletions in the Haptoglobin gene associated with lower blood cholesterol levels. Nat Genet. 2016;48(4):359–366. doi:10.1038/ng.3510
13. Langlois MR, Delanghe JR. Biological and clinical significance of haptoglobin in humans. Clin Chem. 1996;42(10):1589–1600. doi:10.1093/clinchem/42.10.1589
14. Chintagari NR, Nguyen J, Belcher JD, Vercellotti GM, Alayash AI. Haptoglobin attenuates haemoglobin-induced heme oxygenase-1in renal proximal tubular cells and kidneys of a mouse model of sickle cell disease. Blood Cells Mol Dis. 2015;54(3):302–306. doi:10.1016/j.bcmd.2014.12.001
15. Santos MN, Bezerra MA, Dormigues BL, Zachariotto TR, Oliveira DM. Haptoglobin genotypes in sickle cell disease. Genet Test Mol Biomarkers. 2011;15(10):709–713. doi:10.1089/gtmb.2010.0235
16. Santos MN. Haptoglobin: an emerging candidate for phenotypic modulation of sickle cell anemia? Rev Bras Hematol Hemoter. 2015;37(6):361–363. doi:10.1016/j.bjhh.2015.08.009
17. Miranda SRP, Fonseca SF, Figueiredo MS, et al. Hb Köln [α2β298(FG5) val-met] identified by DNA analysis in a Brazilian family. Braz J Genet. 1997;20(4):745–748. doi:10.1590/S0100-84551997000400030
18. Ballas SK, Lieff S, Benjamin LJ, Dampier CD, Heeney MM, Hope C. Definitions of phenotypic manifestations of sickle cell disease. Am J Hematol. 2010;85:6–13. doi:10.1002/ajh.21550
19. Yano A, Yamamoto Y, Miyaishi S, Ishizu H. Haptoglobin genotyping by allele-specific polymerase chain reaction amplification. Acta Med Okayama. 1998;52:173–181. doi:10.18926/AMO/31301
20. Koch W, Latz W, Eichinger M, et al. Genotyping of the common haptoglobin Hp1/2 polymorphism based PCR. Clin Chem. 2002;48:1377–1382. doi:10.1093/clinchem/48.9.1377
21. Pierrot-Gallo BS, Vicari P, Matsuda SS, Adegoke SA, Mecabo G, Figueiredo MS. Haptoglobin gene polymorphisms and interleukins 6 and 8 in patients with sickle cell anaemia. Rev Bras Hematol Hemoter. 2015;37(5):329–335. doi:10.1016/j.bjhh.2015.07.006
22. Ijas P, Melkas S, Saksi J, et al. Haptoglobin Hp2 variant promotes premature cardiovascular death in stroke survivors. Stroke. 2017;48(6):1463–1469. doi:10.1161/Strokeaha.116.015683
23. Moreira HW, Naoun PC. Serum haptoglobin types in patients with hemoglobinopathies. Hereditas. 1990;113:227–331. doi:10.1111/j.1601-5223.1990.tb00088.x
24. Bora K, Natarajan K, Kutlar F, et al. In vitro exploratory studies of haptoglobin polymorphism and their effect on cytokine release from cultured mononuclear cells in sickle disease. Blood. 2008;112:1441. doi:10.1182/blood-2008-03-140830
25. Guetta J, Strauss M, Levy NS, Fahoum L, Levy AP. Haptoglobin genotype modulates the balance of Th1/Th2 cytokines produced by macrophages exposed to free hemoglobin. Atherosclerosis. 2007;191(1):48–53. doi:10.1016/j.atherosclerosis.2006.04.032
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.