Back to Journals » ImmunoTargets and Therapy » Volume 3
Hairy cell leukemia – immunotargets and therapies
Authors Basheer F, Bloxham D, Scott M, Follows G
Received 20 November 2013
Accepted for publication 22 April 2014
Published 24 June 2014 Volume 2014:3 Pages 107—120
DOI https://doi.org/10.2147/ITT.S31425
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Faisal Basheer, David M Bloxham, Mike A Scott, George A Follows
Department of Haematology, Addenbrookes Hospital, University of Cambridge, Cambridge, UK
Abstract: Hairy cell leukemia (HCL) is an indolent low-grade B-cell lymphoproliferative disorder that is reasonably sensitive to standard first-line purine analog therapy. However, in many cases, repeat relapses occur, requiring multiple courses of purine analog therapy, promoting eventual drug resistance. This, coupled with the concerning side effects of repeated purine analog exposure, has prompted the search for alternative targets and therapies that may provide deeper remissions. Novel strategies employing immune-mediated targeting via monoclonal antibody therapies and recombinant immunotoxins appear promising in HCL and are currently under investigation. More recently, the concept of targeted kinase inhibition using small-molecule inhibitors in HCL has emerged as another potentially viable option. As a deeper understanding of the aberrant molecular pathways contributing to the pathogenesis of HCL develops, the landscape of management for HCL, particularly in the relapse setting, may change significantly in the future as a result of these promising immunotargets and therapies.
Keywords: hairy cell leukemia, immunotargets, therapies
Introduction
Hairy cell leukemia (HCL) is a low-grade, chronic B-cell lymphoproliferative disorder accounting for approximately 2% of all leukemias. It comprises 8% of all cases of mature T-cell and B-cell lymphoproliferative diseases.1 The disease is more common in Caucasians and is particularly frequent in male Ashkenazi Jews, with an overall male to female ratio of approximately 4:1 and a median age of 50 years at presentation. There are approximately 1,600 new cases per year in Europe.2,3 HCL was first described in 1958 by Bouroncle et al as a “leukemic reticuloendotheliosis” due to its presentation with pancytopenia, splenomegaly, and malignant “histiocytic” or “reticulum” cells noted on morphology.4 Later, in 1966, Schrek and Donnelly coined the term “hairy cell leukemia” when describing the irregular cytoplasmic projections of the abnormal cells circulating in the blood or originating in the bone marrow.5 Initially, the treatment options were limited to splenectomy and low-dose cytotoxic chemotherapy, with median survivals of 3–4 years. This was significantly improved upon by the advent of interferon (IFN) therapy in 1984, and later enhanced further with the arrival of purine analog therapy, allowing overall 10-year survival rates in excess of 90%. These agents have been the mainstay of therapy for the past three decades and, for the most part, provide good remissions lasting several years. In recent years, however, new targets have been identified, resulting in the development of innovative immunotherapies and targeted kinase inhibitors. This review focuses principally upon these targets and therapies.
Clinical presentation and laboratory diagnosis
Clinical presentation
Patients are commonly asymptomatic at diagnosis and the disease is usually an incidental finding, identified from a full blood count taken for other reasons. However, HCL may present with clinical features of cytopenia, such as recurrent infections or bleeding. Other clinical features include fatigue, weight loss, and right upper quadrant tenderness/abdominal bloating due to splenomegaly. Abdominal lymphadenopathy at presentation, although infrequent, may be associated with a poorer response to treatment and overall outcome.6
Laboratory diagnosis
The World Health Organization recognizes classical HCL and HCL variant (HCL-v). The most common laboratory finding in classical HCL is a cytopenia affecting up to all three lineages, with monocytopenia being a recurrent feature. With HCL-v, a lymphocytosis is more prominent. Hairy cells, typically twice as large as normal lymphocytes and possessing round to oval-shaped nuclei with an open chromatin pattern, are regularly seen in the peripheral blood smear (Figure 1). Separating classical HCL from HCL-v based on morphology can be challenging. Bone marrow aspirate is generally unsuccessful as aspiration is typically dry. Trephine biopsy regularly demonstrates either a diffusely interstitial or focal infiltration of cells with a “fried egg” appearance due to a shrinkage artifact. Background reticulin fibers are also notably increased in association with HCL.
 | Figure 1 Classical hairy cell lymphocyte (arrow) seen in peripheral blood. |
Immunophenotyping of blood or bone marrow aspirate is the mainstay for confirming the diagnosis, with classical hairy cells expressing common B-cell antigens, ie, CD19, CD20, and CD22. The classic immunophenotypic profile consists of bright coexpression of CD11c with these antigens and additional expression of CD25, CD103, and CD123. There is also bright monotypic surface immunoglobulin expression. Immunohistochemistry of the bone marrow biopsy on paraffin sections demonstrates CD20, DBA.44 (which reacts with the antigen CD72), and tartrate-resistant acid phosphatase positivity. HCL is usually cyclin D1-positive, and this can be helpful in assessing residual disease. Coexpression of tartrate-resistant acid phosphatase and DBA.44 has 100% sensitivity for HCL and a high specificity for the disease. Annexin A1 positivity on trephine biopsy is also highly specific for HCL, not being expressed in any other B-cell lymphoproliferative disorders, although it is widely recognized as not being useful for detecting minimal residual disease (MRD) due to its expression on myeloid cells and some T-cells.1 Rarely, the diagnosis of HCL is made on histologic analysis of tissue post splenectomy or via needle core biopsy. This demonstrates an almost exclusive infiltration of the red pulp with an atrophic white pulp. The liver may also demonstrate infiltration in a sinusoidal pattern.
BRAF V600E mutation
V600E mutation of the BRAF gene has recently been identified as an almost universal finding in classical HCL.7 The BRAF gene encodes a protein belonging to the RAF/MIL family of serine/threonine protein kinases and regulates the mitogen-activated protein kinase signaling pathway, which is important for cell division, differentiation, and secretion. BRAF is activated by RAS and subsequently activates ERK via MEK phosphorylation, thereby contributing to increased cell proliferation. An amino acid substitution of glutamic acid for valine at position 600 of the BRAF protein (BRAF V600E) affecting the kinase domain of BRAF constitutes this activating mutation and is the commonest BRAF mutation seen in melanoma and other cancers, including colorectal cancer, thyroid cancer, and non-small-cell lung carcinoma. Employing polymerase chain reaction and direct DNA Sanger sequencing, Tiacci et al7 demonstrated the BRAF V600E mutation in 48 of 48 cases of HCL and its absence in 195 other mature B-cell neoplasms. We have also corroborated this finding with a high-resolution melting analysis assay that detects BRAF exon 15 mutations in HCL samples containing as few as 5%–10% hairy cells.8 All 48 patients demonstrated the BRAF V600E mutation identified by high-resolution melting analysis, and this was verified by sequencing the polymerase chain reaction product and additional confirmation of HCL by independent pathology review. Within the same study, 114 non-HCL malignancies tested using the same method all demonstrated negativity for BRAF V600E. Other studies have also employed high-resolution melting analysis, targeted pyrosequencing, and allele-specific polymerase chain reaction techniques to demonstrate a near 100% sensitivity for BRAF V600E in HCL, with a few rare exceptions.9 Demonstration of BRAF V600E is rapidly becoming standard practice to complete the diagnostic evaluation of classical HCL. Its discovery has also informed the development of further therapies for HCL, which are discussed later in this review.
Hairy cell variant
This separate World Health Organization classification entity comprises approximately 10% of cases of HCL and is typically seen as an aggressive, poorer-prognosis variant that is more resistant to conventional treatments.1 It has features similar to splenic lymphoma as reflected in the World Health Organization classification, and presents with an elevated white blood cell count due to lymphocytosis. Morphology demonstrates unevenly distributed villous projections on large lymphocytes with distinct nucleoli and round nuclei (Figure 2). Other features include pronounced splenomegaly and lack of cytopenias, including an absence of monocytopenia. Bone marrow aspiration is typically easier than with HCL due to lower reticulin marrow content, and the histology of both marrow and spleen is similar to HCL. Immunophenotypically, however, HCL-v characteristically differs due to absence of CD25 and infrequent expression of CD103, while CD11c, CD20 and CD22 usually remain positive with persisting light chain restriction. Additionally, annexin A1 has been reported as negative in 100% of cases in one study.10 BRAF V600E has not been reported as positive in any HCL-v cases thus far.9
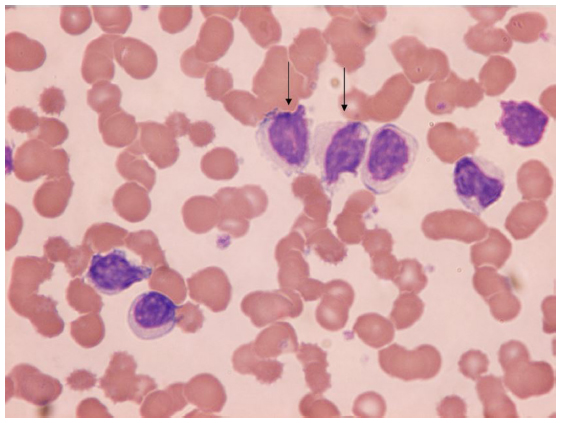 | Figure 2 Atypical hairy cell lymphocytes (arrows) seen in peripheral blood. |
Conventional treatment with splenectomy has been shown to provide a good hematologic and clinical response (74% of 19 patients) in HCL-v, eliciting a median response duration of 4 years in one study.11 However, other studies have failed to replicate this, with only some attaining a partial response and none achieving a complete response.12 Importantly, the condition is poorly sensitive to purine analog therapy, with only partial responses in approximately 50% of patients and complete response rates of <10%.10 Similar poor responses are seen with IFN therapy in HCL-v.10,11 Given its poorer outcomes to conventional therapies, the identification of newer immunologic targets in HCL is also likely to be important for HCL-v.
There is a recognized subset of HCLv associated with immunoglobulin VH chain VH4-34 rearrangements that has a poorer prognosis. These patients typically have lower response rates, poorer progression-free survival, and a shorter overall survival after diagnosis.13 Additionally, this subset has been reported to be negative for the BRAF V600E mutation, suggesting an alternative pathogenesis to that of conventional HCL.14
Conventional therapies for HCL
In general, the majority of patients do not require therapy immediately at presentation and the main indications for treatment are symptomatic disease with fatigue interfering with activities of daily living, symptomatic splenomegaly, or progressive cytopenia that can become symptomatic with bone marrow failure. The latter in particular requires careful monitoring, given that commencing treatment with severe cytopenia may present additional clinical complications.
Interferon
Interferon therapy for HCL was first reported in 1984, with 30% complete remission and 56% partial remission rates.15 Subsequent studies could not demonstrate better responses and highlighted early relapse on discontinuing therapy. As alluded to above, IFN therapy has now largely been superseded given the excellent results of purine analog therapy.
Purine analogs
The adenosine deaminase inhibitor 2-deoxycoformicin (pentostatin) was developed in the 1970s after it was confirmed that adenosine deaminase was required for proliferation of lymphocytes. Hairy cells contain low levels of adenosine deaminase, so pentostatin administered biweekly for 3–6 months in patients with HCL was trialed to test the effect of its inhibition. This produced complete response rates of >70% and significant 10-year disease-free survival rates. A randomized trial versus IFN demonstrated significantly superior outcomes (complete response 76% versus 11%; partial response 79% versus 38%).16
Subsequently, cladribine (2-chloro-2′deoxyadenosine) was developed as another purine analog. Cladribine accumulates selectively as a 5′-triphosphate derivative and contributes to the death of cells rich in deoxycytidine kinase (an enzyme found at high levels in hairy cells) by producing a profound imbalance in deoxyribonucleotide pools and preventing DNA repair. Cladribine causes DNA strand breaks and inhibits DNA synthesis. Given as a 5–7 day infusion, this initial study also showed excellent responses with 11/12 patients treated achieving a complete remission following cladribine therapy, with no relapses observed within the relatively short follow-up documented in this first report.18 The drug was also better tolerated with regard to toxicity. A better side effect profile coupled with ease of administration as a single cycle over 5–7 days has allowed cladribine to become more commonly used as upfront therapy. However, the less frequent dosing schedule of pentostatin may be a more appropriate therapeutic option for patients presenting with severe cytopenia. A randomized multicenter trial in untreated HCL patients demonstrated similar complete remission rates, overall response rates, and overall survival between standard 5-day cladribine and a weekly schedule, with no significant difference in toxicity.19
Longer-term follow-up of patients treated with pentostatin and cladribine was subsequently documented. A large retrospective study of 233 patients treated with either agent demonstrated an overall complete response rate of 80% with a median relapse-free survival of 16 years.6 Patients with refractory disease or relapse received a further course of either drug, with approximately 70% achieving a second complete response. Median relapse-free survival in these two groups was approximately 11 years. A small subgroup in this study required a further third treatment following second relapse, and 50% of these patients attained complete response with a median relapse-free survival of 6.5 years. The risk of relapse in patients still in complete response at 5 years was only 25% by 15 years. This study highlights the decline in time to relapse with subsequent courses of chemotherapy, and although relapse-free durations are lengthy in terms of years, a younger patient at presentation may easily require multiple (more than three) courses of treatment simply to keep the disease in remission during their lifespan.
Another extended follow-up of patients with HCL having received a single first course of cladribine alone showed a 95% complete response rate and 5% partial response rate (n=207).20 The median response duration for all responders in this study was around 8 years. However, 37% percent of patients experienced relapse after their first course of cladribine.
There have been no randomized controlled studies comparing the efficacy of cladribine and pentostatin. However, a retrospective study by Dearden et al examined a group of 210 patients treated with either purine analog.21 An 82% complete response rate and a 15% partial response rate were achieved in the pentostatin-treated group, with a similar 84% complete response rate and a 16% partial response rate in the cladribine-treated group. At a median of 71 months and 45 months for pentostatin and cladribine, relapse rates were 24% and 29%, respectively. The majority of relapsed patients achieved second remissions after further courses of either agent. No evidence of cross-resistance was seen. The 5-year survival rate was 97% for all patients.
Although these data strongly suggest a role for purine analogs in the management of HCL, it is clear that many patients require repeat courses, potentially over decades, and progressive shortening of response duration tends to be the rule. A further significant issue is that patients are potentially left with considerably reduced cellular-based immunity following purine analog therapy. Studies have shown that recovery can take between 9 and 12 months after treatment with initial courses of either drug.22,23 Indeed, CD4 cell recovery has been shown to vary from 3.5 years to just under 5 years after single courses of either purine analog.24 The risk of succumbing to opportunistic systemic infections therefore is increased. Such immunosuppression also theoretically increases the risk of additional malignancies. Of 209 HCL patients followed up by Goodman et al, when treated with an initial course of cladribine, 47 were noted to develop 58 other malignancies, with a doubling in the observed-to-expected ratio.20 Given that other studies have also demonstrated similar figures,24 there remains some controversy over whether these drugs have an oncogenic effect in HCL. Additionally, purine analog therapy has been associated with a 15% neurologic toxicity rate on both agents, ranging from more common adverse effects such as headaches and somnolence to severe irreversible motor weakness of the upper/lower extremities.25
Despite their success in treating HCL, these considerations detract from the utilization of purine analog therapies and have inspired a search for alternative targets and therapies that may improve on responses whilst avoiding such inconveniences.
Novel immunotherapies and targeted kinase inhibition
In the search for more effective therapeutic options to purine analogs, three major alternative strategies have been developed to tackle HCL over recent years, particularly when the disease is refractory or multiply-relapsed. The first strategy uses monoclonal antibodies to target HCL cell surface markers and directly induce cell killing via antibody-dependent cell-mediated cytotoxicity. The second approach uses monoclonal antibodies to HCL cell surface markers to deliver toxins into the cell. The most recent approach uses small molecule inhibitors to target kinases key to B-cell signaling. A summary of these therapies is provided in Table 1.
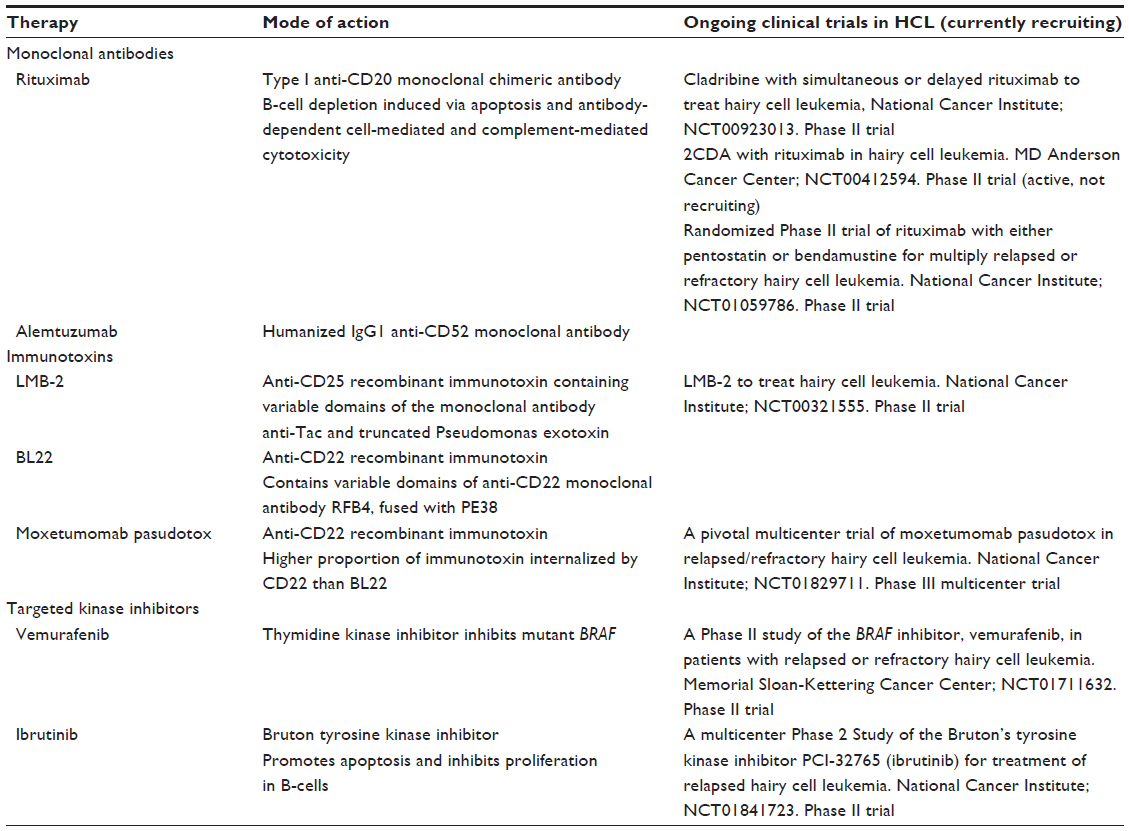 | Table 1 Novel targeted therapies in development for hairy cell leukemia (HCL) |
Monoclonal antibody therapy
The majority of early therapeutic monoclonal antibodies were of rodent origin, resulting in a largely unfavorable side effect profile when attempts were made to apply them clinically in humans. The major issue was the immunogenicity of the nonhuman framework, which would frequently result in strong antirodent antibody responses after only a few treatments. This increased the risk of infusion reactions, toxicity, and diminished therapeutic efficacy considerably. Additionally, unfavorable pharmacokinetics with shortened half-lives and reduced Fc receptor portion binding led to their modification towards a more human-tolerant form using recombinant DNA technology. Chimeric antibodies were developed, which had undergone substitution of up to 70% of the nonhuman sequences within the framework. In efforts to improve this further, “humanized” antibodies were generated, with over 90% of nonhuman sequences having been replaced, being left with only the original nonhuman complementarity determining regions. A further advance allowed the generation of monoclonal antibodies with fully human sequences displaying a high affinity for antigen and minimal immunogenicity. Current efforts are focusing on re-engineering the effector function of such antibodies by switching the Fc subtype to facilitate better complement activation of binding to macrophages and improve cytotoxicity.
As a strategy to treat hematologic malignancies, the efficacy of monoclonal antibody therapies depends largely upon the characteristics of the target antigen. The ideal target antigen would demonstrate expression exclusively on malignant cells. If this antigen expression were to include normal cells, targeting and potential depletion of these cells should not result in serious complications, such as significant immunosuppression or life-threatening cytopenias. Additionally, the antigen would be expressed at a sufficiently high density on the surface of the malignant cells in order to provide the antibody with a sufficient number of binding sites. Finally, exposure to the antigen would not cause downregulation or internalization of antigen expression on the cell surface when exposed to an antibody that relies solely on cell-mediated toxic mechanisms to function appropriately.
Rituximab
CD20 expression is exclusive to mature healthy and malignant B-cells. CD20 is a B-cell-specific differentiating antigen that has been shown to regulate the intracellular calcium influx and mediate B-cell proliferation and activation.26 Amongst all B-cell malignancies, it is most densely expressed on hairy cells.27 Accordingly, there has been much interest in using this as an immunologic target for treating HCL.
Rituximab is a type I anti-CD20 monoclonal chimeric antibody consisting of a human kappa constant region, a human IgG Fc portion, and a murine-derived complementarity determining region. Upon exposure to CD20, B-cell depletion is induced via antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, and a direct antitumor effect employing the induction of internal signaling events within malignant cells and causing suppression of proliferation and apoptosis.28 In antibody-dependent cell-mediated cytotoxicity, granulocytes, macrophages, and natural killer cells bind to the Fc portion of rituximab via their Cfu receptors, subsequently releasing effector molecules such as perforin that cause direct cell death. Complement-dependent cytotoxicity results from complement C1 activation due to binding to the Fc portion of rituximab and results in direct cell lysis via the complement cascade. As a type I anti-CD20 antibody, rituximab functions efficiently by grouping membrane CD20 molecules upon binding and allowing increased clustering, thereby facilitating attachment of C1q and causing antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity. Its mechanism of B-cell depletion is likely to vary by disease.
The typical side effect profile stems from infusion-related reactions, such as pyrexia, rigors, flushing, pruritus, bronchospasm, urticaria, and tumor-related pain. The risk of infection appears to be higher with more intense rituximab regimens. Viral infections, particularly reactivation of hepatitis B virus and cytomegalovirus, are reported reasonably often with rituximab. There is also a recognized but very low risk of developing progressive multifocal leukoencephalopathy with intensive rituximab regimens caused by reactivation of latent John Cunningham virus.29
Rituximab administered with chemotherapy has been shown to significantly improve event-free survival and overall survival in patients with diffuse large B-cell lymphoma,30 and has subsequently become the standard of care in a number of patients with indolent CD20+ B-cell lymphoma. It has been shown to improve progression-free survival and overall survival in these groups, and is also now employed regularly in maintenance regimens as well as at induction.31
Rituximab in combination with purine analogs
In an initial study of eleven patients with HCL, an overall response rate of 64%, including six complete responses and one partial response, was reported when these patients were treated with rituximab (three upfront at diagnosis and eight upon relapse after cladribine).32 The median response duration was 14 months. A subsequent study in 15 patients (refractory to or relapsing after IFN/purine analogs) demonstrated an overall response rate of 80%, with eight patients achieving complete response.33 A median remission duration of 32 months was reported for the responders. Nieva et al performed a Phase II trial in 24 patients relapsing after cladribine.34 The median remission duration in this cohort was 73 months. One third of the patients in that study had received two previous courses of cladribine. Twelve percent of these patients achieved a complete response, 12% had a partial response, and of these, two relapsed at 15 months. All three studies used a typical 4–8-weekly regimen of rituximab 375 mg/m2 given intravenously.
Given that rituximab is used concurrently with chemotherapy in the management of other B-cell malignancies, its use in HCL was investigated retrospectively by Else et al,35 whose results suggested an added benefit of rituximab when compared with single purine analog therapy. Eighteen patients previously treated with 1–6 courses of either purine analog were retreated concurrently with a purine analog followed immediately by rituximab. All patients demonstrated an overall response, with 89% achieving a complete response. Impressively, at a median follow-up of 3 years, all of these patients maintained their complete response status. One partial responder developed recurrent disease at 10 months. In 13 patients in whom MRD status was assessed, all were negative. The responses achieved in this study were considerably improved when compared with purine analogs previously given without rituximab. Current national guidelines in the UK36 support the combination of purine analog therapy with concurrent rituximab at relapse based largely upon these data.
Ravandi et al prospectively treated 36 patients at presentation (31 with HCL and five with HCL-v) with a standard 5-day course of cladribine followed by 8 weeks of rituximab weekly.37 All patients attained a complete response, with only one HCL-v patient relapsing, who later succumbed to the disease. Two other HCL-v patients who achieved a complete response died from unrelated malignancies. For the remainder of the patients, the median complete response duration was not reached during follow-up (range 1–63 months), with a very low rate of adverse events directly related to treatment. These data suggested strongly that rituximab may have a role in improving the complete response rate of cladribine when given in combination, but further work was required to determine if the cost of treatment with additional rituximab was justified for all patients with HCL or only in those with higher-risk disease. In order to address this, the National Institutes of Health are currently running a trial (ClinicalTrials.gov identifier NCT00923013) randomizing de novo or first-relapse HCL patients to cladribine or concurrent rituximab and cladribine with the aim of comparing responses and MRD-free survival. This trial hopes to determine whether addition of rituximab is justified as part of first-line or second-line therapy, and whether patients achieving MRD-negative remission without rituximab fare as well as those achieving MRD-negative status with rituximab.
Rituximab and fludarabine
Fludarabine is a deoxyadenosine derivative that inhibits DNA and protein synthesis, thereby affecting cell replication and growth and leading to apoptosis. It is currently used in a variety of hematologic malignancies, both myeloid and lymphoid, with its use in the nearest homologue to HCL being chronic lymphocytic leukemia. It can be considered as a potentially attractive alternative due to its ease of oral bioavailability, well documented toxicity spectrum, and the ability to deliver multiple courses. Its first use in HCL as monotherapy was documented by two case reports in 1991,38,39 in which it produced a partial response in three patients with HCL. Gerrie et al took this further by assessing the combination of rituximab and fludarabine in a group of 15 patients with multiply relapsed or first-line refractory HCL, and demonstrated that 14 of 15 patients were progression-free at a median follow-up of 35 months, with 5-year progression-free survival and overall survival rates of 89% and 83%, respectively.40 These results suggest a role for rituximab in combination with fludarabine when managing multiply relapsed patients in whom concerns about further courses of cladribine/pentostatin are present due to a high likelihood of refractoriness.
Rituximab in HCL-v
Rituximab has also demonstrated some effects in patients with HCL-v, as documented by several case reports. A complete response of over 19 months was achieved in one patient with an 8-year history of refractory HCL-v following a 4-week course of weekly rituximab (375 mg/m2).41 Additionally, another patient with HCL-v whose previous treatments had resulted in bone marrow aplasia achieved a complete response after treatment with rituximab.42 Finally, a patient with advanced stage HCL-v was treated similarly with complete hematologic recovery documented.43 Although the data are very limited, these reports suggest rituximab may also play a role in the management of HCL-v, particularly when the disease is repeatedly refractory to conventional therapy.
Alemtuzumab
CD52 is a cell surface glycoprotein of unknown function expressed by B-cell and T-cell lymphocytes, natural killer cells, and monocytic cells. Initially, rodent-derived monoclonal antibodies against CD52 were “humanized” by transfer of antigen-specific complementarity determining regions to a human framework in the 1980s. Of these, the most successful in clinical applications is alemtuzumab (CAMPATH 1H). This is a humanized IgG1 anti-CD52 monoclonal antibody active in lymphoid malignancies, and exerts its effect by binding to the cell membranes of normal and malignant lymphocytes. It has been used in the treatment of T-cell and low-grade B lymphomas. Like rituximab, it is associated with infusion reactions, fevers, rashes, and chills. Severe T-cell depletion and lymphopenia with increased risk of opportunistic infections, such as reactivation of pneumocystis and cytomegalovirus, are also associated with alemtuzumab.
Interest in using CD52 as a potential immunologic target in HCL stemmed from the demonstration of CD52 expression on clonal hairy cells at levels of 92%–100% by flow cytometry in a series of nine HCL cases and one HCL-v case.44 CD52 staining on hairy cells was reported to be approximately equal in intensity to that of normal T-cells. This consistent expression across patients with HCL was corroborated in other studies of CD52 expression.45
However, success in achieving a complete response with alemtuzumab therapy for HCL has been modest, with only limited case reports demonstrating any efficacy.46 There are better results in treating patients with HCL-v, with a couple of case reports describing its use. The first case was a 72-year-old male with HCL-v and splenomegaly refractory to cladribine, pentostatin, IFN, and rituximab.47 He was treated with alemtuzumab following splenic irradiation. This resulted in a complete response coupled with complete hematologic recovery and resolution of splenomegaly. An in vitro assay was also done as part of this study and demonstrated a direct cytotoxic effect of alemtuzumab against the patient’s hairy cells in the presence of complement. The second case was of a 58-year-old male with HCL-v and p53 mutation, in whom a complete hematologic remission was induced following 8 weeks of treatment with alemtuzumab.48
These limited data suggest a possible role for alemtuzumab in HCL and its variant, particularly when refractory to other modes of therapy; however, more studies are clearly required to delineate this further.
Immunotoxins
An immunotoxin comprises a monoclonal antibody or antibody fragment that is attached to a cellular protein toxin. This is done by joining the Fv portion of the antibody to the binding-domain of the toxin. The toxin is then directed to the target cell by means of the antibody specificity and receptor binding on the cell surface. Once bound, the immunotoxin-receptor complex is internalized or downregulated. Intracellularly, the toxin is released and causes cell death via mechanisms dependent on the type of toxin, although largely through inhibition of protein synthesis and apoptosis.
Immunotoxins may have a role in targeting HCL for patients in whom conventional therapies and rituximab produce limited responses or treatment failure. Rituximab-treated patients with HCL, in particular, can experience this due to the varying levels of underlying immunosuppression as a result of the disease leading to impaired antibody-dependent cell-mediated cytotoxicity or complement-dependent cytotoxicity.
Sources of the toxin component for a recombinant immunotoxin include fungi, bacteria, and plants. In particular, Pseudomonas exotoxin A and diphtheria toxin have been identified as molecules that effectively cause cell death even when only present intracellularly in very few numbers. These have been shown to inactivate elongation factor 2.49,50 Very low numbers of toxins are required within the target cell to induce cell death. An ideal therapeutic immunotoxin would require high specificity for the target of choice and marked potency once delivered there. As a result, three different immunotoxins, each utilizing a truncated form of Pseudomonas exotoxin A (PE38), have been developed over the past two decades, and have been or are currently being trialed in the treatment of classical HCL.
Targeting CD25 with LMB-2
The CD25 antigen is the α-chain of the interleukin-2 receptor and is a type I transmembrane protein. It is expressed in most B-cell malignancies and is part of the characteristic diagnostic panel for HCL. Using this as a potential target, LMB-2 was developed. This is a single-chain anti-CD25 Fv portion of antibody fused to PE38, originally developed in the 1990s. It achieved one complete response and three partial responses in a cohort of four patients with classical HCL in a Phase I trial at the National Cancer Institute,51 additionally obtaining responses in patients with Hodgkin’s disease, chronic lymphocytic leukemia, or certain types of T-cell lymphoma. Commonly reported adverse events were fever, elevated transaminases, and hypoalbuminemia. The single patient entering complete remission currently remains disease-free. However, LMB-2 has since been superseded by immunotoxins against CD22, due to approximately one tenth of HCL patients lacking CD25 positivity.
The role of LMB-2 in patients following relapse after purine analog therapy (or those unable to receive anti-CD22 immunotoxin treatment as part of a different trial) is now being assessed in a Phase II clinical trial52 (ClinicalTrials.gov identifier NCT00321555), providing adults entering are hairy cell CD25-positive.
Targeting CD22 with BL22
Attention has subsequently shifted to targeting CD22 in HCL, with the realization that not all HCL patients demonstrate CD25 tumor cell positivity. CD22 is a member of the immunoglobulin superfamily, ubiquitous on B-cells, and functions as an inhibitory receptor during B-cell signaling. Several studies have demonstrated its near uniform presence in cases of classical HCL.53–55 CD22 undergoes rapid endocytosis upon antibody binding,56 thus making it an attractive option for intracellular delivery of a toxin bound to an antibody. It is also expressed strongly in follicular, mantle, and marginal zone B-cells.
BL22 is the Fv domain of an anti-CD22 antibody conjugated to PE38. After demonstrating a degree of suppression of CD22-positive tumor xenograft growth, it was tested in a Phase I trial on a subset of 31 patients with HCL refractory to prior standard treatment.57 It was also given to eleven patients with chronic lymphocytic leukemia and four with CD22-positive non-Hodgkin’s lymphoma. Responses were seen in 25 of the HCL patients, with complete remissions observed in 61%.57 Although primary chronic lymphocytic leukemia and non-Hodgkin’s lymphoma cells were found to be sensitive ex vivo, this sensitivity did not translate well in the group of patients with non-Hodgkin’s lymphoma in the trial. Significant toxicities noted in a few patients were a dose-limiting cytokine-release syndrome related to a secondary immune response to BL22 and a marked hemolytic-uremic syndrome that responded completely to plasmapheresis. Other reversible biochemical changes including hypoalbuminemia and elevated transaminases were documented, although not considered dose-limiting. However, around 25% of patients developed immunotoxin-neutralizing antibodies, which had a variable effect on the tumor response.
A further Phase II trial, limiting BL22 to one cycle in an attempt to avoid the complication of hemolytic-uremic syndrome, was performed, with 25% of 36 patients achieving a complete response and 25% achieving a partial response.58 This improved further to a 47% complete response rate and a 25% partial response rate with a further cycle of therapy (reserved only for those who had not met criteria for complete response after the first cycle). However, a further three cases of hemolytic-uremic syndrome were noted. At a follow-up of nearly 7 years, the median time to relapse of cytopenia has not been reached in this cohort of patients, suggesting a potential use in refractory or relapsed HCL.58 However, concerns regarding the risk of hemolytic-uremic syndrome have remained. In an attempt to improve further upon the activity of BL22 in non-Hodgkin’s lymphoma, including chronic lymphocytic leukemia, and potentially avoid hemolytic-uremic syndrome-related toxicity, the immunoconjugate was modified further, leading to development of a more viable prospect, ie, moxetumomab pasudotox.
Moxetumomab pasudotox
Moxetumomab pasudotox comprises a mouse disulfide-stabilized variable fragment, with the Fv heavy domain fused with a linker and the PE38 fragment. It was developed from BL22 by mutating three specific residues of the third complementarity determining region of the heavy domain.59 This generated an Fv domain with higher affinity captured by antibody phage display. The resulting immunoconjugate displayed a greater affinity for and a lower rate of dissociation from CD22.
Cytotoxicities of up to 50-fold over BL22 versus HCL and chronic lymphocytic leukemia cells were described.59 A subsequent preclinical study characterized the efficacy of moxetumomab pasudotoxin both in vitro and in vivo when killing B-cells from different tumor cell lines.60 In the same study, limited toxicity was seen when moxetumomab pasudotoxin was tested in monkeys, and it demonstrated antitumor activity in murine xenograft experiments. Shortly afterwards, these findings were extrapolated into a Phase I trial testing the immunotoxin in 28 patients with relapsed/refractory HCL at varying doses.61 No dose-limiting toxicities were observed, with an overall response rate of 86%. Forty-six percent of patients achieved a complete response, with only one of the 13 cases relapsing at less than one year. Of the 12 patients who received the full dose, 50% attained a complete response and 50% of these were MRD-negative. Two patients demonstrated hemolytic-uremic syndrome, which was of a lower grade than with BL22 and was also reversible. Neutralizing antibodies were seen in 38% of patients, and although these did not prevent a complete response from being reached or cause any adverse effects, they did not allow for further treatments with the immunotoxin. There was no clear association between response to treatment and previous purine analog therapy. Notably, no patients with prior splenectomy achieved a complete response (0 of 7), whilst 13 patients otherwise achieved complete response. The study cited the possibility of disease status being more advanced in the patients with prior splenectomy due to a heavier disease burden within the marrow, and thus further courses of treatment than those given in the trial could have resulted in a complete response. The patients who attained MRD-negative status currently remain so at a median of 48 months.9
Given these favorable results during Phase I testing, the role of moxetumomab pasudotoxin in the treatment of advanced HCL is currently being assessed in a multicenter Phase III trial (ClinicalTrials.gov identifier NCT01829711) with eligible patients including those who have had HCL unresponsive to or relapsing at least twice after standard treatment. In this study, patients will receive doses of the immunotoxin (as informed by the Phase I trial) administered in 28-day cycles until complete response, disease progression, or unacceptable toxicity is observed.
The results of the LMB-2 and Phase III moxetumomab pasudotoxin trial will determine whether these immunotoxins have a future role in the treatment of advanced HCL. The results with moxetumomab pasudotoxin will be of considerable interest, as the broad strategy with this drug represents a novel approach to the treatment of hematological malignancies.
Targeted kinase inhibition
Vemurafenib
The discovery of BRAF V600E as a driver mutation in cancer is driving the development of therapies targeted toward the BRAF/ERK/MEK pathway. An early iteration of the thymidine kinase inhibitor, vemurafenib, was shown to cause apoptosis in malignant melanoma cell lines by inhibiting mutant BRAF.62 Its efficacy in melanoma was validated in Phase I studies that demonstrated tumor regression particularly in individuals harboring the BRAF V600E mutation.63,64
These observations coupled with the almost universal finding of BRAF V600E in HCL patients have led to considerable interest in the role of vemurafenib in HCL therapy. Dietrich et al reported a 51-year-old male with four prior failures on standard HCL therapies (purine analogs and rituximab) achieving a complete response at day 43 of treatment with a dose-escalating regimen of vemurafenib.65 A less than 0.1% clone was detectable via bone marrow immunophenotyping. Six-month follow-up in this patient demonstrated an ongoing complete response with no evidence of MRD or cytopenia.66 Interestingly, this patient showed strong ERK phosphorylation (a downstream effect of BRAF V600E) within HCL cells on trephine biopsies, which was considerably diminished after 6 days of treatment with vemurafenib. Interrupted signaling through the ERK/MEK pathway leading to apoptosis was also demonstrated. This strongly suggests a role for mutated BRAF signaling in maintaining HCL and provides a plausible mechanism for the activity of vemurafenib.
We have reported the first case of nonescalated low-dose vemurafenib therapy for HCL in a 68-year-old male with a biallelic mutation of BRAF V600E following fourth-line standard therapy and intolerance to IFN.67 The only reported side effect (also commonly seen in melanoma patients treated with vemurafenib) was the growth of three biopsy-confirmed benign seborrheic keratoses. His cytopenias improved considerably, and he achieved normalized neutrophil counts and transfusion independence at day 28. This patient achieved a partial response by day 58 of treatment, deriving considerable clinical benefit from therapy. Unfortunately, his disease relapsed within 4 months off therapy, but retreatment has reinduced a complete response and the patient continues on long-term, low-dose vemurafenib.
Peyrade et al have since reported induction of a complete response in a 72-year-old BRAF V600E-positive male with refractory/relapsing HCL when treated with the same regimen of low-dose vemurafenib as used in our study.68 A complete response was attained at day 90, with complete cytologic and molecular responses, although transfusion independence and normalized platelets and white cell counts were not seen until day 56. A further report by Munoz et al demonstrated an ongoing complete response after treatment with a higher-dose regimen of vemurafenib in a 59-year-old male with heavily pretreated HCL (14 relapses in 27 years).69
With multiple isolated case reports of rapid responses following vemurafenib in BRAF V600E HCL patients, a Phase II study (ClinicalTrials.gov identifier NCT01711632) is currently recruiting patients with relapsed or refractory HCL to assess both the efficacy of and overall response rates to vemurafenib following 3 months of treatment. Importantly, evaluation of MRD status following treatment will be assessed, along with biomarkers assessing mitogen-activated protein kinase pathway responses.
The reports above suggest durable remissions may be inducible following a short course of treatment with vemurafenib. Coupled with promising accounts of depths of remission, a limited side effect profile, and ease of administration, vemurafenib and subsequent iterations of BRAF V600E thymidine kinase inhibitors are likely to emerge as front-runners in the management of resistant HCL, provided their use can be validated in robust Phase III clinical trials.
Bruton tyrosine kinase inhibition
Bruton tyrosine kinase (BTK) is a member of the Tec family kinases and has a well defined role in B-cell receptor (BCR) signaling and B-cell activation. Its downstream effects include activation of NFκB and mitogen-activated protein kinase pathways essential for cell survival. It is expressed in multiple hematopoietic lineages, and the primary defect seen in knockout mice is B-cell specific, underlining its importance in B-cell function. In humans, loss-of-function BTK mutations cause an X-linked agammaglobulinemia that is characterized by B-cell-related immunodeficiency with a reduction in both B-cell numbers and immunoglobulin production, again underlining the importance of BTK and its selectivity to B-cells. Initial screens identified BTK as an important kinase for cell survival in a subgroup of diffuse large B-cell lymphomas driven by activated BCR.70 Targeted inhibition of this with an irreversible BTK inhibitor (PCI-32765, later relabeled as ibrutinib) promoted apoptosis. A preclinical study demonstrated overexpression of BTK protein in chronic lymphocytic leukemia cells when compared with normal cells.71 The same inhibitor used in this study resulted in inhibition of BTK phosphorylation and abrogated mitogen-activated protein kinase, phosphatidylinositol 3-kinase, and NFκB pathways within BCR-activated chronic lymphocytic leukemia cells, whilst also inhibiting proliferation of these cells in vitro. These preliminary studies, coupled with further studies in non-Hodgkin’s lymphoma, have led to the development of several BTK inhibitors (ie, ONO-WG-307, LFM-A13, dasatinib, CC-292).72–76
Ibrutinib is the most advanced of these inhibitors in clinical development and is an orally available, potent, irreversible BTK inhibitor. Several Phase II studies have strongly suggested a role for BTK inhibition in relapsed/refractory chronic lymphocytic leukemia, mantle cell lymphoma, and diffuse large B-cell lymphoma.77
Recently, there has been considerable interest in the role of ibrutinib in HCL. An abstract presented at the 2012 meeting of the American Society of Hematology reported significant inhibition of HCL cell line growth and proliferation in the presence of varying concentrations of ibrutinib in a dose-dependent fashion. Primary patient HCL cells were also tested for apoptosis in the presence of ibrutinib, and viability was reduced significantly after 48 hours. Consequently, a multicenter Phase II trial addressing the role of ibrutinib in patients with relapsed HCL has opened (ClinicalTrials.gov identifier NCT01841723) and will assess toxicity, progression-free and overall survival, MRD, and the changes in biomarkers in HCL.
Conclusion
SEER database data show clearly that the outcomes for patients diagnosed with HCL has improved over the last three decades.78 During this period, we have refined our diagnostic techniques, culminating in the recent discovery of BRAF V600E. With respect to therapeutics, the development of purine analogs was a significant milestone in improving the survival of HCL patients. Combining purine analogs with rituximab has now become standard practice for relapsing disease, although prospective trial data in this setting remain limited. Immunotoxins have significant potential, but their development has been hindered by significant toxicities. The era of targeted kinase inhibition has dawned. Since the advent of imatinib for the treatment of chronic myeloid leukemia, the idea of small-molecule inhibition across a number of hematologic malignancies targeting specific kinases or proteins within key pathways for cell signaling and survival has remained an attractive one. With the promising case reports seen thus far in HCL with vemurafenib and biological studies with ibrutinib, targeted kinase inhibition may have potential to transform the landscape of HCL management in the near future.
Disclosure
The authors declare no competing financial interests in this work.
References
Foucar K, Falini B, Catovsky D, Stein H. Hairy cell leukaemia. In: WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. Swerdlow SH, Campo E, Harris NL, et al, editors. Lyon, France: IARC Press, Lyon; 2008. | |
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. | |
Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–1403. | |
Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood. 1958;13:609–630. | |
Schrek R, Donnelly WJ. “Hairy” cells in blood in lymphoreticular neoplastic disease and “flagellated” cells of normal lymph nodes. Blood. 1966;27:199–211. | |
Else M, Dearden CE, Matutes E, et al. Long-term follow-up of 233 patients with hairy cell leukaemia, treated initially with pentostatin or cladribine, at a median of 16 years from diagnosis. Br J Haematol. 2009;145:733–740. | |
Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukaemia. N Engl J Med. 2011;364:2305–2315. | |
Boyd EM, Bench AJ, van’t Veer, et al. High resolution melting analysis for detection of BRAF exon 15 mutations in hairy cell leukaemia and other lymphoid malignancies. Br J Haematol. 2011;155:609–612. | |
Kreitman RJ. Hairy cell leukaemia – new genes, new targets. Curr Hematol Malig Rep. 2013;8:184–195. | |
Sainati L, Matutes E, Mulligan S, et al. A variant form of hairy cell leukemia resistant to alpha-interferon: clinical and phenotypic characteristics of 17 patients. Blood. 1990;76:157–162. | |
Matutes E, Wotherspoon A, Brito-Babapulle V, Catovsky D. The natural history and clinico-pathological features of the variant form of hairy cell leukemia. Leukemia. 2001;15:184–186. | |
Robak T. Hairy-cell leukaemia variant: recent view on diagnosis, biology and treatment. Cancer Treat Rev. 2011;37:3–10. | |
Arons E, Suntum T, Stetler-Stevenson M, Kreitman RJ. VH4-34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy. Blood. 2009;114:4687–4695. | |
Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood. 2012;119:3330–3332. | |
Quesada JR, Reuben J, Manning JT, Hersh EM, Gutterman JU. Alpha interferon for induction of remission in hairy-cell leukemia. N Engl J Med. 1984;310:15–18. | |
Grever M, Kopecky K, Foucar MK, et al. Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: an Intergroup study. J Clin Oncol. 1995;13: 974–982. | |
Cheson BD, Sorensen JM, Vena DA, et al. Treatment of hairy cell leukemia with 2- chlorodeoxyadenosine via the Group C protocol mechanism of the National Cancer Institute: a report of 979 patients. J Clin Oncol. 1998;16:3007–3015. | |
Piro LD, Carrera CJ, Carson DA, Beutler E. Lasting remissions in hairy-cell leukemia induced by a single infusion of 2-chlorodeoxyadenosine. N Engl J Med. 1990;322:1117–1121. | |
Robak T, Jamroziak K, Gora-Tybor J, et al. Cladribine in a weekly versus daily schedule for untreated active hairy cell leukemia: final report from the Polish Adult Leukemia Group (PALG) of a prospective, randomized, multicenter trial. Blood. 2007;109:3672–3675. | |
Goodman GR, Burian C, Koziol JA, Saven A. Extended follow-up of patients with hairy cell leukemia after treatment with cladribine. J Clin Oncol. 2003;21:891–896. | |
Dearden CE, Matutes E, Hilditch BL, Swansbury GJ, Catovsky D. Long-term follow-up of patients with hairy cell leukaemia after treatment with pentostatin or cladribine. Br J Haematol. 1999;106: 515–519. | |
Kraut EH, Neff JC, Bouroncle BA, Gochnour D, Grever MR. Immunosuppressive effects of pentostatin. J Clin Oncol. 1990;8: 848–855. | |
Saven A, Piro LD. 2-Chlorodeoxyadenosine: a newer purine analog active in the treatment of indolent lymphoid malignancies. Ann Intern Med. 1994;120:784–791. | |
Tadmor T. Purine analog toxicity in patients with hairy cell leukemia. Leuk Lymphoma. 2011;52 Suppl 2:38–42. | |
Cheson BD, Vena DA, Foss FM, Sorensen JM. Neurotoxicity of purine analogs: a review. J Clin Oncol. 1994;12:2216–2228. | |
Beers SA, Chan CH, French RR, Cragg MS, Glennie MJ. CD20 as a target for therapeutic type I and II monoclonal antibodies. Semin Hematol. 2010;47:107–114. | |
Ginaldi L, De Martinis M, Matutes E, et al. Levels of expression of CD19 and CD20 in chronic B cell leukaemias. J Clin Pathol. 1998;51: 364–369. | |
Reff ME, Carner K, Chambers KS, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83:435–445. | |
Avivi I, Stroopinsky D, Katz T. Anti-CD20 monoclonal antibodies: beyond B-cells. Blood Rev. 2013;27:217–223. | |
Coiffier B, Lepage E, Briere J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:235–242. | |
Dotan E, Aggarwal C, Smith MR. Impact of rituximab (rituxan) on the treatment of B-cell non-Hodgkin’s lymphoma. P T. 2010;35: 148–157. | |
Hagberg H, Lundholm L. Rituximab, a chimaeric anti-CD20 monoclonal antibody, in the treatment of hairy cell leukaemia. Br J Haematol. 2001;115:609–611. | |
Thomas DA, O’Brien S, Bueso-Ramos C. Rituximab in relapsed or refractory hairy cell leukemia. Blood. 2003;102:3906–3911. | |
Nieva J, Bethel K, Saven A. Phase 2 study of rituximab in the treatment of cladribine-failed patients with hairy cell leukemia. Blood. 2003;102:810–813. | |
Else M, Dearden CE, Matutes E. Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma. 2011;52 Suppl 2:75–78. | |
Jones G, Parry-Jones N, Wilkins B, Else M, Catovsky D; British Committee for Standards in Haematology. Revised guidelines for the diagnosis and management of hairy cell leukaemia and hairy cell leukaemia variant. Br J Haematol. 2012;156:186–195. | |
Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood. 2011;118:3818–3823. | |
Kantarjian HM, Schachner J, Keating MJ. Fludarabine therapy in hairy cell leukemia. Cancer. 1991;67:1291–1293. | |
Kraut EH, Chun HG. Fludarabine phosphate in refractory hairy cell leukemia. Am J Hematol. 1991;37:59–60. | |
Gerrie AS, Zypchen LN, Connors JM. Fludarabine and rituximab for relapsed or refractory hairy cell leukemia. Blood. 2012;119:1988–1991. | |
Narat S, Gandla J, Dogan A, Mehta A. Successful treatment of hairy cell leukemia variant with rituximab. Leuk Lymphoma. 2005;46: 1229–1232. | |
Quach H, Januszewicz H, Westerman D. Complete remission of hairy cell leukemia variant (HCL-v) complicated by red cell aplasia post treatment with rituximab. Haematologica. 2005;90 Suppl:ECR26. | |
Hadzi-Pecova L, Stojanovik A, Petrusevska G, Panovska I. Rituximab in the treatment of the variant of hairy cell leukaemia: a case report. Prilozi. 2008;29:355–360. | |
Quigley MM, Bethel KJ, Sharpe RW, Saven A. CD52 expression in hairy cell leukemia. Am J Hematol. 2003;74:227–223. | |
Rodig SJ, Abramson JS, Pinkus GS, et al. Heterogeneous CD52 expression among hematologic neoplasms: implications for the use of alemtuzumab (CAMPATH-1H). Clin Cancer Res. 2006;12:7174–7179. | |
Fietz T, Rieger K, Schmittel A, Thiel E, Knauf W. Alemtuzumab (Campath 1H) in hairy cell leukaemia relapsing after rituximab treatment. Hematol J. 2004;5:451–452. | |
Sasaki M, Sugimoto K, Mori T, Karasawa K, Oshimi K. Effective treatment of a refractory hairy cell leukemia variant with splenic pre-irradiation and alemtuzumab. Acta Haematol. 2008;119:48–53. | |
Telek B, Batár P, Udvardy M. [Successful alemtuzumab treatment of a patient with atypical hairy cell leukaemia variant]. Orv Hetil. 2007;148:1805–1907. Hungarian. | |
Carroll SF, Collier RJ. Active site of Pseudomonas aeruginosa exotoxin A. Glutamic acid 553 is photolabeled by NAD and shows functional homology with glutamic acid 148 of diphtheria toxin. J Biol Chem. 1987;262:8707–8711. | |
Van Ness BG, Howard JB, Bodley JW. ADP-ribosylation of elongation factor 2 by diphtheria toxin. Isolation and properties of the novel ribosyl-amino acid and its hydrolysis products. J Biol Chem. 1980;255: 10717–10720. | |
Kreitman RJ, Wilson WH, White JD, et al. Phase I trial of recombinant immunotoxin Anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol. 2000;18:1614–1636. | |
National Cancer Institute. LMB-2 to treat hairy cell leukemia. Available from: http://clinicaltrials.gov/show/NCT00321555. Accessed May 13, 2014. | |
Jasper GA, Arun I, Venzon D, et al. Variables affecting the quantitation of CD22 in neoplastic B cells. Cytometry B Clin Cytom. 2011;80:83–90. | |
Babusikova O, Tomova A, Kusenda J, Gyarfas J. Flow cytometry of peripheral blood and bone marrow cells from patients with hairy cell leukemia: phenotype of hairy cells, lymphocyte subsets and detection of minimal residual disease after treatment. Neoplasma. 2001;48:350–357. | |
Cordone I, Annino L, Masi S, et al. Diagnostic relevance of peripheral blood immunocytochemistry in hairy cell leukaemia. J Clin Pathol. 1995;48:955–960. | |
Stoddart A, Jackson AP, Brodsky FM. Plasticity of B cell receptor internalization upon conditional depletion of clathrin. Mol Biol Cell. 2005;16:2339–2348. | |
Kreitman RJ, Squires DR, Stetler-Stevenson M, et al. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol. 2005;23:6719–6729. | |
Kreitman RJ, Stetler-Stevenson M, Margulies I, et al. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009;27:2983–2990. | |
Salvatore G, Beers R, Margulies I, Kreitman RJ, Pastan I. Improved cytotoxic activity toward cell lines and fresh leukemia cells of a mutant anti-CD22 immunotoxin obtained by antibody phage display. Clin Cancer Res. 2002;8:995–1002. | |
Alderson RF, Kreitman RJ, Chen T, et al. CAT-8015: a second-generation pseudomonas exotoxin A-based immunotherapy targeting CD22-expressing hematologic malignancies. Clin Cancer Res. 2009;15: 832–839. | |
Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. 2012;30:1822–1828. | |
Sala E, Mologni L, Truffa S, Gaetano C, Bollag GE, Gambacorti-Passerini C. BRAF silencing by short hairpin RNA or chemical blockade by PLX4032 leads to different responses in melanoma and thyroid carcinoma cells. Mol Cancer Res. 2008;6:751–759. | |
Flaherty K. Phase I study of PLX4032: proof of concept for V600E BRAF mutation as a therapeutic target in human cancer. Abstract presented at the 45th annual meeting of the American Society of Clinical Oncology, May 29 to June 2, 2009, Orlando, FL, USA. J Clin Oncol. 2000;27:15 Suppl: Abstr 9000. | |
Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. | |
Dietrich S, Glimm H, Andrulis M, von Kalle C, Ho AD, Zenz T. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med. 2012;366:2038–2040. | |
Dietrich S, Hüllein J, Hundemer M, et al. Continued response off treatment after BRAF inhibition in refractory hairy cell leukemia. J Clin Oncol. 2013;31:e300–e303. | |
Follows GA, Sims H, Bloxham DM, et al. Rapid response of biallelic BRAF V600E mutated hairy cell leukaemia to low dose vemurafenib. Br J Haematol. 2013;161:150–153. | |
Peyrade F, Re D, Ginet C, et al. Low-dose vemurafenib induces complete remission in a case of hairy-cell leukemia with a V600E mutation. Haematologica. 2013;98:e20–e22. | |
Munoz J, Schlette E, Kurzrock R. Rapid response to vemurafenib in a heavily pretreated patient with hairy cell leukemia and a BRAF mutation. J Clin Oncol. 2013;31:e351–e352. | |
Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463: 88–92. | |
Herman SE, Gordon AL, Hertlein E, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117: 6287–6296. | |
Kozaki R, Yoshizawa T, Yashuhiro T, et al. Development of a Bruton’s tyrosine kinase (Btk) inhibitor – ONO-WG-307, a potential treatment for B-cell malignancies. Cancer Res. 2012;72: Abstr 857. | |
Uckun F, Dibirdik I, Sarkissian A, Qazi S. In vitro and in vivo chemosensitizing activity of LFM-A13, a dual-function inhibitor of Bruton’s tyrosine kinase and polo-like kinases, against human leukemic B-cell precursors. Arzneimittelforschung. 2011;61:252–259. | |
Hantschel O, Rix U, Schmidt U, et al. The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc Natl Acad Sci U S A. 2007;104:13283–13288. | |
Amrein PC, Attar EC, Takvorian T, et al. Phase II study of dasatinib in relapsed or refractory chronic lymphocytic leukemia. Clin Cancer Res. 2011;17:2977–2986. | |
Evans EK, Tester R, Aslanian S, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346:219–228. | |
Aalipour A, Advani RH. Bruton tyrosine kinase inhibitors: a promising novel targeted treatment for B cell lymphomas. Br J Haematol. 2013;163:436–443. | |
Chandran R, Gardiner SK, Smith SD, Spurgeon SE. Improved survival in hairy cell leukaemia over three decades: a SEER database analysis of prognostic factors. Br J Haematol. 2013;163:407–409. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.