")
Back to Journals » Journal of Inflammation Research » Volume 15
Gut Microbiota Mediates the Susceptibility of Mice to Sepsis-Associated Encephalopathy by Butyric Acid
Authors Zhang H, Xu J, Wu Q, Fang H, Shao X, Ouyang X, He Z, Deng Y , Chen C
Received 9 December 2021
Accepted for publication 4 March 2022
Published 30 March 2022 Volume 2022:15 Pages 2103—2119
DOI https://doi.org/10.2147/JIR.S350566
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Huidan Zhang,1– 3,* Jing Xu,1,2,4,* Qingrui Wu,3,* Heng Fang,1,2,4 Xin Shao,1,2 Xin Ouyang,1 Zhimei He,2 Yiyu Deng,2– 4 Chunbo Chen1,3,4
1Department of Intensive Care Unit of Cardiovascular Surgery, Guangdong Cardiovascular Institute, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Guangzhou, 510080, People’s Republic of China; 2Department of Critical Care Medicine, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Guangzhou, 510080, People’s Republic of China; 3School of Medicine, South China University of Technology, Guangzhou, 510006, People’s Republic of China; 4The Second School of Clinical Medicine, Southern Medical University, Guangzhou, 510515, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yiyu Deng; Chunbo Chen, Tel +86-20-83827812 ext. 61526, Fax +86-20-83827712, Email [email protected]; [email protected]
Purpose: Neuroinflammation plays an important part in the pathophysiology of sepsis-associated encephalopathy (SAE). Gut microbiota and gut brain axis are considered as important mediators in the development of neurological diseases. The aim of this study was to investigate the role of intestinal microbiota in sepsis-related brain injury and to explore the underlying mechanisms.
Methods: Mouse model of SAE was established using cecal ligation and puncture (CLP). Based on the mouse mortality and the associated time of death, light SAE (LSAE) and severe SAE (SSAE) were classified. Fecal microbiota transplantation (FMT) was performed to verify the role of intestinal microbiota. Feces of mice in the two groups which collected before operation were sequenced for 16S and targeted short chain fatty acids.
Results: Intestinal microbiota from SSAE and LSAE mice displayed diverse functions. Interestingly, LSAE mice produced more butyric acid compared with SSAE mice. In the in vivo experiments, sodium butyrate (NaB) reduced the high oxidative stress levels in mice hippocampus and conferred a marked survival superiority to sepsis mice. In addition, NaB prevented the increase in intracellular reactive oxygen species (ROS) generation and inducible nitric-oxide synthase expression in LPS-stimulated primary microglia. The GPR109A/Nrf2/HO-1 signaling pathway was found to be involved in the activation of antioxidant response of primary microglia induced by sodium butyrate.
Conclusion: Our findings indicate a crucial role of gut microbiota in the susceptibility to SAE. Butyrate, a metabolite of intestinal microbiota, may have a neuroprotective effect in the process of sepsis by GPR109A/Nrf2/HO-1 pathway.
Keywords: gut microbiome, butyrate, sepsis-related brain injury, primary microglia, oxidative stress
Introduction
Sepsis-associated encephalopathy (SAE) is a common consequence in seriously ill patients, characterized by neurological manifestations and diffuse brain dysfunction as a result of sepsis.1 Sepsis survivors can develop long-term disability and psychiatric disorders, including cognitive deficit, delayed psychomotor, and visual and hearing impairment.2,3 Neuroinflammation in the central nervous system (CNS) is well considered to perform an essential role in SAE, while the disease’s pathogenetic processes are unknown. Moreover, there is a paucity of effective therapies against SAE.
Gut microorganisms constitute the largest microbial bank in human body. Gut microbiota and its metabolites take a significant part in neurodevelopment, neurodegeneration, and other neurological diseases.4,5 Microbiota plays a part in acute brain dysfunction in sepsis.6 Whether modulating the composition of the microbiota can ameliorate septic brain injury is unknown. Studies have found a link between the type of microorganism and the severity of SAE.7 Further, infected patients with lower gut microbiome diversity have higher mortality from sepsis.8 Therefore, we wondered whether the microbiota of different individuals could lead to the different severity of sepsis after infection. It is worth noting that microbial metabolites are thought to be the primary mediators in brain injury progression. Studies have shown that short chain fatty acids (SCFAs), one of the most important metabolites of gut microbiota, its concentration in the feces of sepsis patients is significantly reduced and affected the physiological function of the body in sepsis.9,10 SCFAs can improve cerebral damage by the blood-brain barrier (BBB).11 The microbiota is a vital regulator of gut-brain function, and the importance of the microbiota-gut-brain axis is becoming more widely recognized. These findings suggest that the gut microbiota and its metabolites may perform a significant role in sepsis and related brain damage pathogenesis. The participation of gut microbiota in SAE development, on the other hand, is unidentified. The current study aimed to look at the function of intestinal microbiota-mediated neuroinflammation in course of sepsis to find new ways to treat SAE.
Materials and Methods
Animals
Male C57BL/6 mice (age: 6–8 weeks, weight: 18–22 g) were included in the study and purchased from the Guangzhou Yongnuo Biotechnology Co. Ltd. (Guangzhou, Guangdong, China). All mice (4–5 per cage) were housed in specific-pathogen free (SPF) environment under a 12/12 h light/dark cycle with ad libitum access to water and food. The procedures for Care and use of animals were approved by the Medical Research Ethics Committee of Guangdong Provincial People’s Hospital and all applicable institutional and governmental regulations concerning the ethical use of animals were followed.
CLP Animal Model
Mice were randomly assigned to three groups including Sham group, CLP group, and CLP+Sodium butyrate (NaB) group with 20 mice in each group. Cecal ligation and perforation (CLP) were performed to cause brain injury. In brief, pentobarbital sodium was used to anesthetize the mice, followed by 2-cm midline laparotomy in aseptic conditions exposing the cecum area. A 21-gauge needle was used, half of the cecum was perforated and ligated, and a little of faeces was then expelled from the puncture area. The laparotomy was closed after the caecum was relocated into the abdominal cavity. The mice in the Sham group only underwent laparotomy without puncture and ligation of the cecum. 1 mL saline was used to resuscitate all the animals. NaB, 500 mg/kg diluted in phosphate-buffered saline (PBS), was administered intragastrically to mice in the NaB treatment group 7 days before the establishment of sepsis models.
Fecal Microbiota Transplantation
The procedure for fecal microbiota transplantation (FMT) was followed as reported elsewhere.12,13 In short, male C57BL/6 mice (6–8-week-old) received intragastric administration of antibiotics, including vancomycin, 100 mg/kg, neomycin sulfate, 200 mg/kg, metronidazole, 200 mg/kg, and ampicillin, 200 mg/kg were administered to deplete the gut microbiota once a day for 5 days. The donors were mice in severe SAE group and light SAE group. Their feces were collected and resuspended in 0.125 g/mL of PBS, followed by administration of this resuspension each day, once per day, into mice by intragastric gavage. After 3 days, CLP was performed and mice were sacrificed after 12 hours, at which tissue and blood specimens were collected.
High-Throughput Sequencing Analyzing Gut Microbiota
As per the manufacturer’s instructions, the fecal microbial genome was extracted using a Fast DNA SPIN Kit (MP Bio., SA, USA). The primers were designed to amplify the V3-V4 regions of the 16S rDNA using PCR. Following that, Illumina Miseq PE300 was used to sequence the samples. The software Usearch (version 7.0) was used clustering the operational taxonomic units (OTUs) with a 97% similarity. Mothur (version v.1.30.2) was employed for the calculation of the Alpha diversity indices (Chao, ACE, and Shannon). Based on unweighted_unifrac distances, principal co-ordinates analysis (PCoA) was performed using R software (version 3.3.1). Wilcoxon rank-sum test was used to test species differences at the genus level between two groups. db-RDA analysis using Euclidean distance. Correlations between differentially presented microbiota and the each SCFAs were calculated by Spearman correlation analysis using the “pheatmap” packages of R software (version 3.3.1).
Quantification of Fatty Acids in Fecal Contents
Gas chromatography (GC) was used to assess the concentration of short-chain fatty acids (SCFA) in feces. In a 2 mL Eppendorf tube, the fecal digesta (80 mg) was mixed with 0.2 mL sulfuric acid (50%, v/v), 0.8 mL distilled water, and five glass beads. The mixture was vortexed for 5 min before being centrifuged at 12,000 rpm for 15 minutes. The supernatant was stirred for 2 minutes with 1.0 mL of precooled ethyl ether before centrifugation (12,000 rpm, 15 min). The material was filtered through a 0.22- µ organic membrane and GC analysis were performed with the injection volume was 8 μL, according to Li et al.14
Reagents and Antibodies
Lipopolysaccharide (LPS, L2630) and sodium butyrate (NaB, B5887) were purchased from Sigma. ML385 was purchased from MCE. PTX was purchased from GlpBio. PGE2 ELISA kit was purchased from Shanghai Ruifan Biological Technology Co. Ltd. Antibodies against i-NOS (ab178945), COX-2 (ab179800), BDNF (ab108319), HO-1 (ab52947), NQO1 (ab80588), Keap-1 (ab227828) and GAPDH (ab181602) were purchased from Abcam. Antibodies against Nrf2 (GTX103322) and Iba-1 (GT10312) were purchased from Genetex. Antibodies against GPR109A (sc-377292) was purchased from Santa cruz.
Morris Water Maze Test
The Morris water maze (MWM) test was performed for assessing spatial learning and memory in mice on the seventh day following surgery. A round pool which height is 50 cm and diameter is 120 cm full of water at 25 °C and fitted with a video recorder and video tracker was included in the MWM device. Adaptive swimming, space exploration, and spatial learning were among the steps in the Morris water maze challenge.
Adaptive swimming: After the underwater fixed platform was removed on day 1 of the mission, in the tank the mice could swim for 2 minutes. This was done so that each mouse would have the same time to acclimatize to the message in the surroundings.
Spatial learning: Mice were treated to a swimming testing from the 4 quadrants at a predetermined time once a day for the next 4 days. The escape latency is the time taken by these mice to arrive at the fixed platform. If a mice could not get to the underwater platform less than 2 minutes, its escape latency was determined to be 2 minutes, and the mice was directed to a regular underwater platform to take a rest for 1 minute.
Space exploration: The underwater platform was detached before the test on the final day (6th day) of the job, and the mice come into the water from different initiation points. The frequency of the mice passed through the platform’s original location in 2 minutes was noted.
Neurobehavioral Scores
Neurobehavioral scores were used to assess the animals’ neurobehavioral alterations. The corneal reflex, righting reflex, auricle reflex, avoid reflex, and tail-flick reflex were the five components of the entire exam. The avoid reflex and the tail-flick reflex both used a quick stimulation of the mouse-tail to see if the mouse would attempt to escape. The method of corneal reflex is to touch the cornea of mice with a cotton swab and observe whether the mice blink or shake their head. The auricle reflex is triggered when the mouse’s plane is touched, causing a significant head rotation. The righting reflex method was retained in the supine position and observed to examine if it could turn into the prone position. Each normal reflex was awarded a score of 2, while hyporeflexia was awarded a score of 1, and areflexia was awarded a score of 0. The overall score was calculated by adding the results of these five tests.
Hematoxylin and Eosin (H&E) Staining
Fixation of brain tissues was performed with 4% paraformaldehyde, dried, embedded, and cut into 4-μm slices for histological examinations. Under a light microscope, hippocampus tissue slices were dyed with hematoxylin and eosin for morphological analysis.
Assessment of Oxidative Stress
Fresh hippocampal tissue was obtained after euthanasia of each mouse on the first and third days after surgery in three groups. Lipid Peroxidation MDA Assay Kit was used to quantify malondialdehyde (MDA) (Beyotime, Shanghai, China). Total Superoxide Dismutase Assay Kit with NBT (Beyotime, Shanghai, China) was used for measuring superoxide dismutase (SOD). Intracellular ROS production was measured using DCFHDA (Abcam, San Francisco, USA). The level of intracellular ROS was determined by microplate reader. The oxidative fluorescent dye dihydroethidine hydrochloride (DHE) was utilized for measuring ROS generation in brain tissues. The fresh brain was removed and was sliced to a thickness of 10-μm, placed on glass slides. Each tissue section was incubated with 5 μM of the fluorescence probe dihydroethidine hydrochloride (DHE) for 30 min at 37°C in a dark box. A fluorescent microscope (Olympus System Microscope Model BX53, Olympus Company, Tokyo, Japan) was used to scan the images of brain sections DHE staining.
Determination of Butyrate Levels in Plasma and Hippocampus
Blood samples were obtained from the auricle of each mouse under anesthesia and placed in EDTA-coated tubes; the blood was allowed to stand for 10–20 minutes before being centrifuged for 20 minutes (5652 g). Each mouse’s hippocampus was removed and weighed. The hippocampus was homogenized followed by centrifugation at 5652 g for 20 minutes after being added PBS. The supernatants were collected carefully. As per the manufacturer’s protocol, the butyrate concentration in plasma and hippocampus were estimated through ELISA (Shanghai Ruifan Biological Technology Co. Ltd.).
Primary Culture of Microglia
The cerebral cortex of 1-3-day-old C57BL/6 mice was meticulously dissected from the brain and mechanically dissociated by repeated pipetting into single-cell suspensions. Mice were provided by Medical Experimental Animal Center of Guangdong Province, China. The mixed glia cells were then plated in Dulbecco’s modified Eagle’s medium/F12 medium (DMEM/ F12, Gibco) containing a supplement of 10% fetal bovine serum (FBS) at a density of 1.2×106 cells/mL on a 75-cm2 flask (Hyclone, Logan, UT). The mixed cells were grown in a humidified 5% CO2 incubator for 7–8 days at 37 °C, with half of the medium replaced every 3–4 days. The microglial cells were extracted after 1 hour of shaking at 180 rpm at 37°C. Purified microglia (5×105 cells per well) were seeded with poly-L-lysine (PLL) in a six-pore plate.
Western Blotting
Proteins were isolated with a protein extraction kit (Pierce Biotechnology Inc, IL, USA) from the hippocampus of mice at various periods from each group or primary microglia exposed to different treatments, as per the manufacturer’s protocol. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate proteins in the supernatant. After then, the proteins were transferred to a nitrocellulose membrane (Hybond-ECL, Amersham BioSciences, Little Chalfont, UK). An overnight blocking and incubation of membranes at 4°C were performed with primary antibodies against cyclooxygenase-2 (COX-2; 1:1000), inducible nitric oxide synthase (i-NOS; 1:1000), brain derived neurotrophic factor (BNDF; 1:1000), or glyceraldehyde-3-phosphate dehydrogenase (1:1000); against Nuclear factor E2-related factor 2 (Nrf2; 1:200), Heme Oxygenase-1 (HO-1; 1:1000), NAD(P)H: quinone oxidoreductase 1 (NQO1; 1:1000), or G-protein-coupled receptor109A (GPR109A; 1:1000). Blots were incubated with secondary antibodies (Sigma-Aldrich, St. Louis, MO, USA) combined with horseradish peroxidase, after adequate washing. Enhanced chemiluminescence (Amersham Biosciences) was employed to detect protein bands, which were then quantitated utilizing Quantity One program (Bio-Rad Laboratories, Hercules, CA, USA). The band’s intensity was compared to that of GAPDH.
Double Immunofluorescence
After perfusion with 4% PFA, the brain was removed and immersed in the same fixative overnight at 4°C. After that, the formalin-fixed brain was dehydrated and stored in a 30% sucrose solution. Frozen pieces were sliced to a thickness of 20 μM, placed on glass slides, and cryoprotected at −20°C. Two groups were formed from coronal brain sections taken at different times in the Sham, CLP, and CLP+NaB groups. Brain sections in group-I were taken from mice sacrificed at days 1 and 3 after CLP, and corresponding Shams and treatment group, and treated with anti-Iba-1 and anti-i-NOS antibodies. Brain sections in group-II were acquired from mice sacrificed at 1 day 1 and 3 after CLP, and corresponding Shams and treatment group, and treated with anti-Iba-1 and anti-COX-2 antibodies. After three PBS washes, the sections were incubated at room temperature for 1 hour with Alexa-conjugated secondary antibodies: Alexa Fluor 594 goat anti-rabbit IgG (1:100, Abcam, ab150080), Alexa Fluor 488 goat anti-mouse IgG (1:100, Abcam, ab150113). Nuclear staining was done with DAPI (Sigma-Aldrich, St. Louis, D9542) after three washes with PBS. Finally, a fluorescent mounting medium was used to mount the sections.
The cells were treated with LPS or LPS+NaB for 12 hours in cultured microglia. Fixation of cells was performed with 4% PFA for 30 minutes after treatment, blocking with 1% BSA for 30 min, followed by incubation overnight at 4°C with primary antibodies. Primary antibodies directed against anti-Iba-1, anti-i-NOS, or anti-COX-2 were used for immunofluorescence labeling. The cells were then treated for 1 hour with Alexa Fluor 594 goat anti-rabbit IgG (1:100, Abcam, ab150080) and Alexa Fluor 488 goat anti-mouse IgG (1:100, Abcam, ab150113). Finally, DAPI was used to counterstain the cells.
A fluorescent microscope (Olympus System Microscope Model BX53, Olympus Company, Tokyo, Japan) was used to scan the images of primary cell staining or brain sections.
Statistical Analysis
The mean ± standard error (SD) was used to express the data unless otherwise stated. Statistical significance was established by GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA). Different statistical methods were applied according to different types of data. The data of survival rate were analyzed by Chi-square test. Comparisons between two groups were analyzed by using a two-tailed Student’s t-test on account of the data were homogeneity of variance. Comparison between more than two groups were analyzed by a one-way ANOVA or two-way ANOVA followed by Tukey’s multiple comparison test. P values < 0.05 were considered statistically significant.
Results
Differentiation of Severe and Light Sepsis Encephalopathy in Mice
First, we collected feces from each mouse. Next, we used CLP surgery to evaluate mice’s response to SAE. The survival rate curve demonstrated that the mice rate of mortality within 48 hours after CLP was very high and tended to be stable after 7 days (Figure 1A). Therefore, mice that died before 48 hours were defined as Severe SAE mice (SSAE), and those that survived to 7 days were declared as Light SAE mice (LSAE). Subsequently, we evaluated neurological impairment in the two groups at 24 hours after CLP. SSAE mice had considerably lower neurobehavioral scores than LSAE mice, which was a statistically significant difference (Figure 1B). Furthermore, the extent of hippocampal injury in these two mouse groups was compared. Histopathological study of hippocampus sections from SSAE mice indicated severe neuronal degeneration, whereas LSAE mice had normal hippocampal neuron morphology (Figure 1C). On Western blot analysis, SSAE mice hippocampus showed higher trends of expressions of i-NOS, COX-2, and lower expression of BDNF (Figure 1D). These findings indicated more severe brain injury in SSAE mice. Our results showed that SSAE mice qualified the definition of injury and infection of brain whereas LSAE mice showed resolution of brain damage.
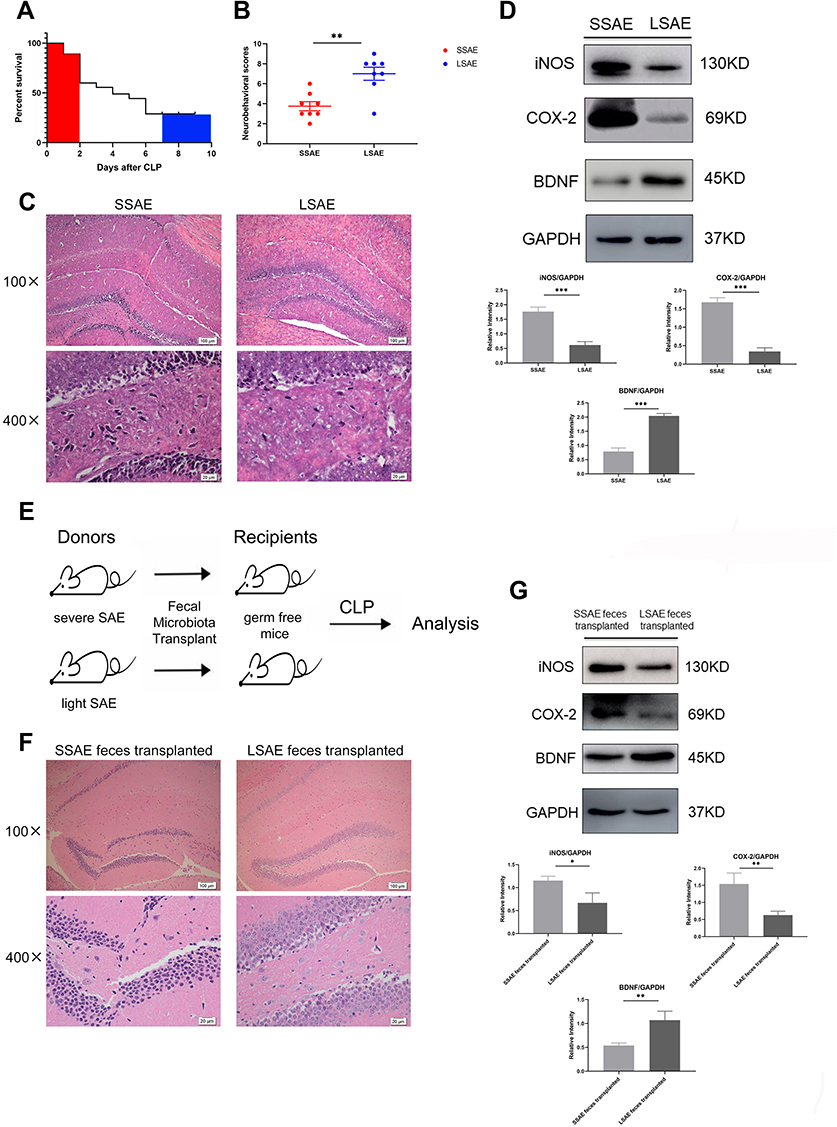 |
Figure 1 Characterization of severe SAE and light SAE mice and dependence of sepsis-induced brain injury on gut microbiota. Survival rates of mice were observed until 7 days post-CLP. Mice that died within 48 hours of CLP were defined as severe SAE (SSAE) because of the high mortality. Mice that survived after 7 days were defined as light SAE (LSAE) because of the mortality rate tends to be flat. (A) Survival rate. (B) Neurobehavioral scores. (C) Representative H&E-stained sections of hippocampus of SSAE and LSAE mice. (D) Western blot analysis of iNOS, COX-2, and BDNF protein expressions in the hippocampus of SSAE and LSAE mice. GAPDH served as the loading control. Bar graphs depict the optical density of iNOS, COX-2, and BDNF expression. (E) Schematic illustration of FMT experiment: Mice were administered intragastric vancomycin (100 mg/kg), neomycin sulfate (200 mg/kg), metronidazole (200 mg/kg), and ampicillin (200 mg/kg) once-daily for 5 days to deplete the gut microbiota; subsequently, they received feces resuspended in PBS from SSAE and LSAE mice for 3 days. CLP surgery was performed, and mice were sacrificed 12 hours after CLP. (F) Representative H&E-stained sections of hippocampus of recipient mice. (G) Western blot analysis of iNOS, COX-2, and BDNF protein expressions in the hippocampus of recipient mice. GAPDH served as the loading control. Bar graphs depict the optical density of iNOS, COX-2, and BDNF expressions. Data presented as mean ± SD (n = 3–8 per group). Scale bars: 100µm and 20µm. *P < 0.05, **P < 0.01, ***P < 0.001 by Student’ t-test. |
Susceptibility to Sepsis-Associated Encephalopathy Was Transmissible by Gut Microbiota
It is hypothesized that gut microbiota regulates brain susceptibility to sepsis to some extent. We conducted an FMT experiment to confirm our hypothesis (Figure 1E). After CLP surgery, mice administered SSAE feces had more severe hippocampal injury than mice given LSAE feces, according to histopathological examination. Moreover, the number of neurons was decreased and nucleus pyknosis was observed in mice that received SSAE feces (Figure 1F). The protein expressions of i-NOS and COX2 were evidently higher in SSAE feces recipients in comparison with LSAE feces recipients, while the converse was true with respect to BDNF level (Figure 1G). These findings revealed that gut microbiota sensitization to sepsis-induced brain damage could be transferred.
Intestinal Microbiota from SSAE or LSAE Mice Displayed Different Functions
We then investigated the intestinal microbiota in SSAE as well as in LSAE animals to determine if it influenced sepsis-induced brain damage. The Chao and ACE indices were determined for community richness, whereas the Shannon index was determined for community diversity. On assessing the alpha diversity between SSAE and LSAE mice, we observed a significant difference in community richness, while community diversity showed no significant difference (Figure 2A–C). In one dimension, the β-diversity reflected by weighted principal coordinates analysis (PCoA) study demonstrated mild separation of gut microbiota clusters between these two groups of mice (Figure 2D). This indicated slightly distinct composition of gut microbiota between SSAE and LSAE mice. Community bar-plot analysis showed varies species composition among all mice at the genus level (Figure 2E). SSAE feces exhibited a considerable reduction in the relative abundance of several strains at the genus level (such as Dubosiella) when compared to LSAE feces, and significant increase in the relative abundance of norank_f_ norank_o__Clostridia__UCG-014 (Figure 2F).
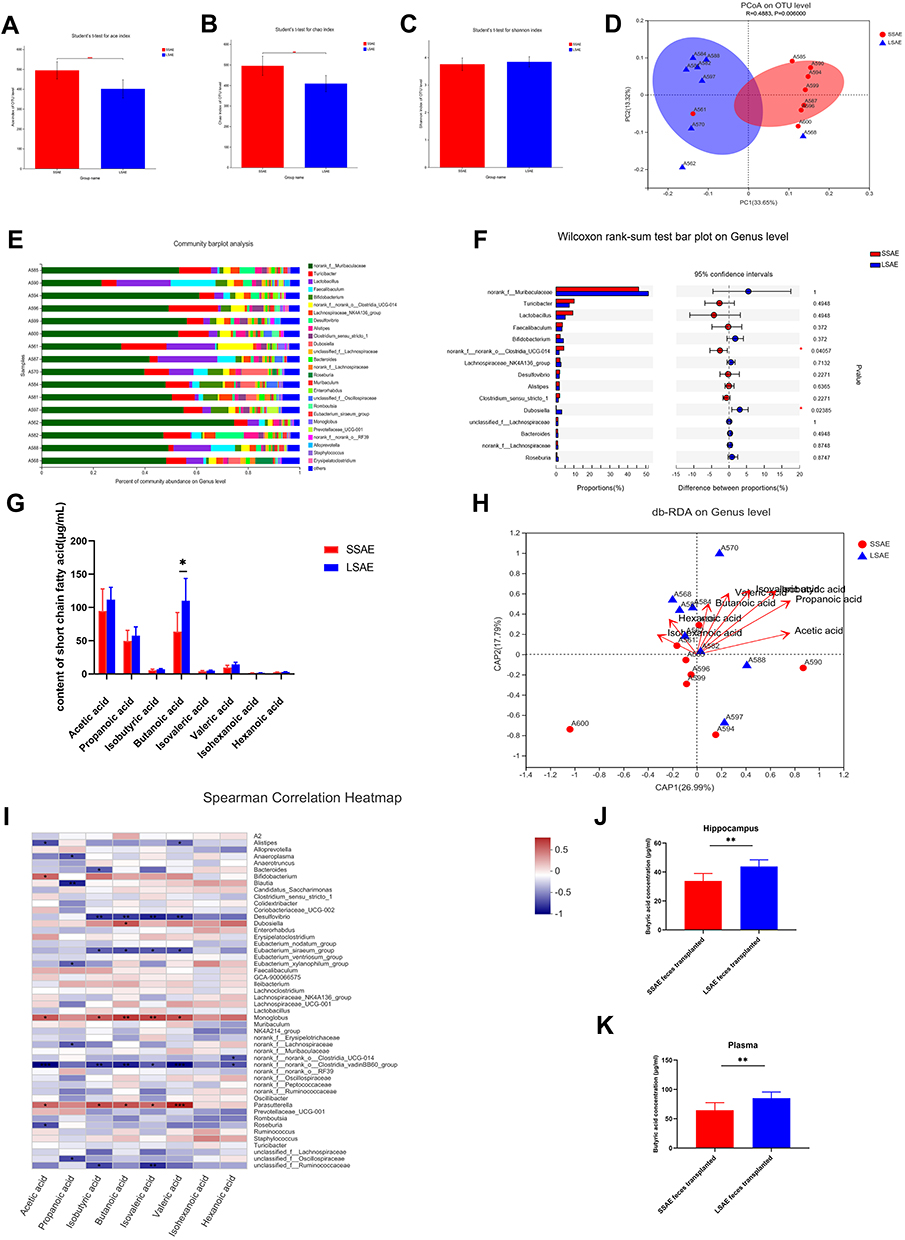 |
Figure 2 Differences in gut microbiota and its metabolites between SSAE group and LSAE group. Feces were collected from SSAE and LSAE mice before CLP. (A–C) α-diversity indices (Shannon, ACE, and Chao). (D) β-diversity assessed by PCoA analysis based on unweighted_unifrac distances. (E) Main bacterial genera. (F) Wilcoxon rank-sum test bar plot of bacterial genera. (G) Concentration of SCFAs in feces of mice in SSAE group and LSAE group. (H) db-RDA at genus level. (I) Spearman Correlation Heatmap. (J) Plasma butyric acid concentrations in sterile mice after SSAE and LSAE fecal transplantation measured 12 hours after CLP. (K) Butyric acid concentrations in the hippocampus of sterile mice after SSAE and LSAE fecal transplantation measured 12 hours after CLP. Data expressed as mean ± SD (n = 8). *P < 0.05, **P < 0.01 in A, B, C, G, J, K by Student’ t-test. |
For further conducted targeted SCFAs metabolomics analysis of the gut microbiota (feces) of SSAE and LSAE mice to explore the changes in acetic acid, propanoic acid, butanoic acid, valeric acid, and hexanoic acid (Figure 2G). SSAE mice were found to generate significantly less butyric acid than LSAE mice (P < 0.05). Spearman correlation heatmap showed positive correlation of several microbiota with production of butyric acid, including Dubosiella, Monoglobus and Parasutterella (Figure 2I). It has been reported that Parasutterella is related to butyric acid production.15 The db-RDA at the Genus level suggested that SCFAs were positively related to LSAE mice (Figure 2H). More interestingly, in the FMT experiment, butyric acid levels in plasma and hippocampus of SSAE feces recipients were both lower than those in LSAE feces recipients, indicating lesser production of butyric acid in the gut of SSAE feces recipients and its subsequent release in the circulatory system to the brain (Figure 2J–K). Our findings indicate distinct functional differences in gut microbiota between SSAE and LSAE mice, especially with respect to the production of metabolites.
NaB Reduces Mortality and Prevents Behavioral Failure in Septic Mice
From the first to the seventh day, the survival rate of sepsis mice gavaged with NaB was considerably higher than that of sepsis mice administered with saline (Figure 3A). To determine whether NaB entered blood and brain after intragastric administration, we identified the butyric acid level in the hippocampus and blood of mice. Butyric acid levels in plasma and hippocampus of NaB-treated CLP mice were significantly higher than those in saline-treated CLP mice (Figure 3B–C). We further assessed whether NaB improved behavioral abnormalities using the Morris Water Maze behavioral test. On the third day, the swimming speed of the NaB-treated group was faster than that of the CLP group; however, there was no significant between-group difference on the other days (Figure 3D). After CLP, the latency duration was much longer than in the Sham group, which was significantly reduced by NaB therapy (Figure 3E). We counted the number of platform crossings after training and after the platform was removed. The frequency of CLP mice crossing the platform was much lower than in the Sham group, whereas the number of crossings was significantly higher in the NaB group (Figure 3F). Mice in the CLP group followed a longer route to the platform than mice in the Sham and NaB groups, according to representative swimming pathways (Figure 3G). NaB-treated group showed alleviation of sepsis-induced cognitive deficit in learning and memory. The morphology of neurons in the hippocampi was normal in the Sham group, whereas significant neuronal degeneration was seen in CLP animals, according to H&E staining. The above histopathological changes were dissipated by treatment with NaB (Figure 3H). These results suggested that butyrate may have conferred a survival and behavioral benefit in SAE progression.
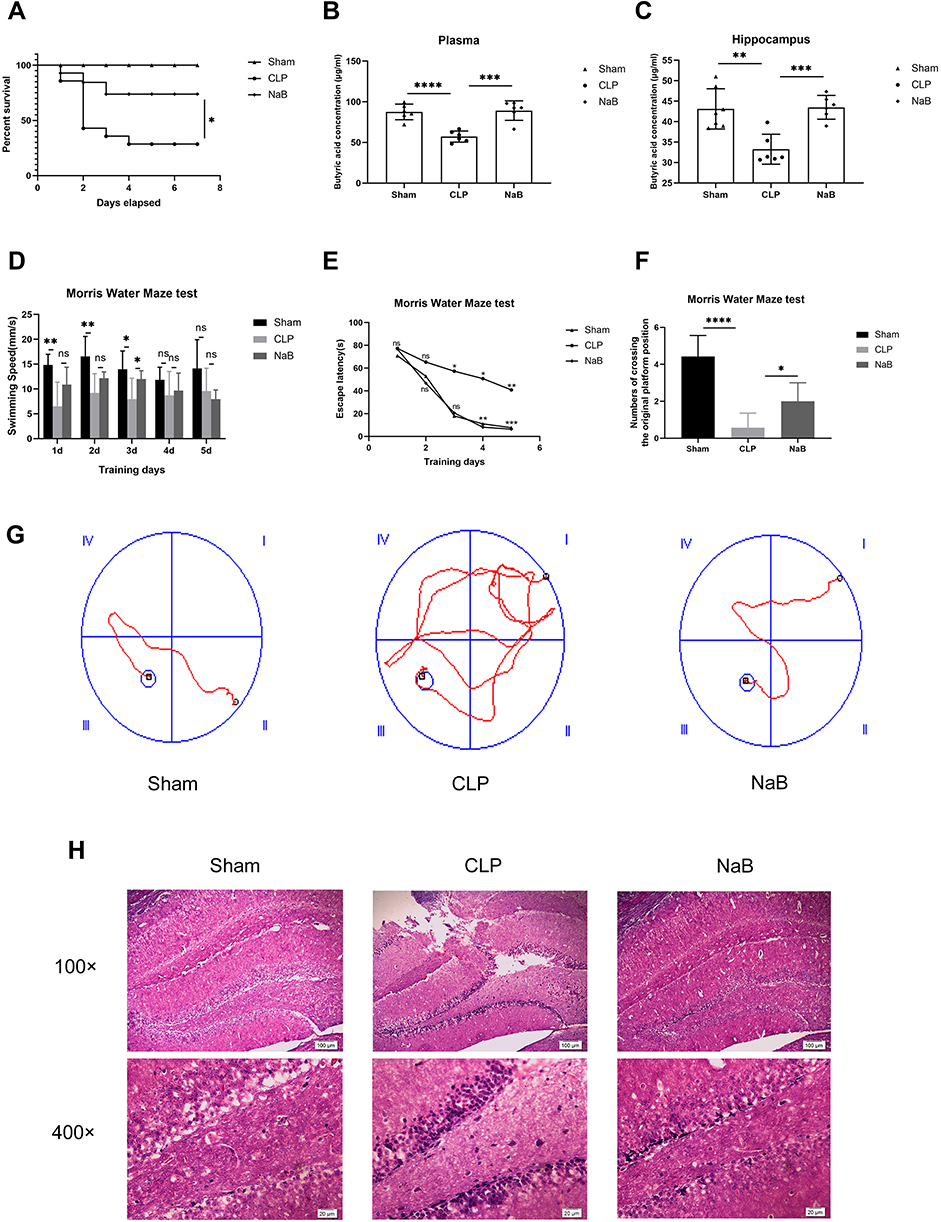 |
Figure 3 NaB promotes survival of CLP mice and alleviates cognitive dysfunction of CLP surviving mice. Intragastric administration of NaB increased the butyrate level in blood and hippocampus of CLP mice. (A) Changes in the survival rate. There were 20 animals in Sham group, CLP group, and CLP+NaB treatment (NaB) group. (B) Butyric acid concentration in plasma of mice on the 1st day. (C) Butyric acid concentration in the hippocampus of mice on the 1st day. (D and E) The escape latency and swimming speed on the 1st, 2nd, 3rd, 4th, and 5th day of the learning trials. (F) The frequency of crossing the original platform location during the probe trial on the 6th day. (G) Representative swimming paths of mice during the probe trial on day 6 are depicted. (H) Representative H&E-stained sections of hippocampus of mice on the 1st day. Data presented as mean ± SD (n =3–7 per group). Scale bars: 100µm and 20µm. *P < 0.05 in A by Chi-square test. * P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 in B, C, D, F by one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001 in E by two-way ANOVA. ns: no significance. |
NaB Protected Mice Against Sepsis-Induced Brain Damage
This study evaluated the NaB effect on oxidative stress. In comparison to the age-matched Sham group, CLP-induced sepsis reduced the SOD activity, and promoted the production of MDA and PGE2 in hippocampus. After intragastric administration of NaB, CLP mice showed increased SOD activity, along with a decrease in the MDA and PGE2 content (Figure 4B–D). The oxidative fluorescent dye DHE was used to assess ROS generation in brain tissues. When compared to age-matched Shams, CLP-induced sepsis leads to an obvious increase in fluorescence intensity in hippocampal tissues. The sepsis-induced rise in fluorescence intensity was dramatically reduced when NaB was administered (Figure 4A). IBA-1 labeling was performed to identify microglia in the Sham, CLP, and CLP+NaB (NaB) groups. IBA1+ cells in the hippocampus were distinctly amplified in the CLP group at day 1, however, were decreased in the NaB group. In the Sham group, immunofluorescence labeling revealed modest expressions of i-NOS and COX-2 in microglia. The CLP group, on the other hand, had a significant rise in i-NOS and COX-2 immunofluorescence. When compared to the CLP group, i-NOS and COX-2 immunofluorescence in microglia were substantially reduced in the NaB group (Figure 4E and F). On day 1 and day 3 following CLP surgery, Western blotting revealed a significant rise in the expressions of i-NOS and COX-2, but the expressions were decreased in the NaB group. Besides, we explored the effect of NaB on neurological impairment. CLP-induced sepsis inhibited the protein expression of BDNF, while NaB treatment significantly reversed the inhibitory effects (Figure 4G). Collectively, these findings indicated that butyrate attenuated sepsis-induced oxidative stress injury and neuronal injury in hippocampus.
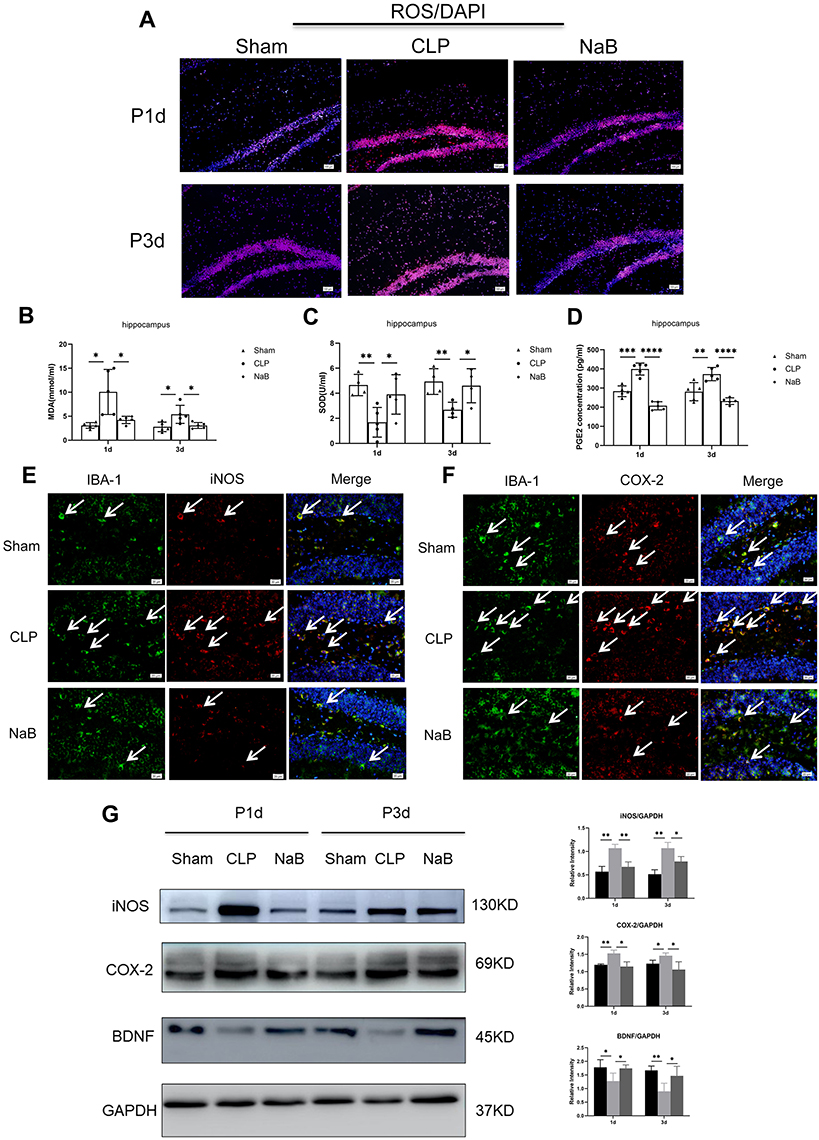 |
Figure 4 NaB treatment alleviates oxidative stress and nerve injury in hippocampus of mice with CLP. (A) Generation of reactive oxygen species (ROS) in brain tissues of mice. The oxidative fluorescence dye dihydroethidine hydrochloride (DHE) was used for measuring ROS production. (B) Malondialdehyde (MDA) content. (C) Superoxide dismutase (SOD) activity. (D) Expression of PGE2 in the hippocampus, as detected by ELISA. (E) Double immunofluorescence staining showing the distribution of IBA-1 labeled (green), iNOS (red), and DAPI (blue) immunoreactive microglial cells in the hippocampus at 1 day after CLP and their matching groups. Co-localized expression of IBA-1 and iNOS in microglia can be seen. (F) Double immunofluorescence staining showing the distribution of IBA-1 labeled (green), COX-2 (red), and DAPI (blue) immunoreactive microglial cells in the hippocampus at 1 day after CLP and their matching groups. Co-localized expression of IBA-1 and COX-2 in microglia can be seen. Arrows (→) indicate the cell nucleus. (G) Western blot analysis of iNOS, COX-2, and BDNF protein expression levels in the hippocampus of mice at 1st and 3rd day after CLP or NaB intragastric administration and their corresponding groups. GAPDH served as the loading control. Bar graphs depicting the optical density of iNOS, COX-2, and BDNF expressions. Data presented as mean ± SD (n = 3–5 per group). Scale bars: 100 μm and 20µm. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 by two-way ANOVA. |
NaB Inhibits Oxidative Stress by Activating Nrf2/HO-1 Pathway in Primary Microglia
The primary microglia were used in an in vitro investigation. IBA-1 labeling was exploited to identify primary microglia in the control, LPS, and LPS+NaB (NaB) groups. Immunofluorescence staining showed marked enhancement of the immunoreactivity of i-NOS and COX-2 at 12 hours after LPS challenge compared with the control group (Figure 5A and B). NaB treatment reversed this trend. The DCFHDA experiment revealed that when primary microglia were exposed to LPS, the amount of intracellular ROS increased significantly. In LPS-stimulated cells, treatment with NaB inhibited an increase in intracellular ROS generation (Figure 5C). To confirm the molecular mechanism of NaB’s anti-oxidative stress impact, we used Western blot analysis. After LPS treatment, the expression of Nrf2, HO-1, and NQO-1 was modestly up-regulated. After treatment with LPS+NaB, however, levels of HO-1, Nrf2, and NQO-1were increased significantly. The use of the Nrf2 inhibitor, ML385 indicated that the Nrf2 pathway is involved in mediating the action of NaB. Treatment with ML385 reversed the up regulation of Nrf2, NQO1, and HO-1 expressions induced by NaB (Figure 5D). We measured the levels of BDNF, COX-2, and i-NOS in primary microglia to see if NaB had a favorable effect on the Nrf2 pathway. The LPS-treated group showed marked up-regulation of i-NOS and COX-2 expressions and down-regulation of BDNF expression. Treatment with NaB significantly attenuated the up-regulation of expression of the i-NOS and COX-2 and increased the down-regulation of BDNF after LPS-treatment. Moreover, administration of ML385 reversed the trend (Figure 5D). These results suggested that butyrate exerts antioxidant activity in primary microglia through the Nrf2/HO-1/NQO1 pathway.
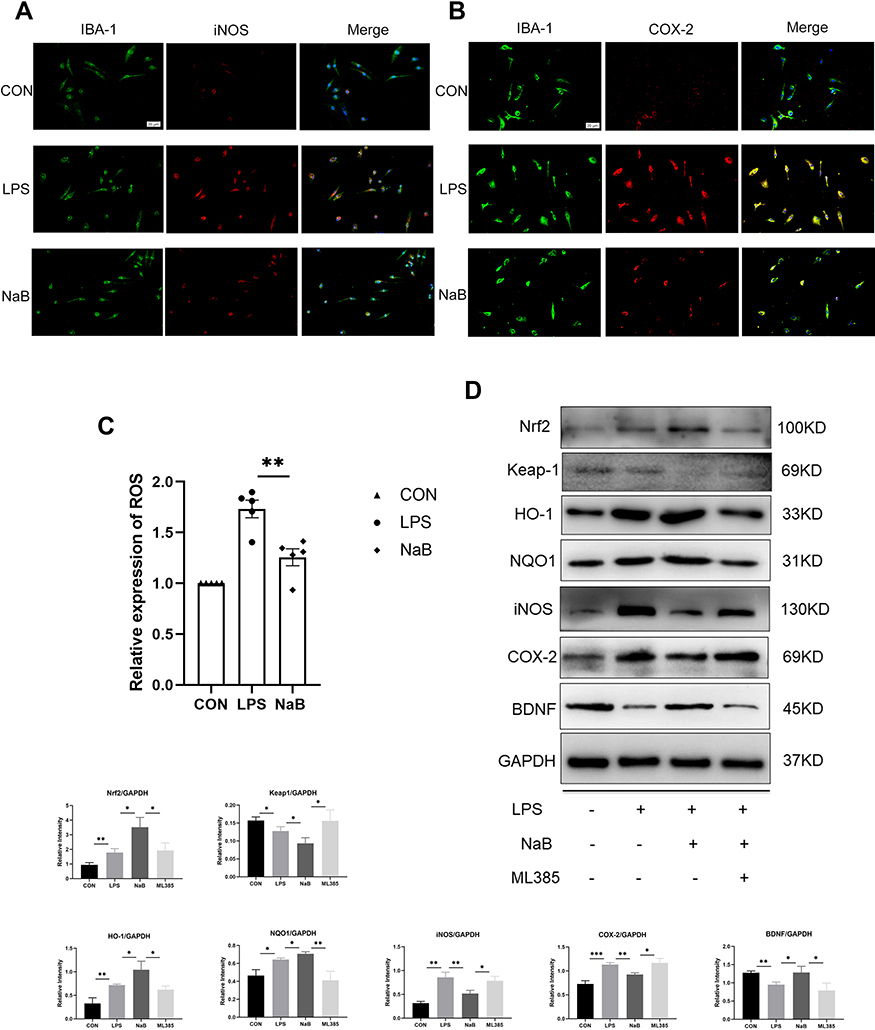 |
Figure 5 Effect of NaB on neuroinflammatory molecules and Nrf2/HO-1 signaling in LPS-stimulated primary microglia. Primary microglia were incubated with LPS, LPS+NaB, or LPS+NaB+ML385 for 12 hours. (A and B) Immunofluorescence images of cultured primary microglia showing expressions of IBA-1 (green), iNOS (red) or COX-2 (red) and DAPI (blue) at 12 h after LPS or LPS+NaB treatment when compared with the corresponding control. (C) NaB treatment inhibits the LPS-induced ROS production in primary microglia. Cells were analyzed with microplate reader. (D) Western blot analysis of Nrf2, Keap1, HO-1, NQO1, iNOS, COX-2, and BDNF protein expressions in the primary microglia at 12h after LPS, LPS+NaB, and LPS+NaB+ML385 and their corresponding controls. GAPDH served as the loading control. Bar graphs depicting the optical density of Nrf2, Keap1, HO-1, NQO1, iNOS, COX-2, and BDNF expressions. Data presented as mean ± SD (n = 3 per group). Scale bars: 20µm. *P < 0.05, **P < 0.01 by one-way ANOVA. |
NaB Ameliorates LPS-Induced Oxidative Stress in Primary Microglia Through GPR109A
Microglia have been confirmed to express GPR109A,16 which is the functional receptor of SCFAs. We investigated the expression of GPR109A in hippocampus by immunohistochemistry (Figure 6A). We further used G protein coupled receptor inhibitors PTX to investigate whether NaB works through this receptor. Western blotting showed downregulation of protein levels of Nrf2, HO-1, and NQO1 after use of PTX. This suggests that the protective effective of NaB may be partly mediated by binding to GPR109A (Figure 6B). Collectively, these findings suggest that the action of butyrate on antioxidant signaling may be mediated by the GPR109A receptor.
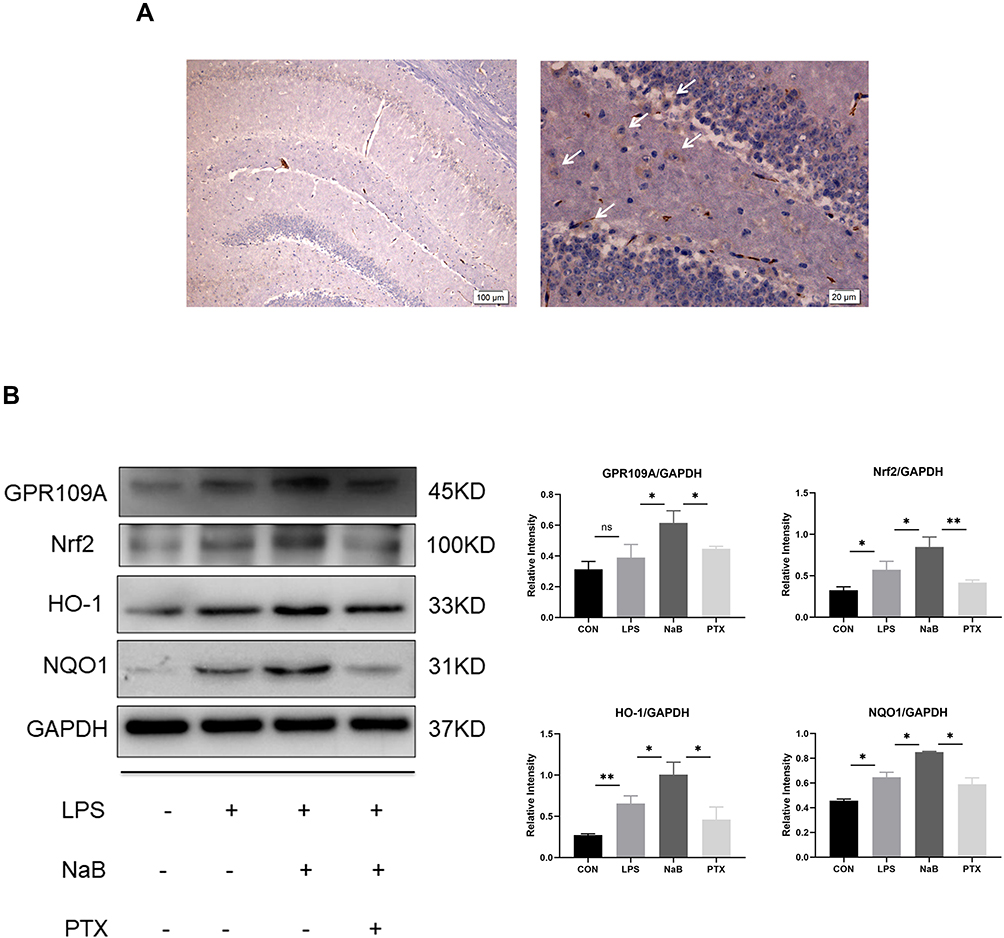 |
Figure 6 Effect of NaB on GPR109A/Nrf2/HO-1 signaling in LPS-stimulated primary microglia. (A) Immunohistochemical stained sections showing expression of GPR109A in hippocampus. Arrows (→) indicate the cell nucleus. (B) Western blot analysis of GPR109A, Nrf2, HO-1, and NQO1 protein expressions in the primary microglia at 12h after LPS, LPS+NaB, and LPS+NaB+PTX and their corresponding controls. GAPDH served as the loading control. Bar graphs depicting the optical density of Nrf2, GPR109A, HO-1, and NQO1 expressions. Data presented as mean ± SD (n = 3 per group). Scale bars: 100µm and 20µm. *P < 0.05, **P < 0.01 by one-way ANOVA. ns: no significance. |
Discussion
The goal of this research was to find out which metabolites from the host gut microbiota affect the susceptibility of mice to SAE. Mice with greater production of butyrate, one of the SCFAs, showed milder brain damage after CLP-induced sepsis, which was probably attributable to the existence of butyric acid-related bacteria and the protective effect of butyrate produced by these bacteria. The main mechanism is that butyrate partially activates GPR109A receptor on microglia, leading to activation of downstream Nrf2/HO-1 signaling pathway, thereby reducing oxidative stress response and neural lesion, ultimately alleviating the long-term cognitive impairment of SAE. Our findings provide a microbial connection to neuroprotective processes, which may be used as a curative target for SAE treatment and prevention.
Studies have revealed that gut microbiota transplantation can transfer host traits and even promote neurogenesis in the brain.17,18 In the FMT experiment, we transplanted the feces of SSAE or LSAE mice into the sterile mice treated with antibiotics. The changed morphology of hippocampal neurons demonstrated severe neuronal degeneration in the brains of germ-free mice transplanted with SSAE feces, as revealed by histopathological investigation (swollen cell with deeply stained and pyknotic nuclei). However, these findings were not obvious in LSAE mice. These histopathological changes point towards presence of critical factors in the gut bacteria of LSAE mice that protect against the development of SAE. The FMT experiment confirmed a close relationship between gut and brain in the setting of SAE. In other words, the difference of gut microbiota and metabolites affects the host susceptibility to sepsis-related brain injury.
We uniformly collected fecal samples from each mouse prior to CLP. One of the most interesting findings of the current research was that mice were housed in the same environment, the results of 16S sequencing showed the diversity index (Shanno) did not change much, while significant differences in the richness index (Chao and ace) in α-diversity. This indicated that the species of microbiota of each group of mice were similar, but their proportion and composition was markedly diverse. Similar to the previous research results, gut microbiota clusters between SSAE and LSAE mice were slightly different at the β-diversity level, but all characteristics of the microbiota may not be necessarily identical.19 For example, the proportion of each microbe and its metabolomics profile may not be parallel, which imply that there were different metabolic end-products for diverse microbial composition in mice. Our results showed that some mice have strong resistance to SAE, while others were very intolerant. This phenomenon may be attributable to the presence of key factors in microbial components, resulting in differential response to inflammation in each mouse. The sequencing results indicated that SSAE group had less Dubosiella colonies and produced less butyric acid than LSAE group. Furthermore, the heatmap analysis showed a positive correlation of Dubosiella with the production of butyric acid. Butyric acid is generated in the gut, absorbed in the blood circulation, enters the hippocampus tissue by means of the BBB, and acts directly on the hippocampus. Reviewing the FMT experiments, which provided germ-free mice with all kinds of SCFAs through fecal transplants. We do not deny the possible role of other SCFAs, however based on the above-mentioned sequencing analysis results of preoperative feces sample, it was found that only the content of butyrate was high and significantly different. Therefore, we speculate that the butyrate may have more than just the proven therapeutic effects of SCFAs, difference in butyric acid content in mice may have led to the inconsistent susceptibility of mice to SAE, ie, mice with lower butyric acid content developed more severe cerebral damage. This implies that existence of specific microbiota and the production of butyrate ultimately lead to different outcomes in mice after SAE.
Butyrate has been showed to inhibit the inflammatory reaction associated with diseases including inflammatory bowel disease and uveitis.20,21 Also sodium butyrate (NaB) has been reported to prevent the lethality of severe sepsis by inhibiting HMGB1 expression.22 Our study further explores the pivotal role of butyrate in sepsis-related neuroinflammation and its unknown mechanism. After 7 days of gavage NaB in mice, CLP was used to establish a sepsis model to verify whether a large amount butyric acid could increase the body’s resistance to sepsis, resist neuroinflammatory damage and cognitive impairment caused by sepsis. Intragastric administration of NaB to CLP-induced sepsis mice inhibited oxidative stress response and neuro-degradation, which is consistent with the beneficial effect observed in germ-free mice receiving gut microbiota transplant from LSAE mice. In addition, in our study, NaB treatment conferred a significant survival advantage and alleviated subsequent cognitive impairment in CLP mice, which may be related to the amelioration of sepsis-related hippocampal injury by NaB. Fluorescence double staining of brain tissue sections revealed that microglial marker protein IBA-1 co-localized with oxidative stress related protein i-NOS or COX-2, supporting microglia are the main source of oxidation-related genes. Western blotting results quantified sepsis-induced increased expression of oxidative products and decreased expression of neurotrophic factors, which were reversed by NaB pretreatment in vivo. These results indicate a critical role of butyrate in the microbe - mediated phenotypes, which is in accord with previous study that showed butyrate-induced improvement in neural lesions in brain.23,24
Butyrate was shown to act on microglia in the present study. Microglia are CNS resident cells that, as peripheral macrophages, exert a significant role in immune surveillance and homeostasis.25 In our previous study, we found that sepsis-induced brain injury has a bearing on microglia activation, diffuse reactive astrocyte proliferation, and oligodendrocyte progenitor cell apoptosis.26–28 It is worth mentioning that hippocampal microglia activation has been connected to cognitive impairment.29,30 Activated microglia have been shown to exhibit increased secretion of ROS, nitric oxide (NO), and superoxide radical.31,32 We found that NaB significantly inhibited LPS-induced ROS generation, high expression of i-NOS and COX-2 expressions, and low expression of BDNF expression in vitro. It has been reported that butyrate inhibits the secretion of IL-6 and TNF-α from primary brain-derived microglia via NF-κB complex.33 Our experiments further confirmed the relationship between the butyrate response and ROS production.
One of the fundamental factors in the pathophysiology of SAE is oxidative stress (OS).34,35 The Nrf2 pathway is involved in the regulation of oxidative stress-induced nerve injury.36–39 It has an antioxidant impact because it upregulates several protective genes like HO-1 and NQO-1. GPR109A, a seven-transmembrane G-protein-coupled receptor of Gi family, has been discovered as an endogenous ligand of SCFAs.40,41 Therefore, we speculate that butyrate may act directly on primary microglia through the GPR109A/Nrf2/HO-1 pathway for inhibition of oxidative stress associated protein proteins that may contribute to its neuroprotective results in vivo. Interestingly, compared with LPS group, NaB treatment significantly activated Nrf2 expression, further increasing the expressions of HO-1 and NQO1, while inhibiting the expression of Keap1. In line with expectations, the Nrf2 inhibitor, ML385, reduced the protection of NaB on LPS-stimulated primary microglia when administered. Furthermore, NaB treatment led to significant activation of GPR109A. PTX, an inhibitor of G-protein coupled receptor, inhibits the Nrf2, HO-1 activation, and NQO1 caused by NaB by blocking GPR109A. Briefly stated, these findings prove that the positive affect of NaB is mediated through the GPR109A/Nrf2/HO-1 pathway.
In conclusion, to the best of our knowledge, this is the first study to demonstrate that gut microbiota is related to the occurrence and development of SAE, especially its metabolite butyric acid plays a major protective role. Butyrate is a key metabolic factor of oxidative and nitrosative stress in CLP-induced sepsis model and LPS-stimulated primary microglia. These findings enrich our comprehension of “gut-brain axis” in the development of sepsis and provide novel insights for the treatment of SAE.
Abbreviations
SAE, sepsis-associated encephalopathy; LPS, lipopolysaccharide; IBA1, Ionized calcium binding adapter molecule 1; i-NOS, inducible nitric oxide synthase; COX-2, cyclooxygenase-2; BDNF, brain-derived neurotrophic factor; Nrf2, nuclear factor E2-related factor 2; Keap-1, Kelch like ECH associated protein 1; HO-1, heme oxygenase-1; NQO1, NAD(P) H:quinone oxidoreductase 1; GPR109A, G-protein coupled receptor 109A; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NaB, sodium butyrate; SOD, superoxide dismutase; MDA, malondialdehyde; ROS, reactive oxygen species; PGE2, prostaglandin E2; DHE, dihydroethidine hydrochloride; CLP, cecal ligation and perforation; FMT, fecal microbiota transplantation; BBB, blood-brain barrier; CNS, central nervous system; SCFAs, short chain fatty acids; DMEM, Dulbecco’s modified eagle medium; FBS, fetal bovine serum; BCA, bicinchoninic acid; BSA, bovine serum albumin; DAPI, 4′6-diamidino-2-phenylindole; TBS, Tris-buffered saline; TBST, Tris-buffered saline Tween-20.
Ethics Statement
The method of animal handling in this experiment conforms to the standards of animal ethics. Approved by the Medical Research Ethics Committee of Guangdong People’s Hospital (Guangdong Academy of Medical Sciences) (Ethics number: GDREC2018199A) (Animal Certificate No.: SCXK2020-0054).
Acknowledgments
Huidan Zhang, Jing Xu and Qingrui Wu dedicated themselves to the same degree to this study and are co-first authors. This study was fiscally assisted by the Science and Technology Program of Guangzhou City (No. 201803010058), Maoming Science and Technology Project (No. 2020255), the major program of Summit Project, Guangdong Province High-level Hospital Construction Project of Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences (No. DFJH2020028).
Disclosure
The authors report no conflicts of interest for this work.
References
1. Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol. 2012;8(10):557–566. doi:10.1038/nrneurol.2012.183
2. Feng Q, Ai YH, Gong H, et al. Characterization of sepsis and sepsis-associated encephalopathy. J Intensive Care Med. 2019;34(11–12):938–945. doi:10.1177/0885066617719750
3. Andonegui G, Zelinski EL, Schubert CL, et al. Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI Insight. 2018;3(9). doi:10.1172/jci.insight.99364
4. Benakis C, Martin-Gallausiaux C, Trezzi JP, Melton P, Liesz A, Wilmes P. The microbiome-gut-brain axis in acute and chronic brain diseases. Curr Opin Neurobiol. 2020;61:1–9. doi:10.1016/j.conb.2019.11.009
5. Cerdo T, Dieguez E, Campoy C. Impact of gut microbiota on neurogenesis and neurological diseases during infancy. Curr Opin Pharmacol. 2020;50:33–37. doi:10.1016/j.coph.2019.11.006
6. Singer BH, Dickson RP, Denstaedt SJ, et al. Bacterial dissemination to the brain in sepsis. Am J Respir Crit Care Med. 2018;197(6):747–756. doi:10.1164/rccm.201708-1559OC
7. Sonneville R, de Montmollin E, Poujade J, et al. Potentially modifiable factors contributing to sepsis-associated encephalopathy. Intensive Care Med. 2017;43(8):1075–1084. doi:10.1007/s00134-017-4807-z
8. Morrow LE, Wischmeyer P. Blurred lines: dysbiosis and probiotics in the ICU. Chest. 2017;151(2):492–499. doi:10.1016/j.chest.2016.10.006
9. Valdes-Duque BE, Giraldo-Giraldo NA, Jaillier-Ramirez AM, et al. Stool short-chain fatty acids in critically ill patients with sepsis. J Am Coll Nutr. 2020;39(8):706–712. doi:10.1080/07315724.2020.1727379
10. Hecker M, Sommer N, Voigtmann H, et al. Impact of short- and medium-chain fatty acids on mitochondrial function in severe inflammation. J Parenter Enteral Nutr. 2014;38(5):587–594. doi:10.1177/0148607113489833
11. Liu J, Jin Y, Ye Y, et al. The neuroprotective effect of short chain fatty acids against sepsis-associated encephalopathy in mice. Front Immunol. 2021;12:626894. doi:10.3389/fimmu.2021.626894
12. Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. doi:10.1038/nature09922
13. Gregory JC, Buffa JA, Org E, et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem. 2015;290(9):5647–5660. doi:10.1074/jbc.M114.618249
14. Li L, Yao H, Li X, et al. Destiny of dendrobium officinale polysaccharide after oral administration: indigestible and nonabsorbing, ends in modulating gut microbiota. J Agric Food Chem. 2019;67(21):5968–5977. doi:10.1021/acs.jafc.9b01489
15. Sun S, Yang Y, Lin X, et al. Qiweibaizhu decoction treats diarrheal Juvenile rats by modulating the gut microbiota, short-chain fatty acids, and the mucus barrier. Evid Based Complement Alternat Med. 2021;2021:8873294. doi:10.1155/2021/8873294
16. Fu SP, Wang JF, Xue WJ, et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J Neuroinflammation. 2015;12(1):9. doi:10.1186/s12974-014-0230-3
17. Kundu P, Lee HU, Garcia-Perez I, et al. Neurogenesis and prolongevity signaling in young germ-free mice transplanted with the gut microbiota of old mice. Sci Transl Med. 2019;11(518):518. doi:10.1126/scitranslmed.aau4760
18. Diao H, Yan HL, Xiao Y, et al. Erratum to: intestinal microbiota could transfer host Gut characteristics from pigs to mice. BMC Microbiol. 2016;16(1):253. doi:10.1186/s12866-016-0879-z
19. Gong S, Yan Z, Liu Z, et al. Intestinal microbiota mediates the susceptibility to polymicrobial sepsis-induced liver injury by granisetron generation in mice. Hepatology. 2019;69(4):1751–1767. doi:10.1002/hep.30361
20. Chen G, Ran X, Li B, et al. Sodium butyrate inhibits inflammation and maintains epithelium barrier integrity in a TNBS-induced inflammatory bowel disease mice model. EBioMedicine. 2018;30:317–325. doi:10.1016/j.ebiom.2018.03.030
21. Chen X, Su W, Wan T, et al. Sodium butyrate regulates Th17/Treg cell balance to ameliorate uveitis via the Nrf2/HO-1 pathway. Biochem Pharmacol. 2017;142:111–119. doi:10.1016/j.bcp.2017.06.136
22. Zhang LT, Yao YM, Lu JQ, Yan XJ, Yu Y, Sheng ZY. Sodium butyrate prevents lethality of severe sepsis in rats. Shock. 2007;27(6):672–677. doi:10.1097/SHK.0b013e31802e3f4c
23. Val-Laillet D, Guerin S, Coquery N, et al. Oral sodium butyrate impacts brain metabolism and hippocampal neurogenesis, with limited effects on gut anatomy and function in pigs. FASEB J. 2018;32(4):2160–2171. doi:10.1096/fj.201700547RR
24. Bayazid AB, Jang YA, Kim YM, Kim JG, Lim BO. Neuroprotective effects of sodium butyrate through suppressing neuroinflammation and modulating antioxidant enzymes. Neurochem Res. 2021;46(9):2348–2358. doi:10.1007/s11064-021-03369-z
25. Fu R, Shen Q, Xu P, Luo JJ, Tang Y. Phagocytosis of microglia in the central nervous system diseases. Mol Neurobiol. 2014;49(3):1422–1434. doi:10.1007/s12035-013-8620-6
26. Huang P, Zhou Q, Lin Q, et al. Complement C3a induces axonal hypomyelination in the periventricular white matter through activation of WNT/beta-catenin signal pathway in septic neonatal rats experimentally induced by lipopolysaccharide. Brain Pathol. 2020;30(3):495–514. doi:10.1111/bpa.12798
27. Zhou Q, Lin L, Li H, et al. Melatonin reduces neuroinflammation and improves axonal hypomyelination by modulating M1/M2 microglia polarization via JAK2-STAT3-telomerase pathway in postnatal rats exposed to lipopolysaccharide. Mol Neurobiol. 2021;58(12):6552–6576. doi:10.1007/s12035-021-02568-7
28. Deng Y, Xie D, Fang M, et al. Astrocyte-derived proinflammatory cytokines induce hypomyelination in the periventricular white matter in the hypoxic neonatal brain. PLoS One. 2014;9(1):e87420. doi:10.1371/journal.pone.0087420
29. Michels M, Danielski LG, Dal-Pizzol F, Petronilho F. Neuroinflammation: microglial activation during sepsis. Curr Neurovasc Res. 2014;11(3):262–270. doi:10.2174/1567202611666140520122744
30. Feng X, Valdearcos M, Uchida Y, Lutrin D, Maze M, Koliwad SK. Microglia mediate postoperative hippocampal inflammation and cognitive decline in mice. JCI Insight. 2017;2(7):e91229. doi:10.1172/jci.insight.91229
31. Kawakami M, Hattori M, Ohashi W, et al. Role of G protein-coupled receptor kinase 2 in oxidative and nitrosative stress-related neurohistopathological changes in a mouse model of sepsis-associated encephalopathy. J Neurochem. 2018;145(6):474–488. doi:10.1111/jnc.14329
32. Yokoo H, Chiba S, Tomita K, et al. Neurodegenerative evidence in mice brains with cecal ligation and puncture-induced sepsis: preventive effect of the free radical scavenger edaravone. PLoS One. 2012;7(12):e51539. doi:10.1371/journal.pone.0051539
33. Huuskonen J, Suuronen T, Nuutinen T, Kyrylenko S, Salminen A. Regulation of microglial inflammatory response by sodium butyrate and short-chain fatty acids. Br J Pharmacol. 2004;141(5):874–880. doi:10.1038/sj.bjp.0705682
34. Mei X, Feng H, Shao B. Alleviation of sepsis-associated encephalopathy by ginsenoside via inhibition of oxidative stress and cell apoptosis: an experimental study. Pak J Pharm Sci. 2020;33(6):2567–2577.
35. Han YG, Qin X, Zhang T, et al. Electroacupuncture prevents cognitive impairment induced by lipopolysaccharide via inhibition of oxidative stress and neuroinflammation. Neurosci Lett. 2018;683:190–195. doi:10.1016/j.neulet.2018.06.003
36. Liu D, Wang H, Zhang Y, Zhang Z. Protective effects of chlorogenic acid on cerebral ischemia/reperfusion injury rats by regulating oxidative stress-related Nrf2 pathway. Drug Des Devel Ther. 2020;14:51–60. doi:10.2147/DDDT.S228751
37. Yoo JM, Lee BD, Sok DE, Ma JY, Kim MR. Neuroprotective action of N-acetyl serotonin in oxidative stress-induced apoptosis through the activation of both TrkB/CREB/BDNF pathway and Akt/Nrf2/Antioxidant enzyme in neuronal cells. Redox Biol. 2017;11:592–599. doi:10.1016/j.redox.2016.12.034
38. Xiong W, MacColl Garfinkel AE, Li Y, Benowitz LI, Cepko CL. NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J Clin Invest. 2015;125(4):1433–1445. doi:10.1172/JCI79735
39. Lei L, Chai Y, Lin H, et al. Dihydroquercetin activates AMPK/Nrf2/HO-1 signaling in macrophages and attenuates inflammation in LPS-induced endotoxemic mice. Front Pharmacol. 2020;11:662. doi:10.3389/fphar.2020.00662
40. Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. 2014;121:91–119.
41. Guo W, Liu J, Sun J, et al. Butyrate alleviates oxidative stress by regulating NRF2 nuclear accumulation and H3K9/14 acetylation via GPR109A in bovine mammary epithelial cells and mammary glands. Free Radic Biol Med. 2020;152:728–742. doi:10.1016/j.freeradbiomed.2020.01.016
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.