Back to Journals » International Journal of Nanomedicine » Volume 11
Guanidinylated bioresponsive poly(amido amine)s designed for intranuclear gene delivery
Authors Yu J, Zhang J, Xing H, Yang Z, Cai C, Zhang C, Zhao X, Wei M, Yang L, Ding P
Received 28 March 2016
Accepted for publication 19 June 2016
Published 17 August 2016 Volume 2016:11 Pages 4011—4024
DOI https://doi.org/10.2147/IJN.S109406
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Jiankun Yu,1 Jinmin Zhang,1 Haonan Xing,1 Zhen Yang,1 Cuifang Cai,1 Conglu Zhang,1 Xiaoyun Zhao,1 Minjie Wei,2 Li Yang,1 Pingtian Ding1
1School of Pharmacy, Shenyang Pharmaceutical University, 2School of Pharmacy, China Medical University, Shenyang, People’s Republic of China
Abstract: Guanidinylated poly(amido amine)s with multiple disulfide linkages (Gua-SS-PAAs) were designed and constructed as nonviral gene carriers. The main chains of these novel carriers were synthesized based on monomers containing guanidino groups (guanidine hydrochloride and chlorhexidine), which could avoid complicated side-chain-modification reactions while introducing the guanidino groups. The synthesized Gua-SS-PAAs polymers were characterized by 1H nuclear magnetic resonance, molecular weight, and polydispersity. Furthermore, Gua-SS-PAAs polymers were complexed with pDNA, and the properties of the complexes were determined, including entrapment efficiency, particle size, ζ-potential, atomic force microscopy images, stability, DNA complexation ability, reduction sensitivity, cytotoxicity, and transfection efficiency. The new Gua-SS-PAAs carriers exhibited higher transfection efficiency and lower cytotoxicity compared with two widely used gene delivery carriers, polyethylenimine and lipofectamine 2000. Furthermore, the relationship between the side-chain structure and morphological/biological properties was extrapolated, and the results showed that guanidine in the side chain aids in the improvement of transfection efficiency. In addition, the introduction of guanidino group might confer the new carriers with nuclear localization function compared to carriers without it.
Keywords: gene carrier, transfection efficiency, cytotoxicity, nuclear localization, guanidine
Introduction
The main challenge in gene therapy is the design of a suitable carrier for safe and efficient delivery of therapeutic genes.1 Carriers for gene delivery can be divided into two classes: viral and nonviral. Viral carriers are modified viruses that can deliver exogenous genes to the cells. Although viral carriers exhibit high transfection efficiency, they possess some defects that are difficult to overcome, such as low gene loading capacity, difficulty in production and quality control, and, especially, immune responses leading to serious drug safety issues.2 In contrast, nonviral carriers have high gene loading capacity with no or lower host immunogenicity. At the same time, nonviral carriers can be obtained through chemical synthesis with controllable quality, although they normally possess lower transfection efficiency compared to viral carriers.3
Nonviral carriers are mainly defined as various polymers that can connect to or complex with exogenous genes, deliver them to cytoplasm, and release the exogenous gene fragments through the reactions in cell membranes or organelles mediated by the complexes.4 Currently, the two most popular nonviral gene delivery systems have been widely used in laboratory and clinical studies: 1) using electroporation to transfect cells,5–7 and 2) cationic polymers, such as cationic liposomes and polyethylenimines (PEIs).8 Poly(amido amine)s (PAAs) with multiple disulfide linkages (SS-PAAs) are the most promising alternatives for liposomes and PEIs.9 The multi-amino structure confers the complexes with good cell membrane permeability and facilitates them to escape from subcellular structure degradation (such as endosomes and lysosomes) due to the amino proton sponge effect.10 The multiple disulfide linkages in the main chains lead to reduction in sensitivity due to the reductive environment in cytoplasm including lysosomes and endosomes, resulting in the rapid release of gene fragments in the intracellular environment.11 In addition, both quality control and industrial production of SS-PAAs nonviral carriers are easy to achieve because of the concise synthetic routes and mild reaction conditions.
Lin et al12 reported that, compared to linear polymers, branched SS-PAAs show higher transfection efficiency and lower cytotoxicity. Their studies further showed that increased transfection efficiency and decreased cytotoxicity were related to the buffering capacity and charge density in the polymer side chains. Martello et al13 synthesized a series of SS-PAAs using various amino-containing monomers and disulfide-containing cross-linking agents and investigated the relationship between different end groups of SS-PAAs and in vitro transfection efficiency. Zhang et al11 found that the number of disulfide bonds in the main chains of SS-PAAs had an impact on their transfection efficiency. Merdan et al15 and Lin et al16 reported that PEGylated SS-PAAs enhanced the colloidal stability, reduced the interactions with blood components, and decreased the cytotoxicity of the complexes compared with SS-PAAs. However, PEGylated SS-PAAs also showed lower transfection efficiency than SS-PAAs. Salmaso et al17 and Li et al14 investigated the effect of cell membrane thiols and reduction-triggered disassembly on the transfection activity of bioreducible polyplexes, which revealed that the plasma membrane protein thiols played a key role in the observed enhancement of DNA transfection.
Guanidine is one of the strongest organic bases with some biological activities.18 Nitrogen and hydrogen atoms in the guanidino group have a strong affinity to carbonic acid esters, phosphate esters, and peptides, leading to the formation of hydrogen bonds with certain geometric configurations. The amino moiety of guanidine has the ability to react with various acids in the human body to form water-soluble salts.19 As a result, a drug containing guanidino groups could be easily transported with selectivity in in vivo absorption and/or permeation. With good permeability and low toxicity, guanidine and the polymer containing guanidino groups have been widely used as bacteriostatic agents, antibacterial finishing agents, and food additives in pharmaceutical settings, and also in textile industries.17 Compared to cationic liposomes, the guanidinylation of PEIs and poly(β-amino ester)s showed superior transfection efficiency and reduced cytotoxicity, as reported by Nimesh and Chandra20 and Kim et al.21 Kim et al22 further showed that the improved transfection efficiency of guanidinylated gene carriers was due to, 1) the good cell membrane permeability mediated by the charged nature of guanidino groups, and 2) the peptidomimetic nuclear localization effect of guanidinylated polymeric gene carriers.
To better address these issues in the search for nonviral gene carriers, guanidinylated SS-PAAs (Gua-SS-PAAs) gene carriers were designed and constructed in our laboratory. The main chains of the novel carriers were constructed based on monomers containing guanidino groups, which were quite different from the existing carriers without guanidino groups. This approach could avoid complicated side-chain modification reactions while introducing the guanidino groups.23 These newly synthesized Gua-SS-PAAs showed high transfection efficiency and low cytotoxicity, suggesting that these polymers could serve as potential delivery carriers in gene therapy.
Materials and methods
Materials
Branched PEI (water free) with a molecular weight of 25 kDa, L-glutathione reduced (GSH), and thiazolyl blue tetrazolium bromide (MTT) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Lipofectamine 2000 (Lipo) reagent, Dulbecco’s Modified Eagle’s Medium, fetal bovine serum (FBS), penicillin, streptomycin, and trypsin were purchased from Thermo Fisher Scientific (Waltham, MA, USA). N, N′-Cystamine bisacrylamide (CBA) was purchased from Alfa Aesar (Ward Hill, MA, USA). TIANpure Midi Plasmid Kit was obtained from TIANGEN Biotech (Beijing) Co., Ltd. (Beijing, People’s Republic of China). Agarose was purchased from Thermo Fisher Scientific. Hoechst 33342 and 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) were purchased from Beyotime Institute of Biotechnology (Shanghai, People’s Republic of China). pEGFP-N3 vector strain was kindly provided by the Department of Pharmacology, China Medical University (CLONTECH Laboratories, Palo Alto, CA, USA). Lyso-ID Red detection kit and TOTAL NUCLEAR-ID Green/Red nucleolar/nuclear detection kits were purchased from Enzo Life Sciences (East Farmingdale, NY, USA). The 2,2,4,6,7-Pentamethyldihydrobenzofuran-5-sulfonyl chloride (Pbf-Cl), guanidine hydrochloride (CAR), and chlorhexidine (CHL) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, People’s Republic of China). Acetone, N, N-dimethylformamide, and other common reagents were of analytical grade.
Synthesis of Gua-SS-PAAs
Gua-SS-PAAs were synthesized by the following three reactions: 1) CAR or CHL acetate (10.46/1.60 mmol, 1.00/1.00 g) was dissolved in water (20 mL) and added to a three-necked flask. After the solution was cooled to 0°C–3°C, a Pbf-Cl/acetone solution (10 mL, 10.46/6.58 mmol, 3.02/1.90 g) was added dropwise to the flask. After the solution addition was completed, the reaction mixture was stirred continuously at room temperature for 3 hours. Throughout the course of the reaction, the pH of the system was maintained at 11–12 with an NaOH solution (4 M). Then, the resulting white precipitate was collected through filtration and washed three times with water; 2) Gua-SS-PAAs-Pbf-Cl was synthesized by Michael addition between CBA and CAR/CHL–Pbf-Cl according to a previous report.24 After oven-drying overnight, the white precipitate (CAR–Pbf-Cl/CHL–Pbf-Cl, 0.76 mmol, and 0.24/0.60 g) and CBA (0.38 mmol, 0.1 g) were added to a brown flask and dissolved with N, N-dimethylformamide (5 mL). The reaction solution was then placed in a temperature-controlled water bath at 60°C under nitrogen atmosphere and was stirred continuously in the dark for at least 6 days. Subsequently, 10% excess CAR/CHL–Pbf-Cl was added to consume any unreacted acrylamide groups, and the reaction was allowed to continue for another 2 days; 3) Ten milliliters of trifluoroacetic acid–triisopropylsilane–water was added to the flask with the volume ratio of 95:2.5:2.5 and then stirred at room temperature for 3 hours.25 After that, the solution was diluted with water to 30 mL and then dialyzed against distilled water (MWCO: 1,000) for 2 days to remove the unreacted monomers. Finally, the solution was lyophilized for 36 hours to obtain the product. The synthesis route is shown in Figure 1.
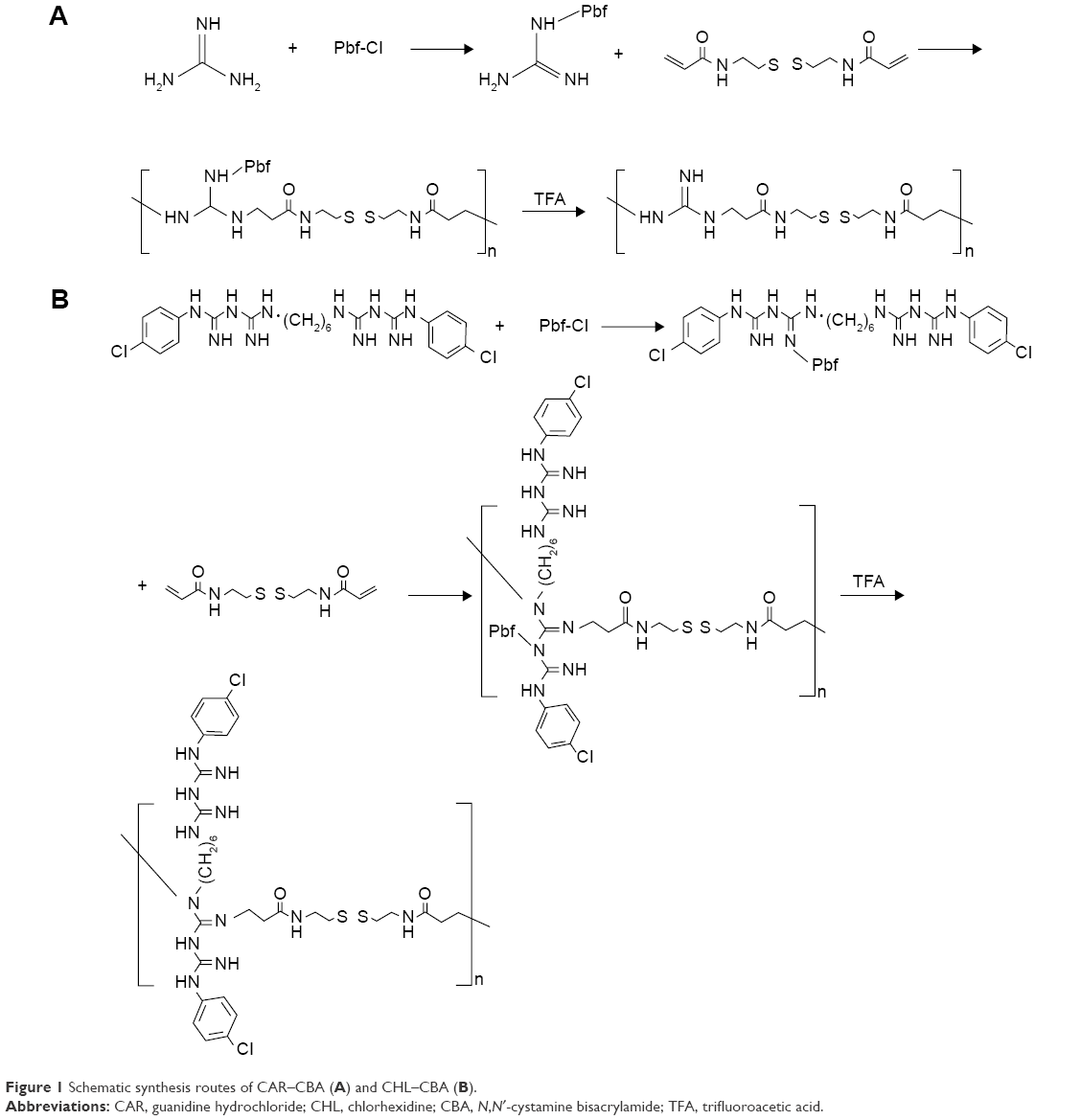 | Figure 1 Schematic synthesis routes of CAR–CBA (A) and CHL–CBA (B). |
Polymer characterization
1H nuclear magnetic resonance (NMR) spectra of the prepared Gua-SS-PAAs were recorded using a Varian Unity 600 MHz NMR spectrometer (Bruker Corporation, Billerica, MA, USA) with deuterated dimethyl sulfoxide (DMSO).
Molecular weights and polydispersity of the Gua-SS-PAAs were measured by gel permeation chromatography with PEG as standard using a Waters 2690D HPLC system equipped with Ultrahydrogel 120 and 250 columns (Waters Corporation, Milford, MA, USA). An HAc–NaAc buffer solution (0.25 M, pH 4.5) was used as the eluent at a flow rate of 0.7 mL/min.
Preparation of Gua-SS-PAAs–pEGFP nanocomplexes
The plasmid DNA (pEGFP-N3) was extracted from the pEGFP-N3 vector strain using the TIANpure Midi plasmid extraction kit and diluted to a final concentration of 20 μg/mL in HEPES buffer solution (HBS; 50 mM HEPES, 750 mM NaCl, pH 7.4). The polymer complexes (Lipo, PEI, and Gua-SS-PAAs-pEGFP) at varying w/w ratios ranging from 1/3 to 1/96 were prepared by adding a certain amount of the polymer (10 μg/μL in HBS) to 1 mL plasmid solution (20 μg/mL in HBS buffer pH 7.4), followed by vortexing for 10 seconds, and the solution was incubated for 20 minutes at room temperature prior to use.
Determination of encapsulation efficiency through Hoechst 33342 intercalation
The pDNA (0.5 μg, pEGFP-N3) condensations with Lipo, PEI, and Gua-SS-PAAs (CAR/CHL-CBA) at different nucleic acid/polymer (N/P, weight ratio) ratios from 1/0.5 to 1/96 were determined by Hoechst 33342 intercalation assay. The pDNA was mixed with 100 μL Hoechst 33342 (0.10 μg/mL) and then incubated with different polymers for 20 minutes at room temperature. The fluorescence of free DNA was used as control. The fluorescence of all samples was measured by a microplate reader (SpectraMax M3; Molecular Devices LLC, Sunnyvale, CA, USA). The encapsulation efficiency (EE) was determined by the following equation:26
|
|

Measurements of particle size and ζ-potential
The size and ζ-potential of the complexes formed by pDNA and PEI or Gua-SS-PAAs (CBA–CAR/CBA–CHL) at varying N/P ratios from 1/6 to 1/96 were measured at 25°C on a Zetasizer Nano system (Malvern Instruments, Malvern, UK) using the dynamic light scattering (DLS) technique. The results were processed using the Dispersion Technology Software V7.01 (Malvern Instruments).
Atomic force microscopy analysis of complex particle formation
Samples were prepared on freshly cleaved mica using a mixed solution of pDNA (final concentration: 20 μg/mL) and different polymers (final concentration: 0.48 mg/mL), resulting in complexes with an N/P weight ratio of 1/48 in HBS. In addition, the Lipo and branched PEI were used at concentrations recommended by their respective providers’ instructions. A total volume of 1 μL of the sample was coated on the mica surface for measuring the images of complexes in the buffer solution. Complex formation was studied using a Picoforce Multimode Nanoscope IIIa (Veeco Instruments, Inc., Town of Oyster Bay, NY, USA) in tapping mode. Tapping mode imaging was performed in situ in HBS using a standard liquid cell and a standard Veeco NP cantilever. Similarly, in order to study different complexes in the solid state, nitrogen was used to blow-dry the samples after they were prepared on the mica surface. Atomic force microscopy (AFM) imaging of the dry samples was then carried out in the same way as described earlier.
Agarose gel electrophoresis assay
Agarose gel retardation assay
The polymer complexes (Lipo, PEI, or Gua-SS-PAAs-pEGFP) at varying N/P ratios from 1/3 to 1/96 were prepared by adding a certain amount of the polymer (10 μg/μL in HBS) to 100 μL plasmid solution (20 μg/mL in HBS buffer pH 7.4), followed by vortexing for 10 seconds and incubating the solution for 20 minutes at room temperature. After the addition of 2 μL of 6× loading buffer to 10 μL of plasmid solution, Lipo–pEGFP, PEI–pEGFP, or CBA–CAR/CBA–CHL–pEGFP, 12 μL of this mixture was loaded onto a 1% agarose gel containing 0.5 μg/mL Goodview™ and run at 100 V for 30 minutes in TAE buffer (40 mM, tris-acetate and 1 mM EDTA). After development of the gel, DNA was visualized with a UV lamp using a Biospectrum AC system W/cooled Gel Camera P/N 97-0154-05 (Upland, CA, USA).
Reduction sensitivity assay
The polymer complexes at varying N/P ratios from 1/6 to 1/48 were prepared by the same method as described in the “Agarose gel retardation assay” section. After the preparation, GSH was added to 10 μL of the plasmid solution (200 ng of pEGFP), including Lipo–pEGFP, PEI–pEGFP, and CBA–CAR/CBA–CHL–pEGFP, at a final concentration of 20 mM. The dispersions were then incubated for 20 minutes. The same polymer complex solutions were also prepared without GSH as negative controls. After mixing with 2 μL of 6× loading buffer, 12 μL of this mixture was loaded onto a 1% agarose gel containing 0.5 μg/mL Goodview™, and electrophoresis was carried out. The electrophoresis conditions were the same as described in the “Agarose gel retardation assay” section.
Heparin replacement assay
The polymer complexes at an N/P ratio of 1/9 were prepared using the same method as described in the “Agarose gel retardation assay” section. Heparin (10 U/μL) was diluted to different concentrations and added to 10 μL of plasmid solution (200 ng of pEGFP), including Lipo–pEGFP, PEI–pEGFP, and CBA–CAR/CBA–CHL–pEGFP. The dispersions with different contents of heparin were then incubated for 30 minutes at 37°C. After the addition of 2 μL of 6× loading buffer, 12 μL of this mixture was loaded onto a 1% agarose gel containing 0.5 μg/mL Goodview™, and electrophoresis was carried out. The electrophoresis conditions were the same as described in the “Agarose gel retardation assay” section.
DNase I degradation assay
Polymer complexes at different N/P ratios (from 1/0.5 to 1/16) were prepared by the same method as described in the “Agarose gel retardation assay” section and incubated with 10 μL DNase I digestion system (DNase I 10 U, 50 mM KCl, 10 mM Tris–HCl, 10 mM MgCl2, 0.1% Triton X-100, pH 9.0) at 37°C for 20 minutes. The mixtures were then incubated with 5 μL termination solution (400 mM NaCl, 100 mM EDTA, pH 8.0) for 5 minutes at room temperature. The DNA integrity was assessed by agarose gel electrophoresis after replacement with heparin solution (4 U/μL). The electrophoresis conditions were the same as described in the “Agarose gel retardation assay” section.
Cell viability assay
MCF-7 (human breast cancer cells) were maintained in RPMI 1640 growth medium supplemented with 10% FBS and penicillin (100 U/mL)–streptomycin (100 μg/mL) at 37°C in a humidified atmosphere containing 5% CO2. MCF-7 cells were seeded in 96-well plates (4×103 cells/well) and cultured overnight. Different N/P weight ratios, ranging from 1/6 to 1/96, were used to prepare the complexes (PEI–pEGFP and CBA–CAR/CBA–CHL–pEGFP). Briefly, polymers and pDNA were dissolved in the serum-containing culture medium. Complexes were prepared by adding 200 μL of polymers at concentrations of 60–960 μg/mL to 50 μL of pDNA at a concentration of 40 μg/mL, followed by vortexing for 10 seconds and incubation at room temperature for 20 minutes. The MCF-7 cells in each well were incubated with the desired amount of complexes for 12 hours at the same cell culture conditions as described earlier, and then the complex solution in each well was removed. After that, 20 μL MTT (5 mg/mL) solution was added to each well and incubated for 4 hours. The medium was removed, and 150 μL DMSO was added. The absorbance was measured at 570 nm using a microplate reader (Spectra Max M3; Molecular Devices LLC), and the absorbance of untreated cells (with cell media only) was taken as 100% cell viability.27 The relative cell viability was calculated as follows: cell viability (%) = (OD570 (samples)/OD570 (control)) ×100, where OD570 (control) was obtained with the medium alone and OD570 (samples) was obtained in the presence of polymers.
In vitro transfection efficiency assay
Transfection experiments were also performed with MCF-7 cells using the plasmid pEGFP-N3. The cell culture and complex preparation were carried out by the same methods as described in the “Cell viability assay” section. Two parallel transfection series – one for the qualitative analysis of transfection efficiency using fluorescence microscopy (FM) and the other for the quantitative analysis using flow cytometry (FCM) – were established. MCF-7 cells were seeded in six-well plates at a density of 1.0×105 cells/well. The cells were cultured in 2 mL RPMI 1640 complete culture medium until the cell confluence reached ~60%. They were then washed with fresh 1× PBS buffer and incubated with 1.5 mL complete medium containing complexes with N/P ratios from 1/6 to 1/96 (the preparation of complexes was the same as described in the “Cell viability assay” section) for 4 hours at 37°C in a humidified atmosphere containing 5% CO2. The medium was then replaced with 2 mL of fresh RPMI 1640 complete medium, and the cells were incubated for another 44 hours. Cells in all wells of the two parallel series were collected, and one series of cells was collected for FCM assay by determining the transfection efficiency using a FACScan flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA), with an excitation wavelength of 488 nm. The cells in other series were observed under a Nikon TE2000U inverted fluorescence microscope (Nikon Corporation, Tokyo, Japan) with an excitation wavelength of 488 nm and a magnification of 200×. The untreated cells were used as a negative control, and Lipo–pEGFP and PEI–pEGFP served as positive controls.
Dynamic uptake assay
The dynamic uptake process of PEI–pEGFP and CBA–CAR/CBA–CHL–pEGFP in MCF-7 cells was recorded at different intervals by laser scanning confocal microscopy. In order to display the intracellular localization of pDNA, Total NUCLEAR-ID Green/Red nucleolar/nuclear detection kit, Hoechst 33342, and DiO were used to stain the nucleolus, nucleus, pDNA, and cell membrane, respectively, according to the protocols provided by the manufacturers. In brief, pEGFP (2 μg/well) and Hoechst 33342 (33.3 μg/mL) were incubated at 37°C for 1 hour. The stained pEGFP was then precipitated with 70% ethanol and resuspended in 1× PBS. MCF-7 cells were seeded at a density of 5×104 cells/well (glass bottom dish, d=35 mm) and cultured overnight in normal cell culture conditions as previously mentioned. After incubation, the cells in each well were washed with PBS thrice and stained with the Total NUCLEAR-ID Green/Red nucleolar/nuclear detection kit and DiO sequentially. Simultaneously, the resuspended pEGFP was incubated with polymers to prepare different complexes at the N/P weight ratio of 1/24 following the method described earlier. After staining, the cells were incubated in complete culture medium containing different complexes, and the localization of complexes was recorded at 5 minutes, 30 minutes, 1 hour, 2 hours, 3 hours, and 4 hours using laser scanning confocal microscopy (Olympus FV1000S-SIM/IX81; Olympus Corporation, Tokyo, Japan) with a 60× objective lens. The intracellular pDNA/polymer, nucleus, nucleolus, and cell membrane appeared in blue, red, green and green, respectively.
Statistics
Statistical comparisons were performed using a one-way analysis of variance, followed by the Dunnett test. Pairwise comparisons between treatments were achieved by using Student’s t-test. A P-value of <0.05 was considered statistically significant.
Results and discussion
Synthesis of CAR–CBA and CHL–CBA
In previous studies involving SS-PAAs as gene delivery carriers, the presence of bioreducible disulfide linkages in these polymers resulted in significant increases in transfection efficiency along with low cytoxicity.28 In this study, two amino monomers, CHL and CAR, were used to introduce guanidino groups into SS-PAAs, leading to the synthesis of CAR–CBA and CHL–CBA following the route shown in Figure 1. CHL is well known and widely used as a bacteriostatic agent in the pharmaceutical field or as a finishing agent in the textile industry with good bioactivity, biocompatibility, and low cytotoxicity. Another guanidine donor, CAR, was chosen in our study to compare it with CHL.
Due to low reactivity of guanidine, Pbf-Cl was used to activate the guanidine group before the Michael addition polymerization occurred between CBA and CHL/CAR. Trifluoroacetic acid was used for deprotecting Pbf-Cl after the Michael addition polymerization. The introduction of the guanidine group was achieved by the polymerization of CBA with the guanidine-based monomer, without any other modification after polymerization happening on the side chains. Compared to traditional methods, which introduce guanidine groups utilizing 1H-pyrazole-1-carboxamidine hydrochloride and N, N-diisopropylethylamine, this new synthetic route requires milder reaction conditions, facilitates product purification and collection, simplifies the entire preparation process, and, not surprisingly, leads to a reduction of the total production cost.
The 1H NMR spectra of CBA in deuterated DMSO solvent confirmed that CAR/CHL–CBA polymers were successfully connected to CBA because the characteristic olefin peak of CBA disappeared (data shown in Figures S1–S5). The molecular weights and polydispersity of two Gua-SS-PAAs were measured by gel permeation chromatography, and the results are listed in Table 1.
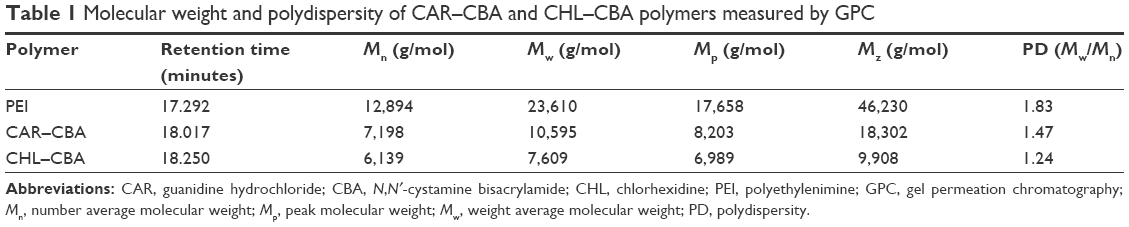 | Table 1 Molecular weight and polydispersity of CAR–CBA and CHL–CBA polymers measured by GPC |
Hoechst 33342 intercalation assay
The DNA EE of two polymers (CAR/CHL-CBA) at different N/P ratios was determined by Hoechst 33342 intercalation and agarose gel assay (Figure 2). As shown in the figure, the EEs of CAR–CBA were all >70% at N/P ratios ranging from 1/1 to 1/96, except at the ratio of 1/0.5, while the EEs of CHL–CBA were ~80%, except at the ratio of 1/96. The EEs of PEI and Lipo used at concentrations recommended by manufacturers (Lipo at 1 μL/μg pDNA; PEI at the N/P ratio of 1/12) were 87.31%±7.21% and 75.23%±6.50%, respectively. These results show that there was no significant difference in EEs between the two polymers, and both of them exhibited equal or even higher EEs than PEI or Lipo. Moreover, pDNA was fully condensed at N/P ratios from 1/1 to 1/48. Notably, CHL–CBA showed a much higher EE (91.58%±5.98%) than CAR–CBA (45.59%±6.63%) at the ratio of 1/0.5, indicating that the former had an enhanced encapsulation capability due to its complicated side-chain structure. In addition, the EE of both polymers reached the peak value at the N/P ratio of 1/24 and did not increase, while the amount of polymers was increased. Instead, a slight downward trend was observed when the ratio was over 1/24, especially for CHL–CBA (falling to 63.66%±6.03% at the ratio of 1/96).
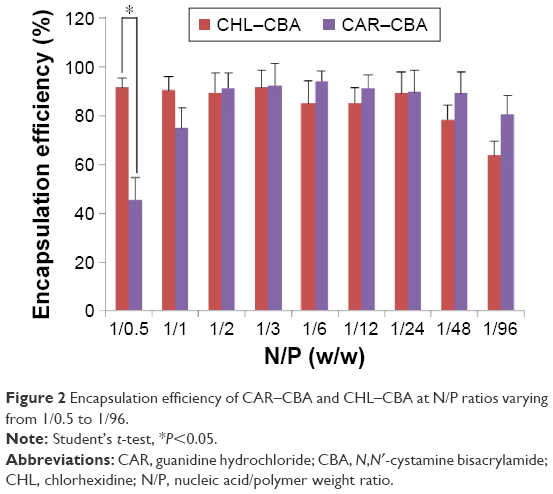 | Figure 2 Encapsulation efficiency of CAR–CBA and CHL–CBA at N/P ratios varying from 1/0.5 to 1/96. |
AFM analysis of complex particle formation
The DNA condensation capabilities of two polymers (CAR/CHL–CBA) were further studied by AFM (Figure 3). Based on the results of a pretest, inadequate polymer dosage (PAAs) could affect the adhesion between the complexes and mica, leading to difficulties in the imaging process of AFM analysis, which was consistent with Piest and Engbersen’s26 report. Therefore, the N/P ratio of the two complexes (CAR/CHL–CBA–pDNA) was set as 1/48 based on that report.26 As the top row in Figure 3 shows, a typical plectonemic conformation of supercoiled plasmids is observed after naked pDNA was deposited onto freshly cleaved mica in the absence or presence of the buffer. Interestingly, we found that there was no particle-like images in CAR–CBA and CHL–CBA groups in the absence or presence of buffer when the N/P ratio was below 1/6 (data not shown), which implied that, though an interaction between pDNA and the polymer was observed in Hoechst 33342 intercalation assay at N/P ratios ranging from 1/0.5 to 1/6, no visible particle-like form condensed by two polymers was actually formed. The images of complexes on dried mica at an N/P ratio of 1/48 (CHL/CAR–CBA) revealed that sizes of these particles were as follows: Lipo (400≤500 nm), CHL–CBA (≤250 nm), CAR–CBA (≤150 nm), and PEI (≤100 nm). The morphological images of complexes on mica in the buffer system were quite different from those of the dried complexes, and the sizes of these particles were smaller than those in dried forms and arranged in a different order: Lipo (≤100 nm), PEI (≤50 nm), CHL–CBA (≤40 nm), and CAR–CBA (≤30 nm). At the same time, in the buffer system, Lipo–pEGFP and PEI–pEGFP exhibited a higher particle density than the other two types of complexes at an N/P ratio of 1/48. This could be caused by the blow-dry process, which might have blown away a part of these particles. The AFM analysis of particle formation also demonstrated that significant morphological difference existed between complexes in the dry form and in the buffer system. However, the complexes in the buffer system were settling on the mica surface, which could make a difference in the results. Since the suspended complexes might exhibit a difference in the particle size and other properties, DLS assay was performed for a quantitative assessment of the complexes.
 | Figure 3 AFM images of Gua-SS-PAAs complexes and naked pDNA in dry form or in buffer system. |
Particle size and ζ-potential measurement
The DNA-condensing ability of Gua-SS-PAAs (CAR–CBA and CHL–CBA) was evaluated by particle size and ζ-potential measurements. Figure 4 shows that the particle size of CAR–CBA–pDNA complexes at N/P ratios ranging from 1/6 to 1/96 was consistently ~50 nm, and the maximum was 62.45±5.82 nm. The ζ-potential of CAR–CBA–pDNA was ~20 mV and reached a maximum value (29.60±1.22 mV) at the N/P ratio of 1/24. The particle size of CHL–CBA–pDNA complexes was <90 nm, which was much larger than that of CAR–CBA–pDNA. When the N/P ratio was varied from 1/12 to 1/96, the particle size increased and reached a maximum of 87.14±3.07 nm. The ζ-potential of CHL–CBA–pDNA showed a similar trend as observed from the measurement of particle size. However, there was no obvious difference between these two types of complexes. Compared to PEI, the average particle sizes of both complexes were larger, but their average ζ-potentials were lower. Along with an increase in the amount of the polymers, the particle size of CAR/CHL–CBA complexes did not continuously grow, which might be a result of using the optimal N/P ratio. When the amount of polymer reached the limit, the repulsive force between the polymers could interfere with the interaction between polymers and DNA, thereby hindering the complexation process. The large particle size at a low N/P ratio indicated that DNA might not have been condensed compactly. Interestingly, the ζ-potential results of CAR/CHL–CBA demonstrated a similar trend, which confirmed that the particulate structure was a prerequisite for generating the ζ-potential. Based on these observations, the particle size and ζ-potential of the two complexes might be affected not only by the N/P ratio but also by the complex structure and/or some other factors.
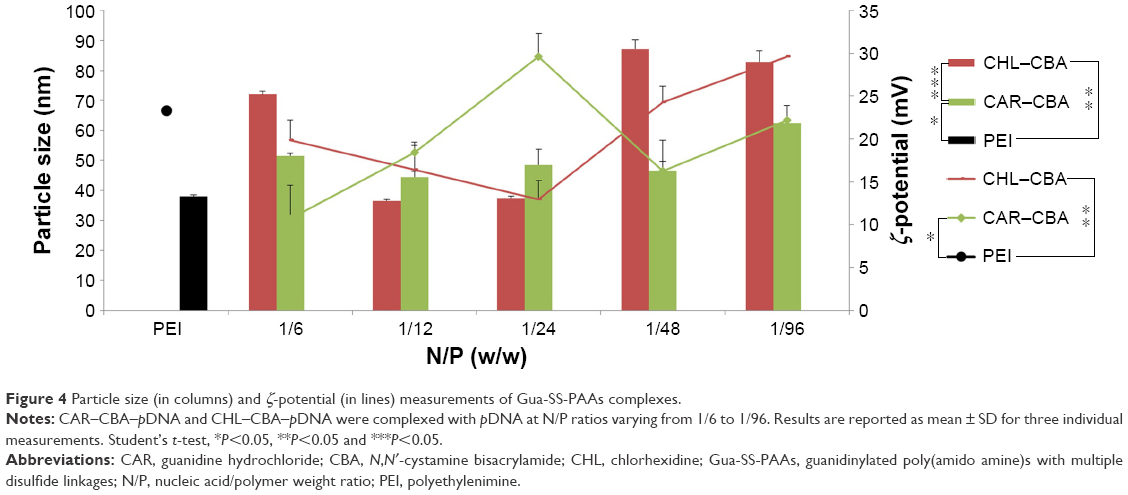 | Figure 4 Particle size (in columns) and ζ-potential (in lines) measurements of Gua-SS-PAAs complexes. |
Agarose gel electrophoresis assay
The agarose gel electrophoresis assay was applied to evaluate the complexation ability and stability of Gua-SS-PAAs. As shown in Figure 5A, no retardation was found with pDNA alone. Retardation mediated by Lipo at the recommended concentration was not complete since a light band was observed. In contrast, PEI showed complete retardation at the recommended concentration. Similarly, complete retardation was observed for CAR/CHL–CBA at N/P ratios from 1/6 to 1/96. There was a slightly higher amount of free pDNA in the channel of CAR–CBA than in CHL–CBA, which indicated that the complexation of CHL–CBA was more capable than CAR–CBA. As shown in Figure 5B, pDNA was not replaced by heparin until the concentration of heparin was >18 U/mL in both CAR–CBA and CHL–CBA complexes. However, in PEI complexes, pDNA was replaced until the concentration of heparin reached 36 U/mL. The pDNA replaced by heparin was more in CAR–CBA complexes than in CHL–CBA complexes. Furthermore, CHL–CBA/CAR–CBA complexes at the N/P ratio above 1/3 were resistant to DNase I digestion, and CHL–CBA showed better resistance compared to CAR–CBA (Figure 5C). Figure 5C also demonstrates the protection of Lipo and PEI against DNase I-mediated DNA degradation.
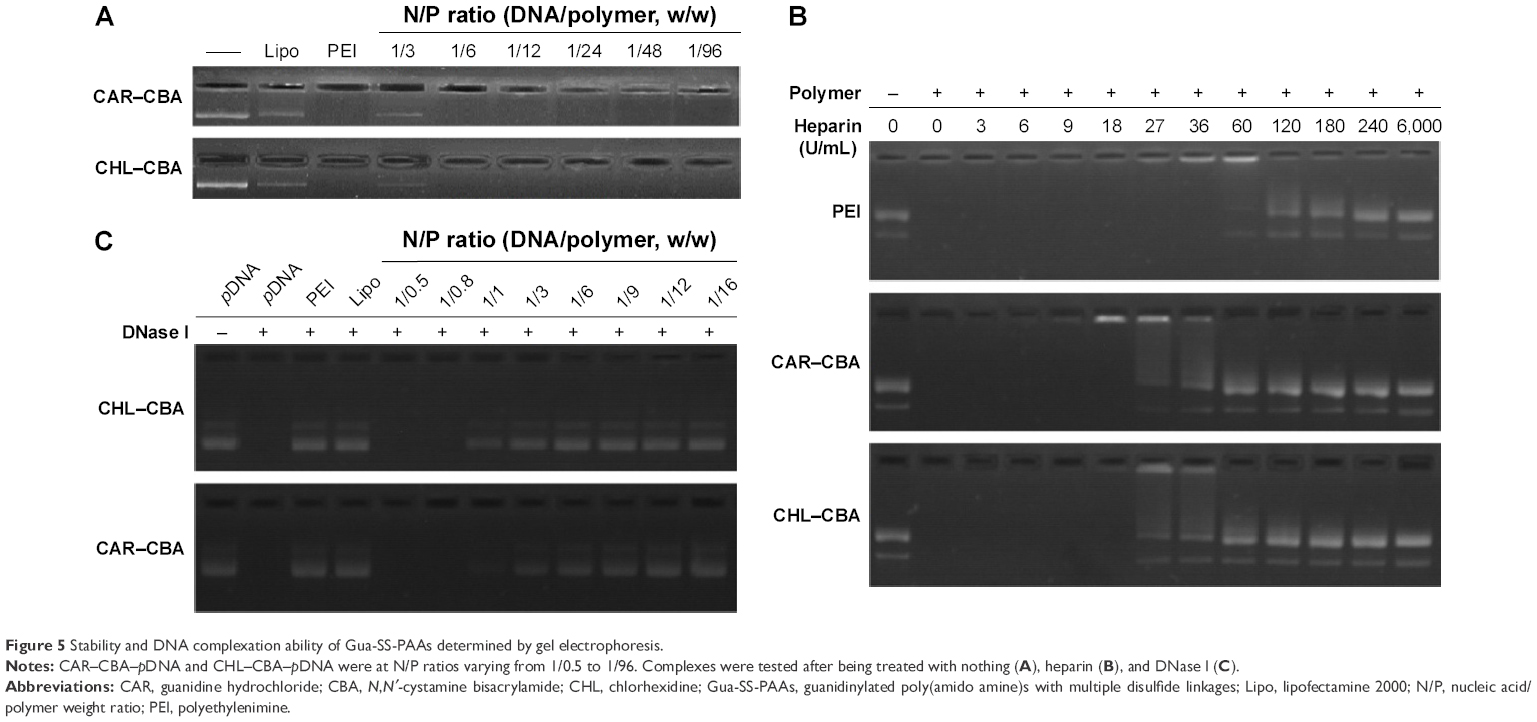 | Figure 5 Stability and DNA complexation ability of Gua-SS-PAAs determined by gel electrophoresis. |
Based on the Hoechst 33342, AFM, DLS, and agarose gel assays, we concluded that pDNA was well encapsulated in CAR/CHL–CBA, and the encapsulation abilities of CAR/CHL–CBA were greater than that of the two positive controls (Lipo and PEI). Furthermore, the complexation mediated by CHL–CBA was more complete and stable than that by CAR–CBA. This might be due to the difference of functional structures and more nitrogen atoms of the guanidino groups in the side chains of CHL–CBA. In addition, different methods were used to evaluate the condensation ability of polymers, and these studies showed diverse results, which should not be simply discarded and will be explored in the future.
Reduction sensitivity of the Gua-SS-PAAs
Reduction-triggered disassembly of Gua-SS-PAAs was evaluated under reducing conditions using agarose gel electrophoresis (Figure 6). Li et al14 reported that 0.75 M NaCl in HBS was indispensable for pDNA release. Incubation with 20 mM GSH resulted in the dissociation of the complexes (CAR/CHL–CBA) and a release of pDNA at N/P ratios from 1/6 to 1/48. In contrast, there was no reduction-triggered disassembly observed in Lipo and PEI complexes under the same conditions. As shown in Figure 6, CAR–CBA was more sensitive to GSH than CHL–CBA since more pDNA was released from the CAR–CBA–pDNA complexes. This might be a result of longer side chains and more guanidine groups in CHL–CBA. In addition, the vast majority of released pDNA was in the close-loop state (with faster electrophoretic speed and higher transfection capability), although a small open-loop pDNA also existed, indicating that plasmid with high transfection capability could be entrapped, protected, and released reduction-triggering from the Gua-SS-PAAs.
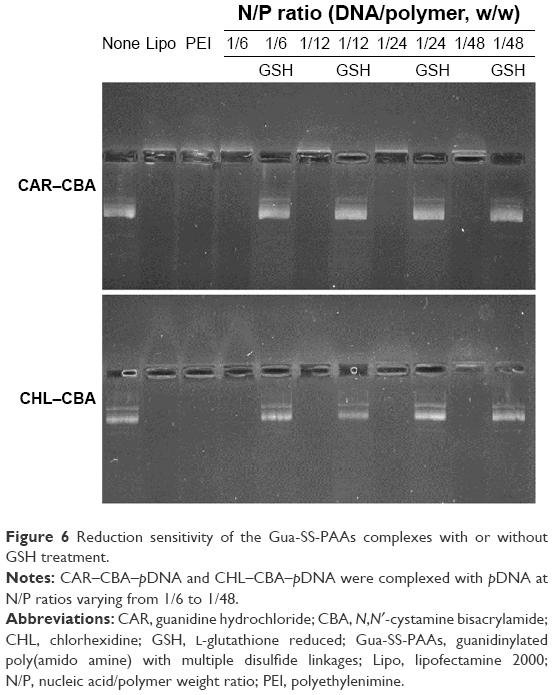 | Figure 6 Reduction sensitivity of the Gua-SS-PAAs complexes with or without GSH treatment. |
Cell viability assay
Cytotoxicity is an important parameter in the development of safe nonviral carriers. High cytotoxicity of a carrier will negatively affect the transfection efficiency. According to the particle size and stability results of complexes from the agarose assay, polymeric carriers were capable of forming stable complexes at N/P ratios from 1/6 to 1/96. In order to investigate the cytotoxicity of Gua-SS-PAAs, MCF-7 cells were treated with the complexes (CAR/CHL–CBA–pDNA) prepared at N/P ratios from 1/6 to 1/96 in the presence of 10% FBS. Cell viability was determined by MTT assays, and Lipo and PEI complexes were used as positive controls. Compared to Lipo and PEI, CAR–CBA and CHL–CBA confer the complexes with significantly lower cytotoxicity, as shown in Figure 7. The cytotoxicity of CAR–CBA complexes was higher than that of CHL–CBA complexes. The stronger cytotoxicity mediated by CAR–CBA could be the result of a strong interaction between the complex and cell membrane due to the high density of guanidine in the main chain of CAR–CBA.29 In contrast, though CHL–CBA possesses more guanidine groups, these groups are located in the side chains of CHL–CBA, leading to less cytotoxicity. As reported by Martello et al,13 cytotoxicity is determined by many factors, including the molecular weight, charge density, type of amines, polymer structure (linear, branched), intracellular degradability, and chain flexibility. In our case, this was a typical example in which the difference in polymer structure caused a discrepancy in the charge density, but not in the ζ-potential, resulting in the difference in cytotoxicity. In addition, though the presence of serum in cell culture medium might result in the enhancement of cell viability,30 this condition was closer to the in vivo working environment of the carriers. Furthermore, the incubation time of complexes was extended to 12 hours, which tripled the in vitro transfection time (4 hours) and further confirmed the low cytotoxicity of these two polymers.
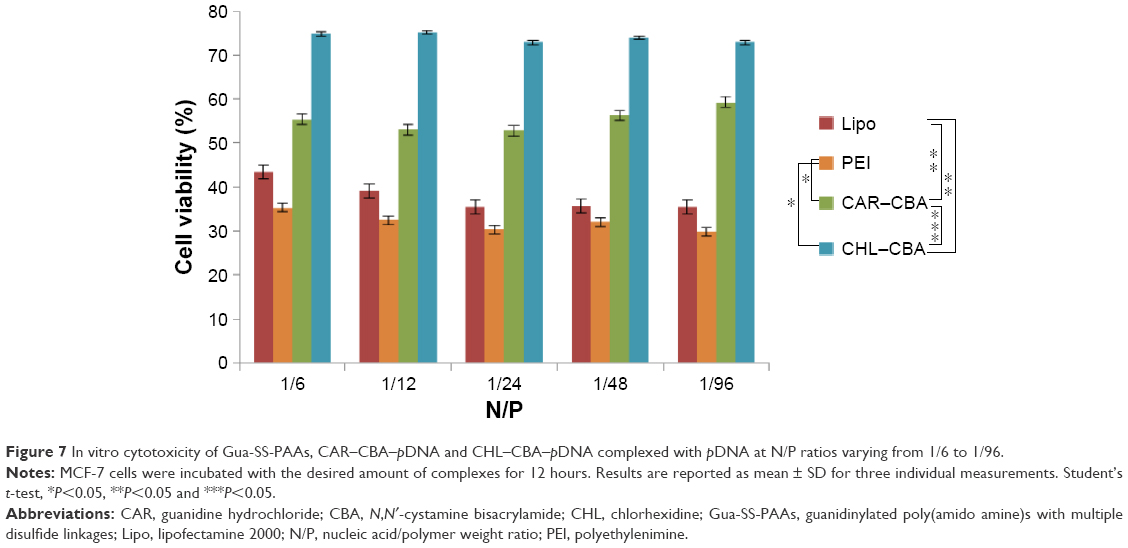 | Figure 7 In vitro cytotoxicity of Gua-SS-PAAs, CAR–CBA–pDNA and CHL–CBA–pDNA complexed with pDNA at N/P ratios varying from 1/6 to 1/96. |
Determination of in vitro transfection efficiency
The transfection efficiencies of the complexes from CAR–CBA and CHL–CBA at different N/P ratios from 1/6 to 1/96 were investigated in MCF-7 cells in the presence of serum. Lipo and PEI served as controls. The fluorescence intensity of green fluorescent protein was analyzed by FM combined with FCM to determine the transfection efficiencies of the carriers (Figure 8). It was found that the transfection efficiencies of CAR–CBA and CHL–CBA increased with the increase in dosage of the polymers and reached a maximum of 78.0% and 92.7%, respectively, at the N/P ratio of 1/96. Compared to those of the two controls, the transfection efficiencies of the two polymers were considerably enhanced when the N/P ratio was >1/48. At the same time, the transfection efficiency of CHL–CBA was much higher than that of CAR–CBA when the N/P ratio was >1/12. These data were consistent with the results from DLS assays, indicating that the transfection efficiency was not related to ζ-potential but affected by other factors. For instance, the transfection efficiency of the two complexes (CHL/CAR–CBA–pDNA) increased with the particle size especially when the N/P ratio was varied from 1/48 to 1/96. Combining the results from cell viability and transfection efficiency, we conclude that the higher cytotoxicity of CAR–CBA might lead to a negative effect on transfection efficiency. In addition, the main reason might be due to the nuclear localization effect (NLE) mediated by the guanidine groups in the side chains of CHL–CBA. This hypothesis was further tested in the following in vitro cellular uptake study.
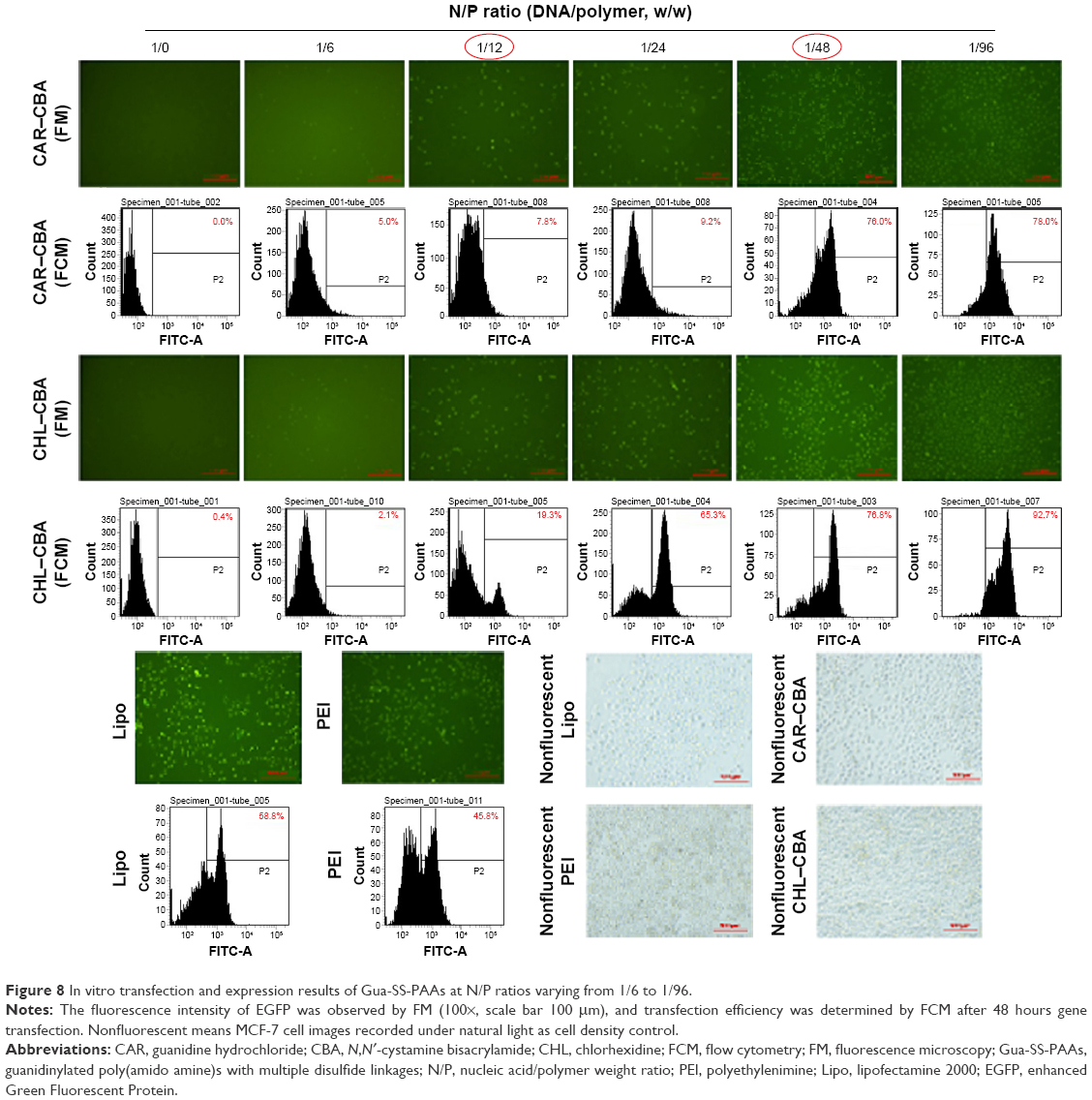 | Figure 8 In vitro transfection and expression results of Gua-SS-PAAs at N/P ratios varying from 1/6 to 1/96. |
In vitro cellular uptake
In order to further understand the effects of polymer/pDNA complexes on the process of transfection, the uptake and subcellular localization of the complexes were investigated. In this study, naked Hoechst 33342-labeled pDNA was used as control. As shown in Figure 9, the pDNA stained with blue could be hardly found at 0 minutes or 5 minutes. CHL–CBA–pDNA complexes were found in the nucleus and cytoplasm of MCF-7 cells, corresponding to the purple and light blue colors, respectively, but no significant color change was observed in the other three groups 30 minutes after the transfection process started. CAR–CBA–pDNA appeared in the nucleus 1 hour after the transfection process started, while Lipo–pDNA and PEI–pDNA just started entering the nuclear region at that time. Four hours after the transfection process started (ie, the ending time point), all treatment groups demonstrated that many of the complexes had already entered the nuclear regions since the red-stained nucleus had turned into dark purple or blue. Moreover, there was more pDNA appearing in the nucleus in the PEI and CHL–CBA groups than in the Lipo and CAR–CBA groups. In addition, more intact cells were observed in the CHL/CAR–CBA groups compared with other two groups, which was consistent with cell viability results, confirming the lower cytotoxicity of CHL/CAR–CBA.
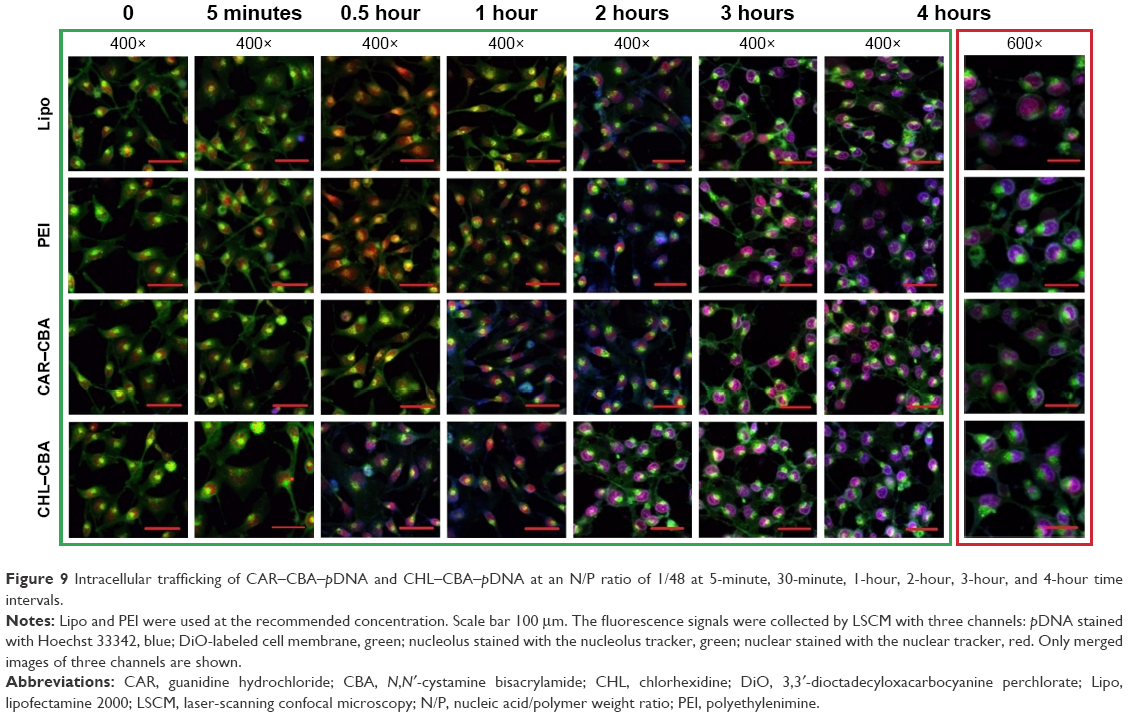 | Figure 9 Intracellular trafficking of CAR–CBA–pDNA and CHL–CBA–pDNA at an N/P ratio of 1/48 at 5-minute, 30-minute, 1-hour, 2-hour, 3-hour, and 4-hour time intervals. |
Conclusion
Two different Gua-SS-PAAs carriers were successfully prepared. Both of them showed higher transfection efficiency and lower cytotoxicity than two widely used carriers (PEI and Lipo). Furthermore, the reasons for the biological difference between these two carriers related to their structural factors were discussed. The results of physical/chemical analysis and biological studies confirmed that both Gua-SS-PAAs carriers possessed promising encapsulation, cell membrane permeability, and nuclear localization effect. In addition, the heparin replacement and DNase I degradation assay suggested that the new carriers might have potential applications as delivery carriers in gene therapy. Certainly, the systemic circulation stability, distribution, and safety issues such as hemolysis or immunogenicity, need to be further investigated through comprehensive in vivo experiments.
Acknowledgments
This work was supported in whole or in part by the Center for Biological Imaging, Institute of Biophysics, Chinese Academy of Sciences, and the School of Pharmacy, China Medical University. The authors are grateful to the Natural Science Foundation Committee of China for financial support (no 81373335).
Disclosure
The authors report no conflicts of interest in this work.
References
Vader P, van der Aa LJ, Engbersen JF, Storm G, Schiffelers RM. Physicochemical and biological evaluation of siRNA polyplexes based on PEGylated Poly (amido amine)s. Pharm Res. 2012;29(2):352–361. | ||
Arote RB, Jiang HL, Kim YK, Cho MH, Choi YJ, Cho CS. Degradable poly(amido amine)s as gene delivery carriers. Expert Opin Drug Deliv. 2011;8(9):1237–1246. | ||
Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15(8):541–555. | ||
Naldini L. Gene therapy returns to centre stage. Nature. 2015;526(7573):351–360. | ||
Chang L, Gallego-Perez D, Zhao X, et al. Dielectrophoresis-assisted 3D nanoelectroporation for non-viral cell transfection in adoptive immunotherapy. Lab Chip. 2015;15(15):3147–3153. | ||
Chang L, Bertani P, Gallego-Perez D, et al. 3D nanochannel electroporation for high-throughput cell transfection with high uniformity and dosage control. Nanoscale. 2016;8(1):243–252. | ||
Gallego-Perez D, Otero JJ, Czeisler C, et al. Deterministic transfection drives efficient nonviral reprogramming and uncovers reprogramming barriers. Nanomedicine. 2016;12(2):399–409. | ||
Maskell DP, Renault L, Serrao E, et al. Structural basis for retroviral integration into nucleosomes. Nature. 2015;523(7560):366–369. | ||
Van der Aa LJ, Vader P, Storm G, Schiffelers RM, Engbersen JF. Optimization of poly (amido amine) s as vectors for siRNA delivery. J Control Release. 2011;150(2):177–186. | ||
Vader P, van der Aa LJ, Engbersen JF, Storm G, Schiffelers RM. Disulfide-based poly (amido amine)s for siRNA delivery: effects of structure on siRNA complexation, cellular uptake, gene silencing and toxicity. Pharm Res. 2011;28(5):1013–1022. | ||
Zhang B, Ma X, Murdoch W, Radosz M, Shen Y. Bioreducible poly(amido amine)s with different branching degrees as gene delivery vectors. Biotechnol Bioeng. 2013;110(3):990–998. | ||
Lin C, Blaauboer CJ, Timoneda MM, et al. Bioreducible poly (amido amine)s with oligoamine side chains: synthesis, characterization, and structural effects on gene delivery. J Control Release. 2008;126(2):166–174. | ||
Martello F, Piest M, Engbersen JF, Ferruti P. Effects of branched or linear architecture of bioreducible poly(amido amine)s on their in vitro gene delivery properties. J Control Release. 2012;164(3):372–379. | ||
Li J, Manickam DS, Chen J, Oupicky D. Effect of cell membrane thiols and reduction-triggered disassembly on transfection activity of bioreducible polyplexes. Eur J Pharm Sci. 2012;46(3):173–180. | ||
Merdan T, Callahan J, Petersen H, et al. Pegylated polyethylenimine-Fab’ antibody fragment conjugates for targeted gene delivery to human ovarian carcinoma cells. Bioconjug Chem. 2003;14(5):989–996. | ||
Lin C, Engbersen JFJ. PEGylated bioreducible poly (amido amine)s for non-viral gene delivery. Mater Sci Eng C. 2011;31(7):1330–1337. | ||
Salmaso S, Caliceti P. Self-assembling nanocomposites for protein delivery: supramolecular interactions of soluble polymers with protein drugs. Int J Pharm. 2013;440(1):111–123. | ||
Wei CM, Zhang YL, Zhu HJ, Wang JT. Crystal structure and hydrogen bond network of a novel 4-guanidinosalicylic acid inner salt. J Chem Crystallogr. 2005;35(7):561–564. | ||
Shi JB, Ouyang J, Li QT, et al. Cell-compatible hydrogels based on a multifunctional crosslinker with tunable stiffness for tissue engineering. J Mater Chem. 2012;22:23952–23962. | ||
Nimesh S, Chandra R. Guanidinium-grafted polyethylenimine: an efficient transfecting agent for mammalian cells. Eur J Pharm Biopharm. 2008;68(3):647–655. | ||
Kim TI, Ou M, Lee M, Kim SW. Arginine-grafted bioreducible poly (disulfide amine) for gene delivery systems. Biomaterials. 2009;30(4):658–664. | ||
Kim TI, Lee M, Kim SW. A guanidinylated bioreducible polymer with high nuclear localization ability for gene delivery systems. Biomaterials. 2010;31(7):1798–1804. | ||
Coue G, Freese C, Unger RE, Kirkpatrick CJ, Engbersen JF. Bioresponsive poly (amidoamine)s designed for intracellular protein delivery. Acta Biomater. 2013;9(4):6062–6074. | ||
Carpino LA, Shroff H, Triolo SA, Mansour E-SME, Wenschuh H, Albericio F. The 2, 2, 4, 6, 7-pentamethyldihydrobenzofuran-5-sulfonyl group (Pbf) as arginine side chain protectant. Tetrahedron Lett. 1993;34(49):7829–7832. | ||
Wang M, Hu H, Sun Y, et al. A pH-sensitive gene delivery system based on folic acid-PEG-chitosan – PAMAM-plasmid DNA complexes for cancer cell targeting. Biomaterials. 2013;34(38):10120–10132. | ||
Piest M, Engbersen JF. Role of boronic acid moieties in poly (amido amine)s for gene delivery. J Control Release. 2011;155(2):331–340. | ||
Kim TI, Kim SW. Bioreducible polymers for gene delivery. React Funct Polym. 2011;71(3):344–349. | ||
Yang W, Pan CY, Luo MD, Zhang HB. Fluorescent mannose-functionalized hyperbranched poly (amido amine)s: synthesis and interaction with E. coli. Biomacromolecules. 2010;11(7):1840–1846. | ||
Wang X, He Y, Wu J, Gao C, Xu Y. Synthesis and evaluation of phenylalanine-modified hyperbranched poly (amido amine)s as promising gene carriers. Biomacromolecules. 2010;11(1):245–251. | ||
Yu Z, Yan J, You Y. Synthesis of bioreducible and acid labile poly(amido amine)s via Michael-addition reactions and their application in gene delivery. J Control Release. 2011;152(suppl 1):e179–e181. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.