Back to Journals » Journal of Multidisciplinary Healthcare » Volume 12
Growth in CHARGE syndrome: optimizing care with a multidisciplinary approach
Authors Dijk DR ![]() , Bocca G, van Ravenswaaij-Arts CM
, Bocca G, van Ravenswaaij-Arts CM ![]()
Received 1 March 2019
Accepted for publication 12 July 2019
Published 1 August 2019 Volume 2019:12 Pages 607—620
DOI https://doi.org/10.2147/JMDH.S175713
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Dieuwerke R Dijk,1 Gianni Bocca,2 Conny M van Ravenswaaij-Arts1
1Department of Genetics, University of Groningen, University Medical Center Groningen, Groningen, The Netherlands; 2Department of Pediatrics, Beatrix Children’s Hospital, University of Groningen, University Medical Center Groningen, Groningen, The Netherlands
Abstract: CHARGE (Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital hypoplasia, Ear anomalies including hearing loss) syndrome is a rare syndrome with an incidence of approximately 1:15,000 newborns. It is caused by pathogenic variants in the CHD7 gene and clinically characterized by a wide range of anomalies with variable expression. Growth retardation affects 60–72% of children with CHARGE syndrome, making it one of the most prominent medical issues in the syndrome. Growth retardation in CHARGE syndrome is thought to be multifactorial and can be influenced by almost all co-morbidities, requiring a multidisciplinary approach to the different medical problems. In this systematic review, we describe what is currently known about growth in CHARGE syndrome and how it is influenced by commonly seen clinical problems including feeding difficulties, hypogonadotropic hypogonadism and growth hormone deficiency. Furthermore, we provide recommendations for a multidisciplinary approach.
Keywords: CHARGE syndrome, growth, short stature, multidisciplinary, hypogonadotropic hypogonadism
Introduction
CHARGE syndrome (OMIM 214800) is a rare disorder with an estimated incidence of 1 in 15,000 to 1 in 17,000 live births.1 It is characterized by a wide spectrum of anomalies that vary among patients. In 1981, Pagon introduced the acronym CHARGE based on some of the most prevalent anomalies in the syndrome: Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital hypoplasia and Ear and hearing abnormalities.2 CHARGE syndrome can be clinically diagnosed by using the Blake or Verloes criteria.3,4
In 2004, variants in the CHD7 gene (OMIM 608892) were identified to be responsible for the CHARGE phenotype.5 Since then, more than 1000 variants in CHD7 have been identified, and a CHD7 variant is found in 83–95% of patients fulfilling Blake or Verloes’ diagnostic criteria.6,7 Next-generation sequencing techniques have led to the identification of an increased number of CHD7 gene variants and to increased detection of these variants in patients with a mild phenotype. The majority of CHD7 gene variants are nonsense or frameshift mutations, while missense and splice site mutations have been detected in a minority of cases, and deletions, duplications and chromosomal rearrangements are rare.1
CHARGE syndrome is a clinically variable syndrome, and there is no clear correlation between genotype and phenotype when focusing on individual cases. However, patients with a missense mutation generally have a milder phenotype, and missense mutations are more frequently found in patients with Kallmann syndrome.8–10 CHARGE syndrome is thought to be caused by a loss of function of CHD7 and has an autosomal dominant inheritance pattern. Most cases are caused by de novo mutations, although some familiar cases have been reported.1,11,12
In 2016, new clinical criteria were proposed that consist of the revised Blake criteria with the addition of a pathogenic variant in the CHD7 gene as a major criterion.13
Growth retardation and hypogonadotropic hypogonadism (HH) are important aspects of CHARGE syndrome in both boys and girls. Short stature is reported in 60–72% of patients with CHARGE syndrome, although the underlying cause is often not well-documented.14–16 HH is also highly prevalent, and 60–88% of patients with CHARGE syndrome do not achieve puberty spontaneously. Nonetheless, there are no syndrome-specific guidelines on how to induce puberty in this group of patients who frequently exhibit challenging behavior and therefore may respond differently to hormone replacement therapy.7,16–19
A number of studies have now been published that describe aspects of growth and puberty in CHARGE syndrome. The aim of this review is to summarize what is currently known about growth in CHARGE syndrome in order to make recommendations for the multidisciplinary approach and identify what future studies are needed to develop evidence-based guidelines for growth and puberty surveillance in CHARGE syndrome.
Methods
For this systematic review, we conducted a literature search on growth and puberty in CHARGE syndrome in PubMED using MeSH terms and in Embase using Emtree terms. We also searched on title and abstract based on keywords related to growth and puberty and included publications regarding CHD7 and Kallmann syndrome because HH is also a feature of Kallmann syndrome and mutations in the CHD7 gene may be found in these patients.20 Our search terms and selection process are described in Figure 1. We excluded all duplicate records and those that were not in English and selected possibly relevant records on title and abstract. The final selection was made after reading the complete publication (DD, GB). The references of the selected articles were checked for any relevant articles that might have been missed.
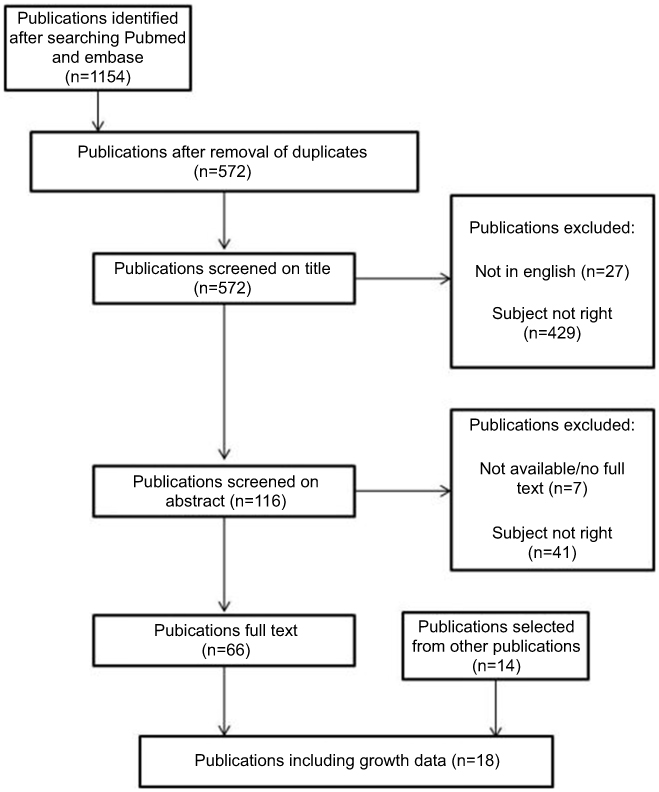 |
Figure 1 Flow chart of publication selection. Entry terms: CHARGE syndrome, CHARGE association, Hall Hittner syndrome, Hall Hittner association, growth, body size, weight, height, body mass, BMI, length, birth weight, head circumference, short stature, birth size, morphometry, anthropometry, puberty, hypogonadism, Kallmann, adolescent and adolescence. MeSH terms for PubMED search: CHARGE syndrome, Body weights and measures, growth and development, puberty, delayed puberty and hypogonadism. Emtree terms for Embase search: syndrome CHARGE, morphometry and growth, development and aging.Abbreviations: CHARGE, coloboma of the eye, heart defects, atresia of the choanae, retardation of growth and/or development, genital hypoplasia, ear anomalies including hearing loss. BMI, body mass index. |
Results
Growth in CHARGE syndrome
Of the studies we found, 22 specifically mentioned growth and 18 of these presented growth data in percentiles or SD values in relation to a reference population. These results are summarized in Table 1. Below, we review the current knowledge about growth in the context of different phases of life.
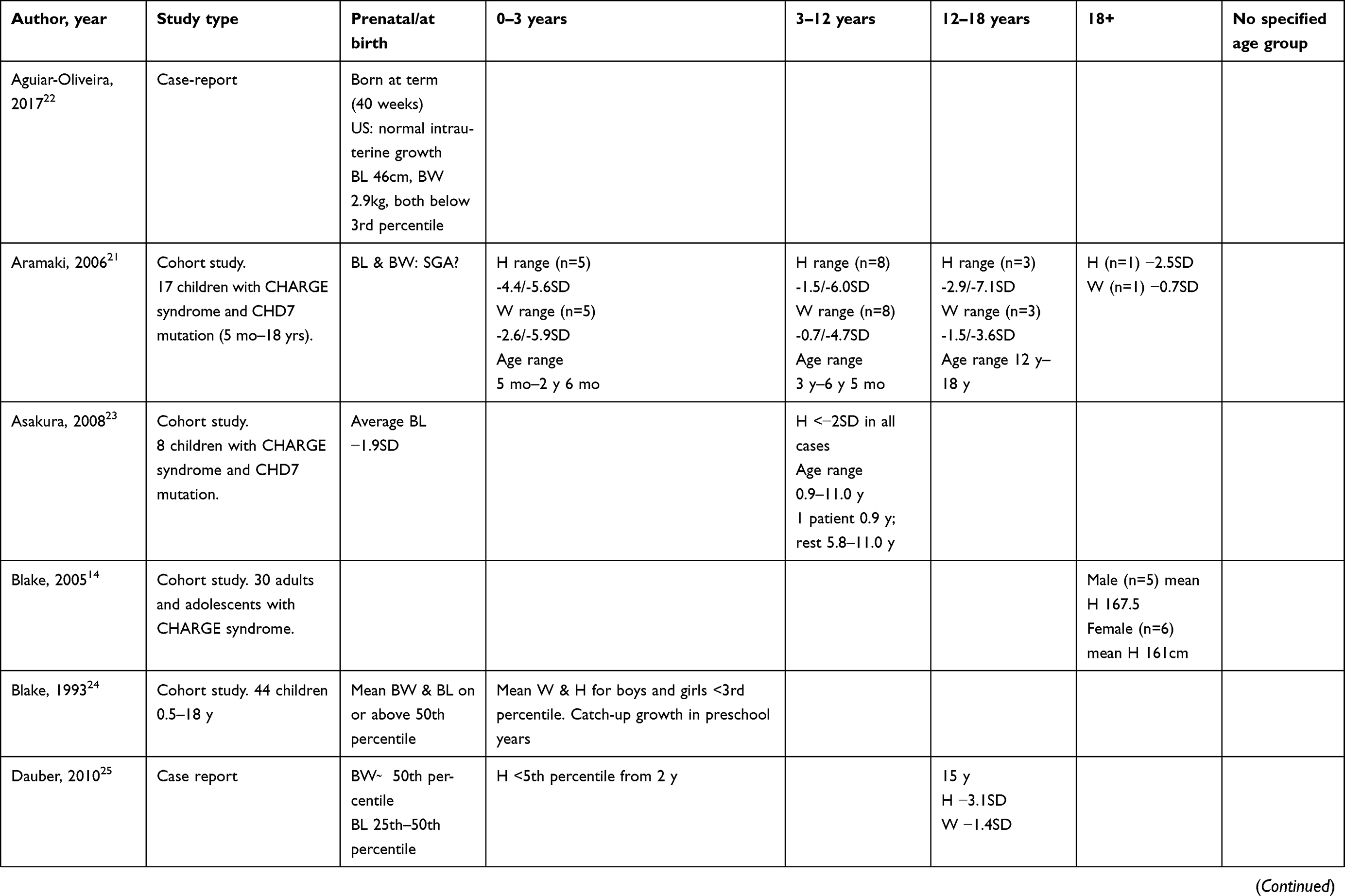 |
Table 1 Growth data from literature |
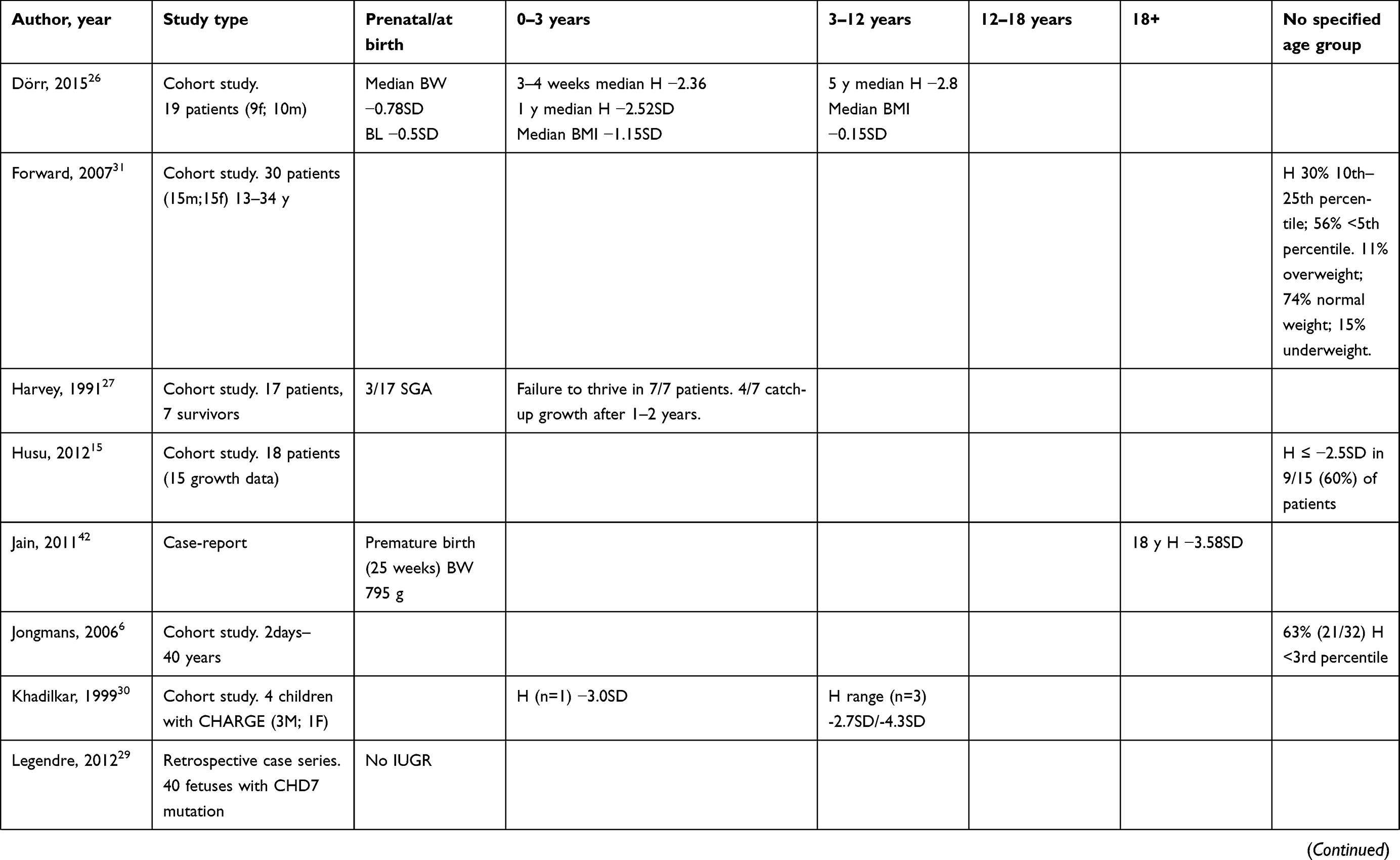 |
Table 1 (Continued). |
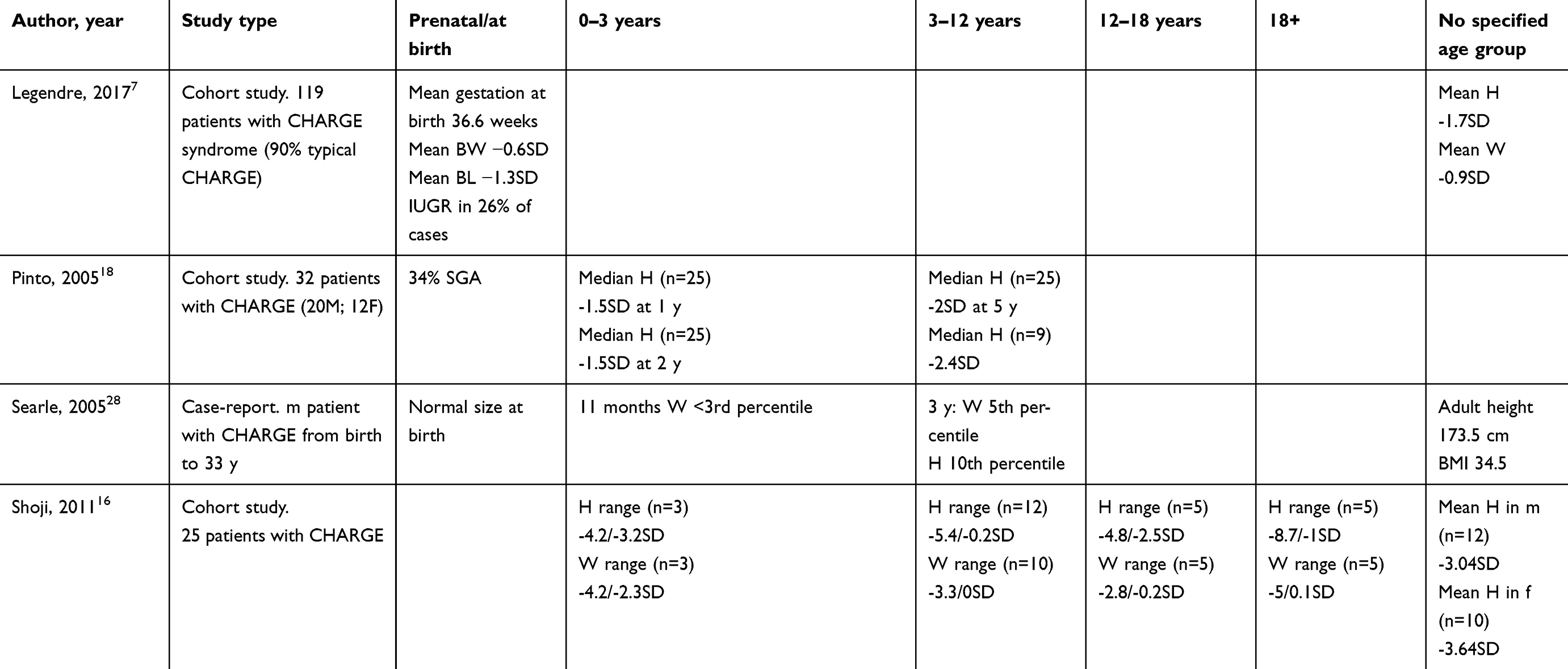 |
Table 1 (Continued). |
Fetal growth
In a cohort of 119 children with CHARGE syndrome, the mean gestational age at birth was 36.6±2.2 weeks.7 In another small cohort, 16 out of 17 children were born at term.21 Birth weight and length were generally normal or slightly reduced when compared to a reference population.7,18,21–28 However, a small proportion of children with CHARGE syndrome are small for their gestational age.21–23,27 According to Legendre and Pinto, this is true for 26–34% of children with CHARGE syndrome.7,18 In another study by Legendre et al that described the characteristics of 40 deceased fetuses with CHARGE syndrome, no intra-uterine growth retardation was found.29 This is remarkable considering that this group probably represents the more severely affected spectrum of CHARGE syndrome. Thus, the largest decrease in growth velocity happens the first period after birth.
Growth from 0–3 years
All the published studies we found showed a sudden decrease in growth rate from early infancy onwards.16,18,21,24–28,30 In a study of 19 German children aged 0–6 years, a sudden decrease in growth rate and body length was documented from as early as 4 weeks of age when compared to a reference population.26 Some authors have described catch-up growth in pre-school years,24,27 while others have described persistently delayed growth with a height between −2.36 and −5.6 SD and no catch-up growth.16,18,21,26,30 In addition to short stature, weight is also frequently below average and BMI is low.26
Growth from 3–12 years
During the early school years, short stature persists in children with CHARGE syndrome, with an average height between −2 to −3 SD below the reference population.16,18,21,23,26,28,30 In these years, the risk of developing scoliosis should warrant attention (see section Factors contributing to growth retardation).
Growth from 12–18 years and later: growth from 12–18 years and later
During the adolescent years, height remains significantly below average,16,21,25 and the majority of children with CHARGE syndrome have absent or delayed puberty due to HH.17 These children will not undergo a pubertal growth spurt unless treated with hormone replacement therapy (HH is discussed in section Factors contributing to growth retardation).
Weight tends to increase in adolescence and adulthood, which poses a risk for the development of obesity, and there is some anecdotal evidence that older individuals with CHARGE syndrome are at risk for developing obesity.14,28 However, other studies are less conclusive, indicating that weight is still below average.7,16,21,25,31 In a study of 30 adolescents and adults with CHARGE syndrome, Forward et al found that 74% had normal weight, 15% were underweight and 11% were overweight.31
Factors contributing to growth retardation
There are many different characteristics in CHARGE syndrome that may negatively influence growth, with feeding difficulties, cardiac malformations, frequent hospitalization and multiple surgeries being particularly important. In addition, endocrinological problems such as growth hormone deficiency, HH and hypothyroidism can negatively influence growth. Below we review the most common factors that may influence growth in CHARGE syndrome per age group.
0–3 years of age
Feeding difficulties
The prevalence of feeding difficulties in CHARGE syndrome is almost 100%. In addition, up to 92% of individuals with CHARGE syndrome have been tube fed at some point in their life.32 These numbers indicate that feeding difficulties are a very important issue in CHARGE syndrome. The decrease in growth rate in early infancy and the slow weight gain during the largest part of childhood strongly suggest that these feeding difficulties are an important factor in growth retardation.
While feeding difficulties can occur at any age, they probably influence growth most in early childhood. There are many different reasons why feeding is at risk in CHARGE syndrome. These include problems with chewing, sucking and swallowing. These issues are mainly due to cranial nerve dysfunction, but they are also influenced by hypotonia and anatomical variations of the facio-oral region, such as atresia of choanae and cleft lip and/or palate. A vascular ring can also impede food passing down through the esophagus. In addition, sensory problems such as hypersensitivity and anosmia can make eating or drinking uncomfortable or unpleasant. Aberrant feeding behavior, like pocketing food or unusual chewing or swallowing patterns are common in CHARGE syndrome.32–35
In addition to problems with ingestion, gastro-intestinal problems can also have a negative influence on feeding. Gastro-esophageal reflux is a common problem in CHARGE syndrome, and abdominal migraine and constipation can cause abdominal pain and reduced appetite.35
Critical illness, multiple surgeries and hospital admissions
Children with CHARGE syndrome often require many medical interventions and hospitalizations, particularly at a young age.36 Research among children from the general population in the pediatric intensive care unit and neonatal intensive care unit has shown that these children, especially infants, have an increased risk of malnourishment and growth retardation that is probably caused by a combination of increased metabolic state and feeding difficulties.37,38
Cardiovascular malformations
Cardiovascular malformations are common in CHARGE syndrome, with 65–92% of patients having a cardiovascular malformation. Many different types of malformations can occur, with the most prevalent being conotruncal defects, septal defects and atrioventricular septal defects.6,7,39 As discussed above, a vascular ring can cause swallowing problems if it occludes the esophagus.33 Cardiovascular malformations are also associated with malnutrition and failure to thrive, probably due to a combination of a hypermetabolic state, inadequate caloric intake, feeding difficulties and gastro-intestinal problems.40 Of note, poor growth in children with cardiovascular malformations has been associated with poorer physical and neurodevelopmental outcomes.40,41
4–12 years of age
Skeletal abnormalities
Scoliosis is a common problem in CHARGE syndrome due to decreased muscle tone. In a survey of 31 older patients with CHARGE syndrome, 19 were reported to have scoliosis with variable severity, and the mean age at which scoliosis was diagnosed was 6.25 years.14 The prevalence of scoliosis was also high in another cohort of older individuals with CHARGE syndrome, where Forward et al reported a prevalence of 50% in a cohort of individuals aged 13–34 years.31 Scoliosis can negatively influence height and usually deteriorates with age. Therefore, when considering growth hormone treatment, a physical examination should be conducted with special attention paid to the presence of scoliosis because of the risk of deterioration with increased growth rate.
Growth hormone deficiency, hypothyroidism and hypoparathyroidism
Because most individuals with CHARGE syndrome have short stature and HH, other pituitary functions in CHARGE syndrome have been frequently studied. Combined pituitary dysfunction is uncommon in CHARGE syndrome. Some authors have reported hypothyroidism among patients with CHARGE syndrome with a prevalence of 12–18%.16,21,23 However, Pinto et al did not report hypothyroidism among 26 patients tested for abnormal levels of TSH and free T4.18 Incidental cases of hypoparathyroidism have been found in patients with typical and atypical CHARGE syndrome.7,16,42 Given their low prevalence, the risk of poor growth due to hypothyroidism or hypoparathyroidism in CHARGE syndrome is probably small.
Growth hormone deficiency is more common in CHARGE syndrome, being reported with a frequency of 12–34% in several studies.7,15,18,23 Growth hormone therapy may be a safe and effective way to increase growth in patients with CHARGE syndrome suffering from growth hormone deficiency, although no randomized controlled trials are available. In a cohort study of 16 patients with CHARGE syndrome who were registered in the Pfizer international growth database for patients treated with growth hormone, growth velocity increased after growth hormone supplementation and adverse events were generally mild, although one patient with kyphoscoliosis was reported.43 Therefore, when considering growth hormone treatment, a physical examination should be conducted with special attention to the possible presence of scoliosis due to its increased prevalence in CHARGE syndrome and the possible risk of deterioration with increased growth velocity.
From 12–18 years
Hypogonadotropic hypogonadism
Delayed or absent puberty due to HH is common in CHARGE syndrome. The prevalence of HH is estimated to be between 60% and 88%, with HH being more common in boys. In boys, cryptorchidism and micropenis may be signs of HH, while girls usually have normally developed external genitalia. However, in a minority of cases, girls present with hypoplastic labia at birth. Other genitourinary malformations related to HH in girls with CHARGE syndrome are rare.6,7,16–18,44
HH can be diagnosed in the first 6 months of life or at pubertal age by the detection of low blood levels of luteinising hormone and follicle stimulating hormone, together with low levels of testosterone in boys and estradiol in girls. Due to a strong correlation between olfactory function and HH in CHARGE syndrome, the presence of anosmia is a strong indicator of HH.17,18 This co-occurrence of anosmia and HH also occurs in Kallmann syndrome, which suggests an overlap between both syndromes. Several studies have shown that approximately 6% of patients previously diagnosed with Kallmann syndrome have a CHD7 gene mutation, and most of these patients appeared to have more CHARGE syndrome characteristics after close examination.20,45–47
HH can be treated by hormone replacement therapy, either with testosterone or human chorionic gonadotropin (which stimulates the production of testosterone in the testis) in boys and estrogens in girls.48 Currently, there is no evidence-based data on what is the preferred therapy for male HH in CHARGE syndrome.
HH may contribute to a short stature because patients with HH lack a pubertal growth spurt. When hormone replacement therapy is not started, growth will slowly continue through adolescent years due to delayed epiphyseal closure.49 This may lead to a taller final height, but usually results in characteristic body proportions with relatively long limbs and a short trunk.
Another important complication of delayed puberty is osteoporosis. The risk for osteoporosis is probably further increased in patients with CHARGE syndrome due to feeding difficulties and a reduced level of physical activity.31 Thus, in addition to inducing the development of secondary sexual physical characteristics, hormone replacement therapy may be important to both prevent osteoporosis and improve growth.
Recommendations for multidisciplinary care
The need for a multidisciplinary approach to the medical problems in children with CHARGE syndrome was already being discussed in 1990 when Blake et al studied 50 patients with CHARGE association, the majority of whom underwent at least one major surgical intervention.50 The authors concluded that outcome could be improved by collaboration between specialist surgical teams and suggested that a pediatric Ear Nose Throat (ENT)-specialist and a general or community pediatrician would be the most appropriate coordinators of the long-term multidisciplinary management of these patients. Since then, only a few articles have been published on the multidisciplinary care of children with CHARGE syndrome,51 which is remarkable given that the expanding phenotype of CHARGE syndrome44 has led to an increased number of medical disciplines becoming involved in the care of children with CHARGE syndrome.
The healthcare transition from pediatrics into adult care systems requires special attention. In the last decades, there has been an increased awareness of the importance of healthcare transition into adulthood of patients with pediatric-onset conditions.52 However, effective transition remains challenging, particularly for patients with rare and complex conditions.53 In addition, many studies have shown that people with intellectual disabilities experience health disparities, partly due to limited access to care.54,55
Inefficient and siloed systems, lack of resources, lack of communication and collaboration between professionals, and a lack of knowledge among adult health care professionals about pediatric-onset conditions, rare diseases and intellectual disability have been recognized as barriers to effective transition of care and targets for improvement of transition.56
There are many different ways to organize healthcare transition. A review by Gabriel et al showed that different types of transition strategies have proved to be successful with regard to population health, consumer experience and utilization of care as long as they provide a systematic approach.57 Thus, depending on the local situation, effective health care transition can be modelled in several ways. However, the following aspects should be incorporated: the transition should be started in a timely manner, it should involve a broad view on care (such as attention to issues like fertility and legal representation), it should involve extensive communication between the pediatric team and the adult team and there should be a coordinating care provider. The American Academy of Pediatrics advises to start planning healthcare transition from 12–14 years of age.52 In CHARGE syndrome, multidisciplinary CHARGE clinics can play an important role in this transition, either by providing adult care themselves or by educating local healthcare providers about CHARGE syndrome and patient-specific topics. During childhood, a pediatrician is usually the coordinating health care provider. In adulthood, this role should be transferred to a physician working with adults, preferably someone with expertise on complex disorders.58,59
Growth monitoring is considered part of standard pediatric care. It is important for identifying children who are at risk of undernutrition or have a medical condition that affects growth, and appears to be cost-effective.60 There is some evidence that retarded growth in early childhood is associated with worse neurodevelopmental outcomes and might also negatively influence adult health parameters. However, this research is complicated by the presence of confounders.61,62 Research among children with congenital heart disease showed that low weight for age was associated with prolonged hospitalization, a higher rate of infections and a higher mortality rate after cardiac surgery.40,41 The largest decrease in growth velocity in CHARGE can be seen in the first years of life. This decrease, which occurs during the most critical period of life from a medical perspective, suggests that at least part of this early growth retardation is influenced by chronic illness, multiple surgeries and feeding difficulties. This provides a challenge for professionals: How can we limit the number of hospital visits and medical procedures and optimize nutritional status? However, there are other health problems common in CHARGE syndrome that can also influence growth. Given the complex nature of the syndrome and the co-occurrence of these problems, a multi-disciplinary approach is essential. In Table 2 we summarize the most important problems and which professionals we suggest should be consulted.
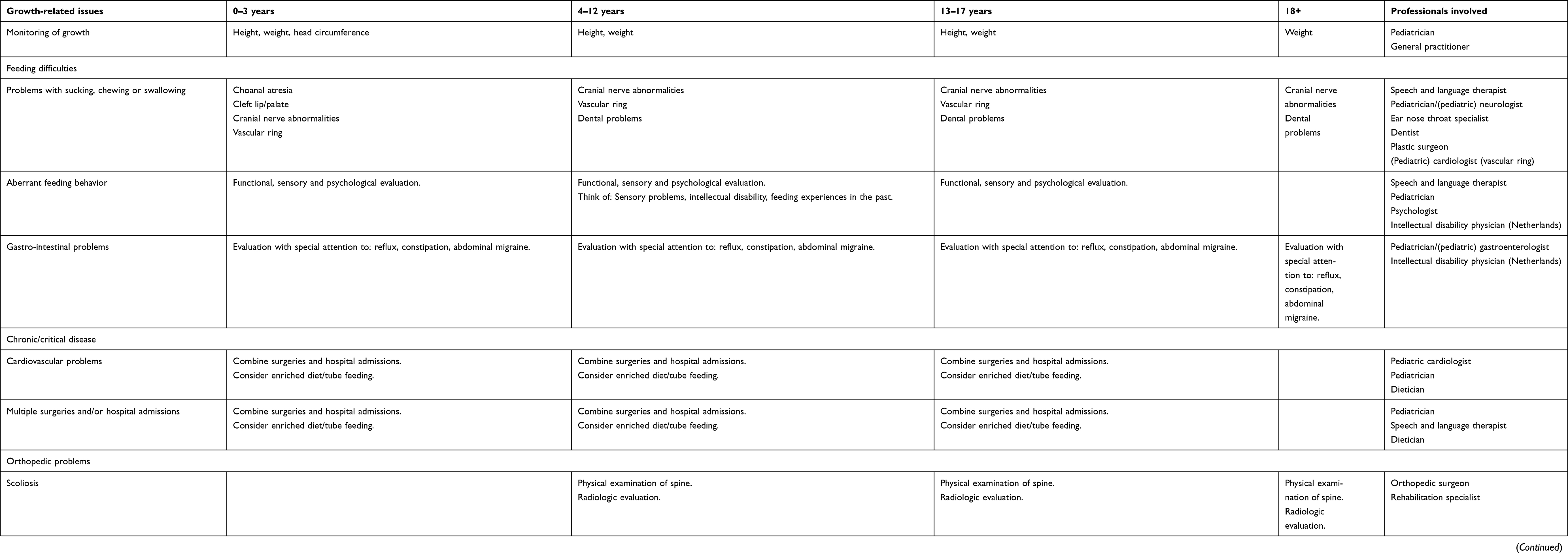 |
Table 2 Expert-based advice for multidisciplinary approach/guidance of medical problems in CHARGE syndrome |
Growth monitoring and health follow-up is preferably done by a pediatrician who has regular contact with the parents and who can decide when consultation with other professionals is necessary. A useful guideline and checklist was provided by Trider et al and supplemented with radiological guidelines by de Geus et al.63,64 The discussion below provides more information with regard to specific growth-related issues.
Feeding difficulties
At birth, children with CHARGE syndrome should be checked for choanal atresia by an ENT-specialist. Orofacial anomalies that require surgery, such as choanal atresia and cleft lip and/or palate, can be treated by an ENT specialist or plastic surgeon.63 When feeding difficulties are present, a speech therapist should be consulted for feeding analysis.63 Based on the nature of the problems, other professionals can be involved (see Table 2). Blake and Hudson have written a useful review of feeding difficulties in CHARGE syndrome that includes practical advice for specific problems.33
Chronic or critical illness
At birth, all children with CHARGE syndrome should be screened for cardiovascular malformations, including a vascular ring, by means of a cardiac ultrasound and chest X-ray.63 When cardiovascular malformations are present, the child has to be referred to a pediatric cardiologist. The number of hospital admissions and anesthesias should be limited as much as possible. While in hospital, and before and after surgical intervention, extra attention should be paid to possible feeding difficulties and undernutrition. A dietician and speech therapist can help to improve feeding and nutritional status.40
Endocrine issues
Given the possibility of diagnosing HH during the so-called mini-puberty of infancy, a pediatric endocrinologist should be consulted in the first weeks or months of life and then again when the patient is reaching pubertal age or when growth hormone deficiency is suspected. Evaluation for hypothyroidism and hypoparathyroidism is only necessary if there are clinical signs or symptoms.
Conclusion
Growth retardation and short stature are present in the majority of individuals with CHARGE syndrome. Height and weight remain low in CHARGE children in all age groups, and adults typically have short stature. However, the largest decrease in growth velocity occurs in the first years of life. Given the broad spectrum of condition-related factors that might influence growth retardation, this complex problem requires a multidisciplinary approach towards diagnostics and treatment.
Strengths and limitations of this study and recommendations for further research
In this review we systematically searched for previously published growth data to present the most extensive overview of growth data published thus far. However, because of the different ways of reporting growth data in the different studies and the low patient numbers per study, it was only possible to get a general overview of growth in CHARGE syndrome. Moreover, interpretation of body weight data is difficult because of the generally short stature. To be truly applicable in daily clinical practice, systematic collection of growth data and construction of CHARGE-syndrome-specific growth charts is necessary.
We have also described many factors that can negatively influence growth in CHARGE syndrome. However, no studies in children with CHARGE syndrome have proven that there is a statistically significant correlation between these conditions and growth retardation. The recommendations we make regarding interventions to improve care are therefore based on expert opinions. An extensive growth study with an analysis of the factors that influence growth would allow us to gain more insight into growth-related problems in CHARGE syndrome. For example, it would be interesting to know whether there is an intrinsic CHD7 gene haploinsufficiency-effect on growth. Of the studies in this review, only Aramaki et al and Shoji et al described patients in sufficient detail to make it possible to compare their growth to their genotype.16,21 Dauber et al also presents one patient with a missense mutation and describes the growth characteristics of this patient.25
Aramaki and Shoji described 12 patients with a nonsense mutation, 13 patients with a frameshift mutation, 6 patients with a splice site mutation and 3 patients with a deletion.16,21 In this small cohort, we could not find a correlation between these different mutation types and growth. Due to the fact that we only had growth data for one patient with a missense mutation, we could not look into the effect of truncating vs non-truncating mutations on growth. In addition, some studies describe familial cases with CHARGE syndrome or cases of unrelated patients with identical CHD7 gene variants. The effect of identical gene variants on growth appears to be variable, which is in line with the wide variation in other characteristics of CHARGE syndrome observed in patients with identical gene mutations.6,7,11,12,65–67
Finally, knowledge about the risks and benefits of growth hormone therapy and hormone replacement therapy for HH in CHARGE syndrome remains limited. More research is needed to be able to make an evidence-based guideline on those therapies.
Acknowledgments
The authors would like to thank Nicole Corsten-Janssen for reviewing and Kate McIntyre for editing the manuscript.
This publication was made possible with funding from SBOH, opleiding arts verstandelijk gehandicapten and stichting vrienden Beatrix kinderziekenhuis.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Janssen N, Bergman JEH, Swertz MA, et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat. 2012;33(8):1149–1160. doi:10.1002/humu.22086
2. Pagon RA, Graham JM
3. Blake KD, Davenport SL, Hall BD, et al. CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila). 1998;37(3):159–173. doi:10.1177/000992289803700302
4. Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet. 2005;133A(3):306–308. doi:10.1002/ajmg.a.30559
5. Vissers LELM, van Ravenswaaij CMA, Admiraal R, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36(9):955–957. doi:10.1038/ng1407
6. Jongmans MCJ, Admiraal RJ, van der Donk KP, et al. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet. 2006;43(4):306–314. doi:10.1136/jmg.2005.036061
7. Legendre M, Abadie V, Attie-Bitach T, et al. Phenotype and genotype analysis of a French cohort of 119 patients with CHARGE syndrome. Am J Med Genet C Semin Med Genet. 2017;175(4):417–430. doi:10.1002/ajmg.c.31591
8. Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome–the clinical overlap with CHARGE syndrome. Clin Genet. 2009;75(1):65–71. doi:10.1111/j.1399-0004.2008.01107.x
9. Marcos S, Sarfati J, Leroy C, et al. The prevalence of CHD7 missense versus truncating mutations is higher in patients with Kallmann syndrome than in typical CHARGE patients. J Clin Endocrinol Metab. 2014;99(10):2138. doi:10.1210/jc.2014-2110
10. Bergman JEH, Janssen N, van der Sloot AM, et al. A novel classification system to predict the pathogenic effects of CHD7 missense variants in CHARGE syndrome. Hum Mutat. 2012;33(8):1251–1260. doi:10.1002/humu.22106
11. Lalani SR, Safiullah AM, Fernbach SD, et al. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am J Hum Genet. 2006;78(2):303–314. doi:10.1086/500273
12. Jongmans MCJ, Hoefsloot LH, van der Donk KP, et al. Familial CHARGE syndrome and the CHD7 gene: a recurrent missense mutation, intrafamilial recurrence and variability. Am J Med Genet A. 2008;146A(1):43–50. doi:10.1002/ajmg.a.31921
13. Hale CL, Niederriter AN, Green GE, Martin DM. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A. 2016;170A(2):344–354. doi:10.1002/ajmg.a.37435
14. Blake KD, Salem-Hartshorne N, Daoud MA, Gradstein J. Adolescent and adult issues in CHARGE syndrome. Clin Pediatr (Phila). 2005;44(2):151–159. doi:10.1177/000992280504400207
15. Husu E, Hove HD, Farholt S, et al. Phenotype in 18 Danish subjects with genetically verified CHARGE syndrome. Clin Genet. 2013;83(2):125–134. doi:10.1111/j.1399-0004.2012.01884.x
16. Shoji Y, Ida S, Etani Y, et al. Endocrinological characteristics of 25 Japanese patients with CHARGE syndrome. Clin Pediat Endocrinol. 2014;23(2):45–51. doi:10.1297/cpe.23.45
17. Bergman JEH, Bocca G, Hoefsloot LH, Meiners LC, van Ravenswaaij-Arts CMA. Anosmia predicts hypogonadotropic hypogonadism in CHARGE syndrome. J Pediatr. 2011;158(3):474–479. doi:10.1016/j.jpeds.2010.08.032
18. Pinto G, Abadie V, Mesnage R, et al. CHARGE Syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J Clin Endocrinol Metab. 2005;90(10):5621–5626. doi:10.1210/jc.2004-2474
19. Hartshorne TS, Stratton KK, Brown D, Madhavan-Brown S, Schmittel MC. Behavior in CHARGE syndrome. Am J Med Genet C Semin Med Genet. 2017;175(4):431–438. doi:10.1002/ajmg.c.31588
20. Bergman JE, de Ronde W, Jongmans MC, et al. The results of CHD7 analysis in clinically well-characterized patients with Kallmann syndrome. J Clin Endocrinol Metab. 2012;97(5):858. doi:10.1210/jc.2011-2652
21. Aramaki M, Udaka T, Kosaki R, et al. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J Pediatr. 2006;148(3):410–414. doi:10.1016/j.jpeds.2005.10.044
22. Aguiar-Oliveira MH, Davalos C, Campos VC, Oliveira Neto LA, Marinho CG, Oliveira CRP. Hypothalamic abnormalities: growth failure due to defects of the GHRH receptor. Growth Horm IGF Res. 2018;38:14–18. doi:10.1016/j.ghir.2017.12.011
23. Asakura Y, Toyota Y, Muroya K, et al. Endocrine and radiological studies in patients with molecularly confirmed CHARGE syndrome. J Clin Endocrinol Metab. 2008;93(3):920–924. doi:10.1210/jc.2007-1419
24. Blake K, Kirk JM, Ur E. Growth in CHARGE association. Arch Dis Child. 1993;68(4):508–509. doi:10.1136/adc.68.4.508
25. Dauber A, Hirschhorn JN, Picker J, Maher TA, Milunsky A. Delayed puberty due to a novel mutation in CHD7 causing CHARGE syndrome. Pediatrics. 2010;126(6):1594. doi:10.1542/peds.2010-0164
26. Dörr HG, Madeja J, Junghans C. Spontaneous postnatal growth is reduced in children with CHARGE syndrome. Acta Paediatr Int J Paediatr. 2015;104(7):e318. doi:10.1111/apa.12980
27. Harvey AS, Leaper PM, Bankier A. CHARGE association: clinical manifestations and developmental outcome. Am J Med Genet. 1991;39(1):48–55. doi:10.1002/ajmg.1320390112
28. Searle LC, Graham JM
29. Legendre M, Gonzales M, Goudefroye G, et al. Antenatal spectrum of CHARGE syndrome in 40 fetuses with CHD7 mutations. J Med Genet. 2012;49(11):698–707. doi:10.1136/jmedgenet-2012-100926
30. Khadilkar VV, Cameron FJ, Stanhope R. Growth failure and pituitary function in CHARGE and VATER associations. Arch Dis Child. 1999;80(2):167–170. doi:10.1136/adc.80.2.167
31. Forward KE, Cummings EA, Blake KD. Risk factors for poor bone health in adolescents and adults with CHARGE syndrome. Am J Med Genet A. 2007;143A(8):839–845. doi:10.1002/ajmg.a.31670
32. Dobbelsteyn C, Peacocke SD, Blake K, Crist W, Rashid M. Feeding difficulties in children with CHARGE syndrome: prevalence, risk factors, and prognosis. Dysphagia. 2008;23(2):127–135. doi:10.1007/s00455-007-9111-6
33. Blake KD, Hudson AS. Gastrointestinal and feeding difficulties in CHARGE syndrome: a review from head-to-toe. Am J Med Genet Part C Semin Med Genet. 2017;175(4):496–506. doi:10.1002/ajmg.c.31586
34. Dobbelsteyn C, Marche DM, Blake K, Rashid M. Early oral sensory experiences and feeding development in children with CHARGE syndrome: a report of five cases. Dysphagia. 2005;20(2):89–100. doi:10.1007/s00455-004-0026-1
35. Hudson A, Macdonald M, Friedman JN, Blake K. CHARGE syndrome gastrointestinal involvement: from mouth to anus. Clin Genet. 2017;92(1):10–17. doi:10.1111/cge.12892
36. Blake K, MacCuspie J, Hartshorne TS, Roy M, Davenport SLH, Corsten G. Postoperative airway events of individuals with CHARGE syndrome. Int J Pediatr Otorhinolaryngol. 2008;73(2):219–226. doi:10.1016/j.ijporl.2008.10.005
37. Valla F, Berthiller J, Gaillard-Le-Roux B, et al. Faltering growth in the critically ill child: prevalence, risk factors, and impaired outcome. Eur J Pediatr. 2018;177(3):345–353. doi:10.1007/s00431-017-3062-1
38. Hulst J, Joosten K, Zimmermann L, et al. Malnutrition in critically ill children: from admission to 6 months after discharge. Clin Nutr. 2004;23(2):223–232. doi:10.1016/S0261-5614(03)00130-4
39. Corsten-Janssen N, Scambler PJ. Clinical and molecular effects of CHD7 in the heart. Am J Med Genet Part C Semin Med Genet. 2017;175(4):487–495. doi:10.1002/ajmg.c.31590
40. Medoff-Cooper B, Ravishankar C. Nutrition and growth in congenital heart disease: a challenge in children. Curr Opin Cardiol. 2013;28(2):122–129. doi:10.1097/HCO.0b013e32835dd005
41. Ravishankar C, Zak V, Williams IA, et al. Association of impaired linear growth and worse neurodevelopmental outcome in infants with single ventricle physiology: a report from the pediatric heart network infant single ventricle trial. J Pediatr. 2013;162(2):
42. Jain S, Kim HG, Lacbawan F, et al. Unique phenotype in a patient with CHARGE syndrome. Int J Pediatr Endocrinol. 2011;2011:11. doi:10.1186/1687-9856-2011-11
43. Dörr HG, Boguszewski M, Dahlgren J, et al. Short children with charge syndrome: do they benefit from growth hormone therapy? Horm Res Paediatr. 2015;84(1):49–53. doi:10.1159/000382017
44. Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet. 2011;48(5):334–342. doi:10.1136/jmg.2010.087106
45. Balasubramanian R, Choi JH, Francescatto L, et al. Functionally compromised CHD7 alleles in patients with isolated GnRH deficiency. Proc Natl Acad Sci USA. 2014;111(50):17953–17958. doi:10.1073/pnas.1417438111
46. Costa-Barbosa FA, Balasubramanian R, Keefe KW, et al. Prioritizing genetic testing in patients with kallmann syndrome using clinical phenotypes. J Clin Endocrinol Metab. 2013;98(5):
47. Xu C, Cassatella D, van der Sloot AM, et al. Evaluating CHARGE syndrome in congenital hypogonadotropic hypogonadism patients harboring CHD7 variants. Genet Med. 2018;20(8):872–881. doi:10.1038/gim.2017.197
48. Dunkel L, Quinton R. Transition in endocrinology: induction of puberty. Eur J Endocrinol. 2014;170(6):R239. doi:10.1530/EJE-13-0894
49. Zirilli L, Rochira V, Diazzi C, Caffagni G, Carani C. Human models of aromatase deficiency. J Steroid Biochem Mol Biol. 2008;109(3–5):212–218. doi:10.1016/j.jsbmb.2008.03.026
50. Blake KD, Russell-Eggitt IM, Morgan DW, Ratcliffe JM, Wyse RK. Who’s in CHARGE? Multidisciplinary management of patients with CHARGE association. Arch Dis Child. 1990;65(2):217–223. doi:10.1136/adc.65.2.217
51. Blake KD, Prasad C. CHARGE syndrome. Orphanet J Rare Dis. 2006;1:34. doi:10.1186/1750-1172-1-34
52. White PH, Cooley WC. Supporting the health care transition from adolescence to adulthood in the medical home. Pediatrics. 2018;142:5. doi:10.1542/peds.2018-2587
53. Van Lierde A, Menni F, Bedeschi MF, et al. Healthcare transition in patients with rare genetic disorders with and without developmental disability: neurofibromatosis 1 and Williams-Beuren syndrome. Am J Med Genet A. 2013;161A(7):1666–1674. doi:10.1002/ajmg.a.35982
54. Hughes-McCormack LA, Rydzewska E, Henderson A, MacIntyre C, Rintoul J, Cooper S. Prevalence and general health status of people with intellectual disabilities in Scotland: a total population study. J Epidemiol Community Health. 2018;72(1):78–85. doi:10.1136/jech-2017-209748
55. Krahn GL, Hammond L, Turner A. A cascade of disparities: health and health care access for people with intellectual disabilities. Ment Retard Dev Disabil Res Rev. 2006;12(1):70–82. doi:10.1002/mrdd.20098
56. Franklin MS, Beyer LN, Brotkin SM, Maslow GR, Pollock MD, Docherty SL. Health care transition for adolescent and young adults with intellectual disability: views from the parents. J Pediatr Nurs. 2019;47:148–158. doi:10.1016/j.pedn.2019.05.008
57. Gabriel P, McManus M, Rogers K, White P. Outcome evidence for structured pediatric to adult health care transition interventions: a systematic review. J Pediatr. 2017;188:
58. Schrander-Stumpel CTRM, Sinnema M, van Den Hout L, et al. Healthcare transition in persons with intellectual disabilities: general issues, the Maastricht model, and Prader-Willi syndrome. Am J Med Genet C Semin Med Genet. 2007;145C(3):241–247. doi:10.1002/ajmg.c.30136
59. Peron A, Canevini MP, Ghelma F, Di Marco F, Vignoli A. Healthcare transition from childhood to adulthood in tuberous sclerosis complex. Am J Med Genet C Semin Med Genet. 2018;178(3):355–364. doi:10.1002/ajmg.c.31653
60. Craig D, Fayter D, Stirk L, Crott R. Growth monitoring for short stature: update of a systematic review and economic model. Health Technol Assess. 2011;15(11):iii.
61. Victora CG, Adair L, Fall C, et al. Maternal and child undernutrition: consequences for adult health and human capital. Lancet. 2008;371(9609):340–357. doi:10.1016/S0140-6736(07)61692-4
62. Martorell R, Horta BL, Adair LS, et al. Weight gain in the first two years of life is an important predictor of schooling outcomes in pooled analyses from five birth cohorts from low- and middle-income countries. J Nutr. 2010;140(2):348–354. doi:10.3945/jn.109.112300
63. Trider C, Arra-Robar A, van Ravenswaaij-Arts C, Blake K. Developing a CHARGE syndrome checklist: health supervision across the lifespan (from head to toe). Am J Med Genet Part A. 2017;173(3):684–691. doi:10.1002/ajmg.a.38085
64. de Geus CM, Free RH, Verbist BM, et al. Guidelines in CHARGE syndrome and the missing link: cranial imaging. Am J Med Genet Part C Semin Med Genet. 2017;175(4):450–464. doi:10.1002/ajmg.c.31593
65. Wincent J, Holmberg E, Stromland K, et al. CHD7 mutation spectrum in 28 Swedish patients diagnosed with CHARGE syndrome. Clin Genet. 2008;74(1):31–38. doi:10.1111/j.1399-0004.2008.01014.x
66. Delahaye A, Sznajer Y, Lyonnet S, et al. Familial CHARGE syndrome because of CHD7 mutation: clinical intra- and interfamilial variability. Clin Genet. 2007;72(2):112–121. doi:10.1111/j.1399-0004.2007.00821.x
67. Pauli S, Pieper L, Häberle J, et al. Proven germline mosaicism in a father of two children with CHARGE syndrome. Clin Genet. 2009;75(5):473–479.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.