")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Greener synthesis of electrospun collagen/hydroxyapatite composite fibers with an excellent microstructure for bone tissue engineering
Authors Zhou Y, Yao H, Wang J, Wang D, Liu Q, Li Z
Received 13 December 2014
Accepted for publication 17 March 2015
Published 29 April 2015 Volume 2015:10(1) Pages 3203—3215
DOI https://doi.org/10.2147/IJN.S79241
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Lei Yang
Yuanyuan Zhou,1,2 Hongchang Yao,1 Jianshe Wang,1 Dalu Wang,1 Qian Liu,1 Zhongjun Li1
1College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, People’s Republic of China; 2Institute of Enviromental and Municipal Engineering, North China University of Water Resources and Electric Power, Zhengzhou, People’s Republic of China
Abstract: In bone tissue engineering, collagen/hydroxyapatite (HAP) fibrous composite obtained via electrospinning method has been demonstrated to support the cells’ adhesion and bone regeneration. However, electrospinning of natural collagen often requires the use of cytotoxic organic solvents, and the HAP crystals were usually aggregated and randomly distributed within a fibrous matrix of collagen, limiting their clinical potential. Here, an effective and greener method for the preparation of collagen/HAP composite fibers was developed for the first time, and this green product not only had 40 times higher mechanical properties than that previously reported, but also had an excellent microstructure similar to that of natural bone. By dissolving type I collagen in environmentally friendly phosphate buffered saline/ethanol solution instead of the frequently-used cytotoxic organic solvents, followed with the key step of desalination, co-electrospinning the collagen solution with the HAP sol, generates a collagen/HAP composite with a uniform and continuous fibrous morphology. Interestingly, the nano-HAP needles were found to preferentially orient along the longitudinal direction of the collagen fibers, which mimicked the nanostructure of natural bones. Based on the characterization of the related products, the formation mechanism for this novel phenomenon was proposed. After cross-linking with 1-ethyl-3-(3-dimethyl-aminopropyl)-1-carbodiimide hydrochloride/N-hydroxysuccinimide, the obtained composite exhibited a significant enhancement in mechanical properties. In addition, the biocompatibility of the obtained composite fibers was evaluated by in vitro culture of the human myeloma cells (U2-OS). Taken together, the process outlined herein provides an effective, non-toxic approach for the fabrication of collagen/HAP composite nanofibers that could be good candidates for bone tissue engineering.
Keywords: collagen, hydroxyapatite, electrospinning, nanofibers, biomaterials
Introduction
Natural bone is an innate example of inorganic–organic biocomposites, with a composition of approximately 70 wt% inorganic crystals hydroxyapatite (HAP) with a chemical formula of Ca10(PO4)6(OH)2 and 30 wt% of organic matrix.1,2 The organic matrix is mainly type I collagen which provides bone with flexibility and resilience, and the inorganic phase is responsible for the stiffness and strength of bone. The unique characteristics of natural bones are the spatial orientation between the inorganic crystals and collagen macromolecules at the nanolevel,3 where nanocrystals (about 50 nm length) of biological HAP are aligned parallel to the collagen fibrils, which is believed to be the source of the mechanical strength of bones.4 Therefore, to a large extent such a collagen/HAP fibrous composite with a similar composition and microstructure to natural bone has been regarded as a prospective candidate for bone tissue engineering and attracted much attention in recent years.
Conventionally, collagen/HAP biocomposites can be prepared by blending or mixing of collagen and HAP, as well as by biomimetic methods.5–16 However, in all blended composites, the crystallite sizes of HAP particles were not uniform and the crystals were often aggregated and randomly distributed within a fibrous matrix of collagen. In recent years, many researchers have turned toward the process of electrospinning in order to engineer a successful bone substitute material. Electrospinning is a process that uses an electric field to produce fibers with a large surface-to-volume ratio, high mechanical strength, and good biomimetic properties. Previous studies have shown that electrospun fibrous architecture is beneficial to the proliferation and differentiation of various stem/progenitor cells.17–39 Therefore, the preparation of collagen/HAP fibrous biocomposites by electrospinning method set off a frenzy of investigation. However, the collagen/HAP composite was usually fabricated via electrospinning the solution prepared by dissolving collagen and dispersing HAP powders simultaneously in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) or 2,2,2-trifluoroethanol (TFE).40–42 Not only can HFIP and TFE induce an apparent loss of the triple helical configuration of collagen, but they can remain a highly volatile and corrosive solvent that poses health risks to humans, as well as its high price.43–46 In consideration of the shortcoming of HFIP, several attempts have recently been made at finding more eco and environmentally benign solvents to prepare collagen solutions for electrospinning. For example, various acids such as formic acid47,48 and acetic acid,49,50 were investigated as less cytotoxic solvents for electrospinning collagen nanofibers. However, the acid-induced partial degradation of collagen and the instability of collagen in acid solutions may adversely affect the structural and material integrity of collagen nanofibers.51 Dong et al have studied a phosphate buffered saline (PBS)/ethanol system as a solvent for collagen electrospinning and investigated the effect of salt concentration on the diameter of collagen fibers,52,53 but a large quantity of salts introduced by PBS were not further removed, which certainly affected the distribution of HAP in collagen matrix and the mechanical properties of the composite materials when co-electrospinning with HAP nanoparticles. Besides, Luciana et al have put forward the ionic liquid as a solvent for dissolving collagen at 100°C, which only emphasized the utility of the green solvent in the dissolution of collagen, but ignored the clinical application of collagen as a biomaterial.54,55 In addition, most of the electrospinning solutions (or dispersions) were prepared by a conventional mixing method in which HAP was normally obtained firstly in powders and followed by mixing it with collagen solutions.40–42,56 Consequently, in the prepared collagen/HAP composites, the particle size of HAP was not uniform and the HAP aggregation was often randomly distributed within the collagen matrix. Such a composite was obviously different from the natural bone microstructure, and only had a compositional similarity to that of natural bone. This inevitably led to weaken mechanical properties of the prepared composites.
Given the above disadvantages in the creation of electrospun collagen/HAP composite, it is desirable to find a more effective and reasonable approach to prepare electrospinning solution with benign solvents. Herein, we report a sol-electrospinning process for greener fabrication of collagen/HAP composite fibrous materials using HAP sol, instead of HAP powder, and PBS/ethanol system as an environmentally friendly solvent for dissolving collagen to prepare electrospinning solutions. After being dialyzed to remove the salts, the collagen solution was mixed with HAP sol and subsequently co-electrospun under appropriate parameters. Followed by the cross-linking with 1-ethyl-3-(3-dimethyl-aminopropyl)-1-carbodiimide hydrochloride/N-hydroxysuccinimide (EDC/NHS), the obtained composite fibers became water-insoluble and exhibited significant enhancement in mechanical properties. Based on the characterization of the related products, the formation mechanism of this green collagen/HAP fibrous composites was proposed and discussed. Moreover, the cytocompatibility of collagen/HAP nanofibers was evaluated through in vitro Cell Counting Kit-8 (CCK-8) viability assay. ALP activity of human myeloma cells (U2-OS) cultured in the leaching liquors of fibrous composites was examined at different time points.
Materials and methods
Preparation of collagen solution and HAP sol
PBS 20× buffer was prepared according to the literature57 by dissolving 160 g sodium chloride (NaCl), 28.8 g disodium phosphate (Na2HPO4), 4 g potassium chloride (KCl), and 4.8 g monopotassium phosphate (KH2PO4) in 1 L distilled water, and the pH was adjusted to 7.4 with hydrochloride (HCl). Lyophilized type-I collagen, extracted from calf skin, was kindly provided by the Tianjin Sannie Bioengineerring Technology Co., Ltd, People’s Republic of China. Collagen solution was prepared by dissolving a 150 mg sample in a 1 mL solvent mixture of PBS buffer and ethanol with a PBS to ethanol ratio of 3:2 (v/v). The HAP sol was synthesized with calcium nitrate (Ca(NO3)2·4H2O, AR, Tianjin Chemical Reagent Factory, People’s Republic of China) and ammonium phosphate ((NH4)3PO4·3H2O, AR, Tianjin Chemical Reagent Factory), using sodium citrate (C6H5Na3O7·2H2O, AR, Shanghai Chemical Reagent Factory, People’s Republic of China) as a dispersant. Briefly, 23.6 g of calcium nitrate and 20 g of sodium citrate were dissolved together in 100 mL of deionized water and the pH value of the resulting solution was adjusted to 12 with NH4OH solution. Subsequently, a certain amount of ammonium phosphate with a molar ratio of Ca/P =1.67 was dissolved in 100 mL of distilled water and then added drop wise to the above solution within a certain time under constant stirring. The pH value of the reacting mixture was maintained in the range of 10–11 by the addition of NH4OH solution. The resultant system was kept at 80°C for 2.5 hours under continuous stirring to produce a light blue translucent HAP sol. After being purified by dialysis, the prepared sol had a HAP concentration of 0.055 g/mL.
Fabrication of electrospun fibers
The HAP sol was mixed with the above mentioned prepared collagen solution at different mass ratios of HAP to collagen. After being vigorously stirred, the solution obtained was fed from a 10 mL syringe with a 6-gauge blunt-tip needle attached. The syringe was mounted onto a syringe pump (LongerPump LSP02-1B, Hebei, People’s Republic of China), and the needle was connected to a high-voltage power supply (Dingtong High Voltage Power Supply, DPS-100 [50 KV/50 w], Dalian, People’s Republic of China). Under 17 kV voltage, the fluid jet was injected out at a rate of 1.0 mL/hour and the resultant nanofibers were collected on aluminum foil which was put at 15 cm distance down from the needle. The electrospinning process was continuously performed for 4 hours at 15°C–20°C with a relative humidity of about 20%. Pure collagen fibers were also electrospun as a control. For comparison, a spinning solution with mass ratio of HAP to collagen equal to 3:7 was also prepared by mixing the collagen solution with the HAP powder prepared without dispersant and then electrospun under the same conditions.
Cross-linking
Electrospun fibers were cross-linked with the EDC/NHS at room temperature. The cross-linking solutions were prepared by introducing 600 mM EDC and 600 mM NHS in a 9:1 (v/v) acetone/water mixture, respectively. The composite fibers were first soaked in the EDC solution for 12 hours, and then fully soaked in the NHS solution for another 12 hours. The cross-linked composite fibers were dried at room temperature overnight, and then rinsed with deionized water three times to remove the residual chemicals.
Characterization of samples
Transmission electron microscopy (TEM) images of HAP nanoparticles in HAP sol and electrospinning solution as well as electrospun nanofibers were obtained via transmission electron microscope (JEM-2010; JEOL, Tokyo, Japan). The viscosity of the spinning solution was measured at 25°C by a rotational viscometer (Model NDJ-79, Shanghai, People’s Republic of China). Morphological characterization of the electrospun nanofibers was performed using a scanning electron microscope (SEM; JSM-5600LV, JEOL) with a beam voltage at 10 kV, and all samples were sputter-coated with gold before SEM observation. The phase structure of HAP in sol and composite nanofibers was analyzed by X-ray diffraction (XRD; PANalytical, Almelo, the Netherlands) using Cu Kα radiation, in the range 20°~60°. Chemical bonding state of collagen/HAP composite nanofibers was analyzed by Fourier-transform infrared spectroscopy (FTIR) using a Thermo Scientific (Nexus 470; Thermo Fisher Scientific, Waltham, MA, USA) spectrometer. Mechanical properties of electrospun fibers were measured using a YG-001N fiber tensile tester at 10 mm/minute crosshead speed with 10 mm gauge length at room temperature. All samples were cut into 30×10 mm rectangles with thickness of 0.05 mm. The thickness of these nanofiber mats was measured using a micrometer. An average of five measurements was reported as the mean ± standard deviation for each sample.
Cell culture
Human myeloma cells (U2-OS) were used in this study, and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific) that was supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific) and incubated at 37°C with 5% CO2. When approximately 70%–90% of the cells was adherent, the culture medium in the pipette bottle was firstly aspirated, and then 5 mL PBS buffer liquid was added into the bottle while gently shaking. Finally, the PBS buffer liquid was poured out, repeating the above steps three times. Cultured cells were digested by trypsin to prepare a cell suspension. The suspension cells were centrifuged for 5 minutes (1,000 rpm/minute). After removing the supernatant, the cells were added into the cell medium to re-prepare the cell suspension at a density of 104 cells/mL.
Cell viability and differentiation
Cell viability was assayed by CCK-8 reagent, and the differentiation of U2-OS cells to osteoblasts was determined by measuring their ALP activity.58 Before cell seeding, the nano-fibrous mats were firstly ground and immersed in 2 mL of physiological saline, respectively. Then treated with sonication, and subsequently sterilized under autoclave at 115°C for 30 minutes. Finally, immersed in medium (DMEM/F12 medium/10%FBS/1% antibiotics) overnight before cell seeding. Then, U2-OS cells were seeded at a density of 1.5×104 cells/well for CCK-8 assay and 2×104 cells/well for ALP activity, respectively. For comparison, polytetrafluoroethylene (PTFE) was used as a control.
For CCK-8, briefly, after incubating at 37°C for 1 hour, 40 mL of CCK-8 reagent was added into each well and incubated for 4 hours, 3, 5, 7 days according to the reagent instruction. Aliquots (150 mL) of incubated medium were pipetted into 12-well and read in a spectrophotometric plate reader at 450 nm (Elx-800, Bio-Tek Instrument Inc., Winooski, VT, USA).
For ALP activity assay, after 1, 2, 3, 4, 5, 6, and 7 days culture, every three wells in twelve wells were made to collect together, and centrifuged at 3,000 rpm/minute. Then, the supernatant was discarded, and washed once with saline to avoid the influence of the medium calf serum enzymes on the experimental results. The precipitate was added into 500 μL of 0.1% Triton for cell lysis overnight at 4°C, and centrifuged for 10 minutes (8,000 rpm/minute). ALP concentration in the supernatant was measured by using automatic biochemical analyzer at the absorbance of 405 nm.
Results and discussion
Characterization of HAP sol
Figure 1A shows that the HAP sol synthesized by the dispersant exhibited a translucent state, which could be stand for up to several months without any obvious segregation of its initial sol state, and the zeta potential of HAP nanoparticles was determined to be about −30.76 mV. From Figure 1B, it can be seen that the HAP nanoparticles had a needle-like morphology with a uniform width of about 10 nm and length of 50 nm or so.
 | Figure 1 Characterization of HAP sol. |
The crystallographic structure of the HAP nanocrystals was investigated by XRD as shown in Figure 2, which shows the peak intensity was proportional to the standard spectrum of reference pattern HAP 00-001-1008. The crystallite size was calculated according to the following Scherrer’s equation:
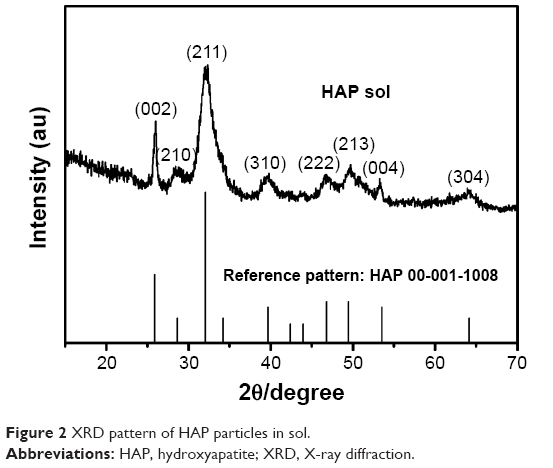 | Figure 2 XRD pattern of HAP particles in sol. |
| (1) |
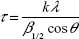
where τ is the average diameter in Å, β is the broadening of the diffraction line measured at half of its maximum intensity in radian, λ=0.1542 nm, k=0.9, and θ is the Bragg’s diffraction angle. Taking into account the broadening of each peak in XRD, the crystallite size of the (002) and (310) planes of the HAP nanoparticles was calculated to be 43.6 nm and 14.2 nm, respectively.
Morphology of the electrospun fibers
Figure 3 shows the morphology of the electrospun pure collagen and collagen/HAP composite fibers obtained with different HAP contents. Pure collagen fibers exhibited a highly smooth surface with an average diameter of 700 nm or so (Figure 3A). The shape of the collagen/HAP composite fibers was not affected by the addition of HAP nanoparticles to the collagen matrix. The collagen/10 wt% HAP composite fibers were continuous and fairly uniform (Figure 3B). When increasing HAP content up to 20%–30%, the obtained collagen/HAP composite fibers had the same homogeneity as the pure collage fibers (Figure 3C, D). In particular, it was noticeable that the spinning solution containing up to 40% HAP nanoparticles could be electrospun into the continuous fibers (Figure 3E). The average size diameter of the collagen/HAP composites shifted from 630 nm to 500 nm, and a gradual decrease in the average fiber size occurred with the increase of HAP nanoparticles. The decrease in the fiber diameter was associated with the reduction in viscosity of the spinning solution. The more HAP the collagen/HAP composite fibers contained, the more volume of HAP sol was added in the spinning solution, which resulted in the decrease of solution viscosity. For the comparative study, a control sample of composite fibers with collagen/30% HAP was also produced by electrospinning the collagen solution with conventional HAP powders being dispersed. As illustrated in Figure 3F, the control composite fibers exhibited a broader average diameter of 820 nm and were less homogeneous compared with the collagen/30% HAP composite fibers prepared using HAP sol. Additionally, some bead-on-string structures were observed, which may be due to the agglomeration of HAP particles. Some HAP aggregates had diameters larger than the average fiber’s diameter and could not be embedded inside the fiber, but protruded from the fiber. Therefore, the control sample was not smooth and uniform as the above five mentioned samples.
 | Figure 3 The morphology of electrospun fibers with different HAP contents. |
Figure 4 presents the TEM images of the fibers and the related spinning solutions. As shown in Figure 4A, the collagen fibers, before desalination, displayed much larger particles inside the fibers, which may be the salt accumulation introduced by PBS buffer. This phenomenon was consistent with the results previously reported.52,53 After blending the collagen with HAP sol, the collagen/HAP composite nanofiber before desalination showed that most of the particles were held along inside the collagen fibers, without any particle exposure on the surface of the fibers (Figure 4B). Our attempts had been performed to figure out the distribution of HAP in the collagen matrix by means of TEM. Unfortunately, the HAP nanoparticles seemed to be covered by the salt accumulation, which made it quite difficult to differentiate where the HAP particle was and where the salt was. Although the mechanisms involved in the composite fiber formation were still unclear, the negative effects of salts existing in collagen or collagen/HAP fibers in the potential application were of no doubt. By the way, if these materials containing high salts were implanted inside the body, the excessive salt concentrations would result in an ionic imbalance and increase the osmotic pressure in the extracellular fluid, which could cause cell dehydration and lead to plasmolysis or loss of cells activity. More seriously, the blood volume would increase, causing hypertension. Moreover, these large particles inside the collagen fibers could probably have a weak interaction with the collagen matrix, resulting in poor mechanical properties. Therefore, the use of PBS/ethanol system for collagen only was insufficient for manufacturing the collagen/HAP fibers, a further desalination for collagen solution was necessary. In this study, removing the salts was effectively achieved by dialysis of collagen solution, and the collagen fibers, after desalination, had a more uniform structure without any sign of particle formation (Figure 4C). More to the point, the collagen/HAP fibers after desalination were totally different from those before desalination. As clearly seen from Figure 4D, the HAP needles were well distributed within the electrospun fibers, and the HAP nanoparticles seemed to have a directional arrangement. The TEM of collagen/HAP spinning solution was presented in Figure 4E, it demonstrated that the needle-like HAP crystallites with a clear boundary were co-embedded in the collagen solution. After electrospinning, the HAP nanoparticles could completely retain their needle-shape in the collagen matrix, and were well-aligned along the axis of the electrospun fiber, which can be seen from the TEM image with a higher resolution (Figure 4F). The HAP crystallites had a crystal particle size range from 20 to 60 nm, close to those found in mammalian bone,59 and the microstructure of the collagen/HAP composite fiber was similar to natural bone at the nano-level.3,4
 | Figure 4 TEM images of the fibers and spinning solutions. |
For the comparative study, the spinning solution containing collagen with dispersed HAP powders and the related composite fiber were also produced with the same process. As illustrated in Figure 4G, it showed a rather poor dispersion of HAP particles in collagen solution with a large degree of aggregation. This poor dispersion could also influence the distribution of HAP particles in the composite fibers, resulting in serious HAP particle agglomeration in some places and no HAP nanoparticles at all in other places (Figure 4H). This aggregation of HAP particles in collagen matrix had basically occurred as reported elsewhere,40,60,61 and did not benefit the formation of smooth, uniform and bead-free fiber morphology (as shown in Figure 3F). The above comparison further highlighted the advantages of using the desalted collagen solution and HAP sol to fabricate electrospun collagen/HAP composite fibers with an excellent microstructure.
Oriented alignment of HAP nanoparticles within collagen matrix
Herein, we have established a schematic illustration to explain how the HAP particles were oriented along the collagen fibers by electrospinning as shown in Figure 5. In this study, sodium citrate was chosen as the dispersing agent for preparing HAP sol. Obviously, the surfaces of HAP particles were negatively charged due to the adsorption of citrate ions, and the diffusion layers of HAP particles accordingly had a positive charge. Due to the double electrical layers of colloidal particle, the HAP particles could maintain good dispersion capability in the spinning solution. During electrospinning, when a high electric field was applied to the spinning solution, the positive diffusion layers began to move in the direction of electric field, which caused the HAP nano-needles to be positively and negatively charged in the two tips, respectively. Under the action of electric field, both the charges on the two tips of HAP particle would receive the opposite forces F1 and F2, which led the HAP particle to rotate to be parallel with the long axis. When the applied electric field force overcame the surface tension of the droplet, a charged jet of spinning solution containing HAP particles was ejected. With the solvent evaporating continuously, the jet including HAP particles extended through the nozzle and grew longer and thinner until it was solidified and collected on the target. At the same time, the needle-shape HAP particles also tended to align along the direction of the fluid jet. Consequently, it was of interest to note that the needle HAP particles were oriented along the long axes of the collagen fiber and were nearly parallel in each electrospun fiber. Therefore, the electrospun composite of collagen and HAP nano-needles not only had the similar chemical composition to natural bone, but also mimicked the microstructure of natural bone to a large extent.
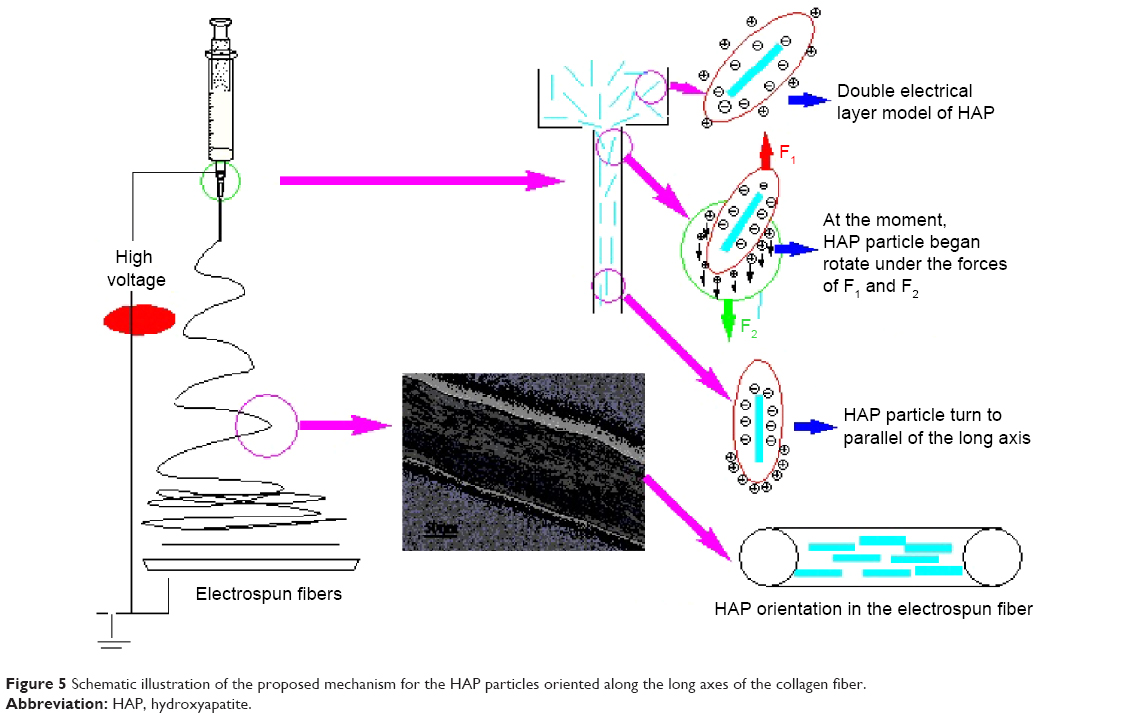 | Figure 5 Schematic illustration of the proposed mechanism for the HAP particles oriented along the long axes of the collagen fiber. |
Chemical analysis of electrospun nanofibers
The representative FTIR absorption spectra of the pure HAP, electrospun collagen, and collagen/HAP composite were shown in Figure 6. The FTIR spectrum of HAP nano-needles corresponding to PO4−3 in the range 500–700 cm−1 was presented in Figure 6A. As shown in Figure 6B and C, the FTIR spectra of electrospun collagen/HAP composite and collagen displayed characteristic absorption peaks at 3,274 and 2,918 cm−1, which represented the amide A and B bands of collagen, respectively. The amide I, amide II, and amide III adsorption peaks typical of collagen were also found at 1,640, 1,540, and 1,241 cm−1. These FTIR data indicated the presence of the triple-helical structure of collagen both in electrospun collagen and collagen/HAP fibers. In addition, the characteristic peaks of HAP also appeared in the FTIR spectrum of collagen/HAP composite fibers, indicating that the collagen/HAP composite had been successfully fabricated by the sol-electrospinning method. Comparing the spectrum of collagen/HAP composite fibers with that of pure collagen fibers, it can be seen that the addition of HAP to the collagen resulted in the red-shift of the peaks of amide I and amide II bands, and the intensity of the three mainly amide peaks of collagen/HAP composite fibers decreased obviously, which probably indicated that there was a certain degree of interaction between the HAP nanoparticles and collagen.
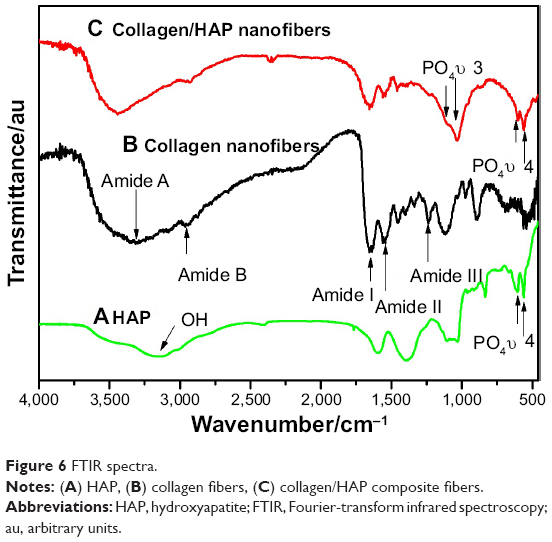 | Figure 6 FTIR spectra. |
Figure 7 shows the XRD patterns of pure collagen fibers and collagen/HAP composite fibers. As can be seen, the collagen fibers apparently contained NaCl (Figure 7A). After removal of salts by dialysis the diffraction peak of sodium chloride completely disappeared (Figure 7B), only an extensive broadening peak in the 2θ range of 15°–30° was observed, which was the characteristic diffraction peak of collagen. Figure 7C was the XRD result of the electrospun collagen/HAP composite fibers with 30% HAP content. Except for the characteristic diffraction peak of collagen in the 2θ range of 15°–30°, the diffraction peaks of HAP in collagen/HAP composite were shown to be slightly broader and weaker as compared to those of original HAP nanoparticles (shown in Figure 2), implying that HAP crystals in the composite had a smaller size and a lower crystallinity.
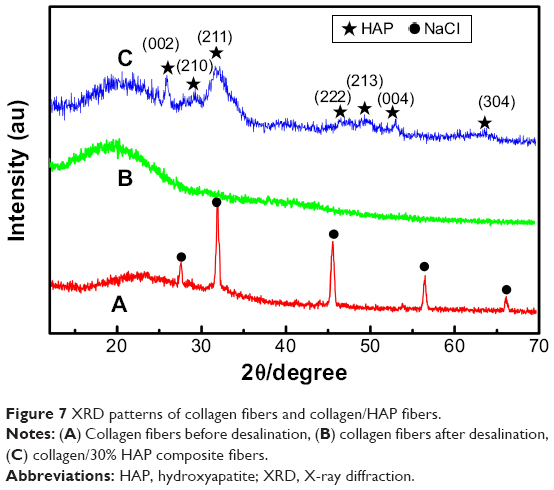 | Figure 7 XRD patterns of collagen fibers and collagen/HAP fibers. |
Cross-linking of electrospun fibers
Figure 8 shows SEM images of the cross-linked collagen fibers and the cross-linked collagen/HAP composite fibers. According to the previous literature,62 the electrospinning technology could not fully recover the original, crystalline supramolecular structure of collagen, which led to the solubility of electrospun collagen and collagen/HAP fibers in water. In order to use electrospun collagen and collagen/HAP fibers as a substitute for tissue engineering, it is necessary to cross-link the electrospun fibers. To our knowledge, a great deal of chemical agents such as formaldehyde, glutaraldehyde, and epoxy compound have been used in the cross-linking of collagen, but the greatest concern is cytotoxicity.63 In this study, EDC/NHS were used as a less problematic alternative. After being cross-linked, all the electrospun fibers became strongly interconnected, forming an apparently robust network and being insoluble in water, which can be used as a substitute for tissue engineering.64 In the process of cross-linking, the carboxyl groups of the aspartic acid and glutamic acid residues in the collagen structure can react with EDC to form a precipitating agent-unstable urea derivative. Then, a more stable carbodiimide product was formed with the help of NHS, which evidently enhanced stability of a cross-linked product. Both EDC and NHS did not enter the matrix of collagen, but was converted into a water-soluble urea derivative, which had very low cytotoxicity.65
 | Figure 8 SEM photographs of the electrospun fibers after cross-linking with EDC/NHS. |
Mechanical properties of collagen and collagen/HAP composite fibers
The effect of HAP on the mechanical properties of the composite nanofibers was tested with both cross-linked and un-cross-linked states as well as before and after desalination. For comparison, the pure collagen nanofibers were evaluated as control. As shown in Table 1, the tensile strength of pure collagen nanofibers after desalination was 74±0.31 MPa and the elongation was 36.3%±1.98%. Without desalination, both of them were lower, which was due to the existence of a large amount of salt. By adding HAP nanoparticles to collagen matrix, the tensile strength increased with the increasing HAP contents. The collagen/HAP composite fibers containing 30% HAP nanoparticles displayed better mechanical properties with a tensile strength of 147±0.12 MPa and elongation of 72.3%±1.48%, in contrast with the other samples. The increase in tensile strength was attributed to an increase in rigidity with the addition of HAP and the strong interaction between collagen and HAP nanoparticles. However, further increasing the amount of HAP up to 40% resulted in a slight decrease in the tensile strength to 124±0.14 MPa. In addition, the composite fibers fabricated with HAP sol showed a significant enhancement in the mechanical properties compared to the control composite fibers prepared by using conventional HAP powders, which was the result of the alignment of the HAP needles in the collagen matrix. After cross-linking with EDC/NHS, the electrospun fibers all showed a significant increase in tensile strength. This is not surprising, because the cross-linked electrospun fibers became strongly interconnected, as noted earlier. Among them, the collagen/HAP composite fibers containing 30% HAP nanoparticles displayed the highest tensile strength compared to the other samples, it showed a tensile strength of 209±0.16 MPa, which was 40 times higher than that reported with a similar method.40
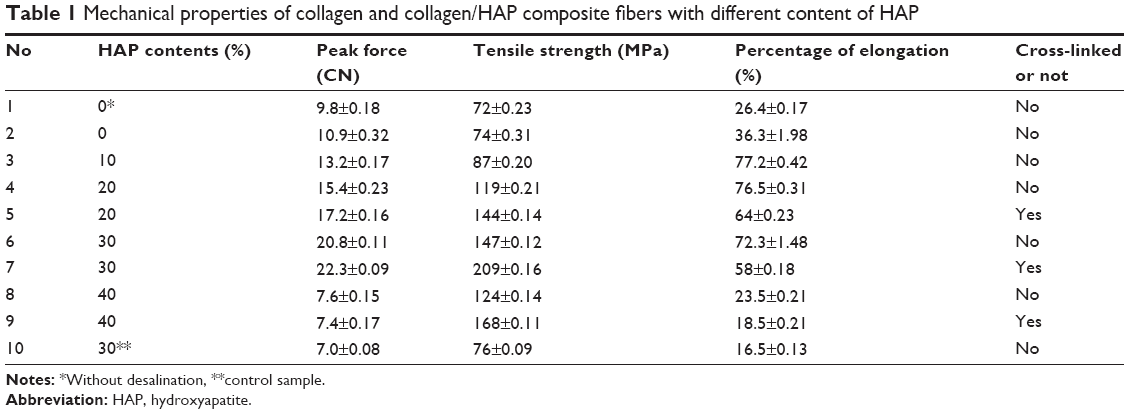 | Table 1 Mechanical properties of collagen and collagen/HAP composite fibers with different content of HAP |
Compatibility assay
Cell proliferation
To determine the effect of the prepared composite fibers on supporting cell growth, we measured cell proliferation ability on days 1, 3, 5, and 7 by CCK-8 assay. Using the pure PTFE culture as a control, the relative cell viability in every medium containing material extracts was shown in Figure 9. Apparently, the number of cells in all samples studied increased with the culture time. At the earlier time point of day 3, these three kinds of samples supported cell proliferation at a similar level. On day 5, due to the addition of HAP particles, more cells were found in the medium containing collagen/HAP composite fiber extracts than in the other two samples, which indicated that the collagen/HAP composite fibers supported cell proliferation better than the control samples. On day 7 this difference was relatively reduced, which may be attributed to the further proliferation of the U2-OS cells as to cover the full cultured well, and the growth of cells would be inhibited with the consumption of nutrients in cell culture fluid, and then the cells began to die under the high density. These results indicated that the prepared collagen/HAP composite fibers had essentially no in vitro cytotoxicity and supported cell attachment and proliferation better than the other samples.
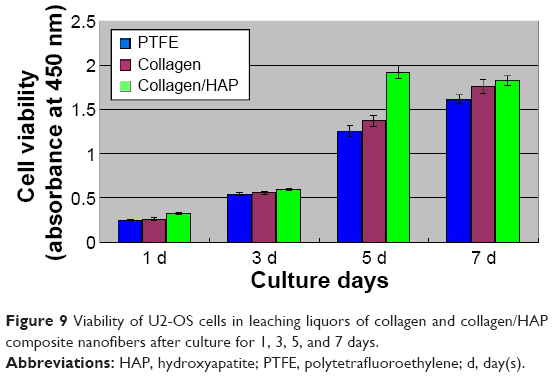 | Figure 9 Viability of U2-OS cells in leaching liquors of collagen and collagen/HAP composite nanofibers after culture for 1, 3, 5, and 7 days. |
Cell differentiation
The differentiation of U2-OS cells on the fibrous composite was studied by evaluating cell ALP activity in every medium containing material extraction from 1 day to 7 days. ALP activity is an early osteogenic marker, which indicates the osteo-inductive potential of the samples. Using PTFE as the control, the relative ALP data for different samples were calculated and illustrated in Figure 10. As shown in Figure 10, U2-OS ALP activity in the three mediums containing material extractions maintained slow growth over 4 days. By day 5, U2-OS ALP activity in three samples increased significantly, then peaked at day 6 and declined thereafter. Furthermore, U2-OS ALP activity in the medium containing collagen/HAP composite fiber extracts had higher ALP activity than that in the other two samples at day 6, which remained at a constant low level. Expression of this early osteogenic marker decreased in the three mediums containing material extractions by day 7, which may be attributed to the further maturation of the U2-OS cells, as stated above in cell proliferation assessment. When compared to the control samples, these data indicated that the presence of HAP particles enhanced the differentiation of U2-OS cells.
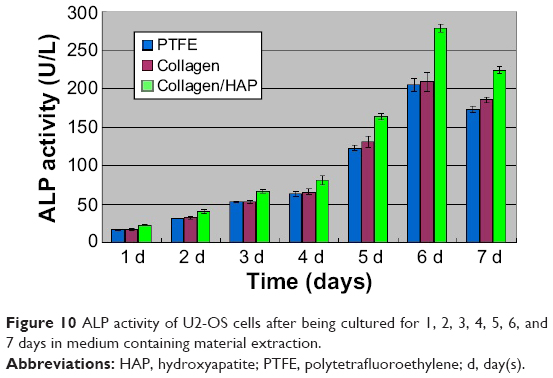 | Figure 10 ALP activity of U2-OS cells after being cultured for 1, 2, 3, 4, 5, 6, and 7 days in medium containing material extraction. |
Conclusion
This work developed an effective and greener strategy to generate collagen/HAP composite fibers using buffer/ethanol/water system as a replacement for HFIP or TFE via sol-electrospinning technique. The desalination of collagen solution together with the use of HAP sol was the key point of the research. Desalting salt from the collagen solution and using HAP sol not only ensured good dispersion of HAP particles in spinning solution and the oriented alignment of HAP particles in the electrospun collagen/HAP composites, but also improved the mechanical properties of the collagen/HAP composites obtained. Cross-linking with EDC/NHS further increased the mechanical properties. The prepared collagen/HAP composites exhibited 40 times higher tensile strength than that previously reported. Bioactivity of the composite fibers was demonstrated in vitro using U2-OS cells, it showed that the prepared collagen/HAP fibrous composites had essentially no in vitro cytotoxicity and could promote cell viability as well as proliferation. Our results have provided clues to the design of potential candidates for bone tissue engineering. Further studies will focus on the animal experiments in vivo by implanting the prepared collagen/HAP composite fibers in muscle pouches of rats to repair the damaged bone. Undoubtedly, this greener collagen/HAP composite fiber with an excellent microstructure should be suitable substrates for bone regeneration and this study highlights the current state of the art techniques in fabricating bone substitutes.
Acknowledgment
This work was supported by the Project Number 21001096 of National Natural Science Foundation of China.
Disclosure
The authors have no conflicts of interest to disclose.
References
Olszta MJ, Cheng XG, Jee SS, et al. Bone structure and formation: A new perspective. Mater Sci Eng. 2007;58:77–116. | ||
Kong XD, Cui FZ, Wang XM, Zhang M, Zhang W. Silk fibroin regulated mineralization of hydroxyapatite nanocrystals. J Cryst Growth. 2004;270:197–202. | ||
Dorozhkin SV. Calcium orthophosphate-based biocomposites and hybrid biomaterials. J Mater Sci. 2009;44(9):2343–2387. | ||
Zhang YZ, Venugopal JR, Turki AE, Ramakrishna S, Su B, Lim CT. Electrospun biomimetic nanocomposite nanofibers of hydroxyapatite/chitosan for bone tissue engineering. Biomaterials. 2008;29(32):4314–4322. | ||
Itoh S, Kikuchi M, Koyama Y, Takakuda K, Shinomiya K, Tanaka J. Development of an artificial vertebral body using a novel biomaterial, hydroxyapatite/collagen composite. Biomaterials. 2002;23(19):3919–3926. | ||
Chan CK, Kumar TS, Liao S, Murugan R, Ngiam M, Ramakrishnan S. Biomimetic nanocomposites for bone graft applications. Nanomedicine (Lond). 2006;1(2):177–188. | ||
Du C, Cui FZ, Zhu XD, Groot K. Three-dimensional nano-HAp/collagen matrix loading with osteogenic cells in organ culture. J Biomed Mater Res. 1999;44(4):407–415. | ||
Kikuchi M, Itoh S, Ichinose S, Shinomiya K, Tanaka J. Self-organization mechanism in a bone-like hydroxyapatite/collagen nanocomposite synthesized in vitro and its biological reaction in vivo. Biomaterials. 2001;22(13):1705–1711. | ||
Liao SS, Cui FZ, Zhang W, Feng QL. Hierarchically biomimetic bone scaffold materials: Nano-HA/collagen/PLA composite. J Biomed Mater Res B Appl Biomater. 2004;69(2):158–165. | ||
Tampieri A, Celotti G, Landi E, Sandri M, Roveri N, Falini G. Biologically inspired synthesis of bone-like composite: Self-assembled collagen fibers/hydroxyapatite nanocrystals. J Biomed Mater Res A. 2003;67(2):618–625. | ||
Scharnweber D, Born R, Flade K, Roessler S, Stoelzel M, Worch H. Mineralization behaviour of collagen type I immobilized on different substrates. Biomaterials. 2004;25(12):2371–2380. | ||
Roveri N, Falini G, Sidoti MC, et al. Biologically inspired growth of hydroxyapatite nanocrystals inside self-assembled collagen fibers. Mater Sci Eng C. 2003;23:441. | ||
Tampieri A, Celotti G, Landi E. From biomimetic apatites to biologically inspired composites. Anal Bioanal Chem. 2005;381(3):568–576. | ||
Cui FZ, Li Y, Ge J. Self-assembly of mineralized collagen composites. Mater Sci Eng R. 2007;57:1–27. | ||
Kikuchi M, Ikoma T, Itoh S, et al. Biomimetic synthesis of bone-like nanocomposites using the self-organization mechanism of hydroxyapatite and collagen. Composites Science and Technology. 2004;64:819–825. | ||
Rovira A, Amedee J, Bareille R, Rabaud M. Colonization of a calcium phosphate/elastin-solubilized peptide-collagen composite material by human osteoblasts. Biomaterials. 1996;17(15):1535–1540. | ||
Jin L, Feng ZQ, Wang T, et al. A novel fluffy hydroxylapatite fiber scaffold with deep interconnected pores designed for three-dimensional cell culture. J Mater Chem B. 2014;2:129–136. | ||
Fan ZJ, Wang JQ, Wang ZF, et al. Casein phosphopeptide-biofunctionalized graphene biocomposite for hydroxyapatite biomimetic mineralization. J Phys Chem C. 2013;117(20):10375–10382. | ||
Smith LA, Liu XH, Ma PX. Tissue engineering with nano-fibrous scaffolds. Soft Matter. 2008;4(11):2144–2149. | ||
Peng F, Shaw MT, Olson JR, Wei M. Hydroxyapatite Needle-Shaped Particles/Poly(l-lactic acid) Electrospun Scaffolds with Perfect Particle-along-Nanofiber Orientation and Significantly Enhanced Mechanical Properties. J Phys Chem C. 2011;115(32):15743–15751. | ||
Olmo N, Turnay J, Herrera JI, Gavilanes JG, Lizarbe MA. Kinetics of in vivo degradation of sepiolite-collagen complexes: Effect of glutaraldehyde treatment. J Biomed Mater Res. 1996;30(1):77–84. | ||
Liu WY, Thomopoulos S, Xia YN. Electrospun Nanofibers for Regenerative Medicine. Adv Healthc Mater. 2012;1(1):10–25. | ||
Lu WJ, Sun JS, Jiang XY. Recent advances in electrospinning technology and biomedical applications of electrospun fibers. J Mater Chem B. 2014;2:2369–2380. | ||
Chen DW, Liao JY, Liu SJ, Chan EC. Novel biodegradable sandwich-structured nanofibrous drug-eluting membranes for repair of infected wounds: an in vitro and in vivo study. Int J Nanomed. 2012;7:763–771. | ||
Agarwal S, Wendorff JH, Greiner A. Progress in the Field of Electrospinning for Tissue Engineering Applications. Adv Mater. 2009;21(32–33):3343–3351. | ||
Wang S, Mo XM, Jiang JB, et al. Fabrication of small-diameter vascular scaffolds by heparin-bonded P(LLA-CL) composite nanofibers to improve graft patency. Int J Nanomed. 2013;8:2131–2139. | ||
Frohbergh ME, Katsman A, Botta GP, et al. Electrospun hydroxyapatite-containing chitosan nanofibers crosslinked with genipin for bone tissue engineering. Biomaterials. 2012;33(36):9167–9178. | ||
Sun HL, Zhu F, Hu QG, Krebsbach PH. Controlling stem cell-mediated bone regeneration through tailored mechanical properties of collagen scaffolds. Biomaterials. 2014;35(4):1176–1184. | ||
Place ES, George JH, Williams CK, Stevens MM. Synthetic polymer scaffolds for tissue engineering. Chem Soc Rev. 2009;38(4):1139–1151. | ||
Wang YL, Guo G, Chen HF, et al. Preparation and characterization of polylactide/poly(ε-caprolactone)-poly(ethylene glycol)-poly(ε-caprolactone) hybrid fibers for potential application in bone tissue engineering. Int J Nanomed. 2014;9:1991–2003. | ||
Rieger KA, Birch NP, Schiffman JD, Rieger KA, Birch NP, Schiffman JD. Designing electrospun nanofiber mats to promote wound healing–a review. J Mater Chem B. 2013;1:4531–4541. | ||
Ajalloueian F, Zeiai S, Fossum M, Hilborn JG. Constructs of electrospun PLGA, compressed collagen and minced urothelium for minimally manipulated autologous bladder tissue expansion. Biomaterials. 2014;35(22):5741–5748. | ||
Fu W, Liu ZL, Feng B, et al. Electrospun gelatin/PCL and collagen/PLCL scaffolds for vascular tissue engineering. Int J Nanomed. 2014;9:2335–2344. | ||
Kim HM, Chae WP, Chang KW, et al. Composite nanofiber mats consisting of hydroxyapatite and titania for biomedical applications. J Biomed Mater Res B. 2010;94(2):380–387. | ||
Hong SG, Kim M, Kim GH. Collagen-β-TCP conjugated PCL biocomposites for bone tissue regeneration: fabrication, physical properties, and cellular activities. J Mater Chem. 2012;22:22565–22574. | ||
Rajeswari R, Jayarama RV, Subramanian S, Shayanti M, Seeram R. Precipitation of nanohydroxyapatite on PLLA/PBLG/Collagen nanofibrous structures for the differentiation of adipose derived stem cells to osteogenic lineage. Biomaterials. 2012;33(3):846–855. | ||
Zhang L, Yan JW, Yin ZW, et al. Electrospun vancomycin-loaded coating on titanium implants for the prevention of implant-associated infections. Int J Nanomed. 2014;9:3027–3036. | ||
Zhao Y, Chen J, Chou AK, Li G, LeGeros RZ. Nonwoven silk fibroin net/nano-hydroxyapatite scaffold: Preparation and characterization. J Biomed Mater Res A. 2009;91(4):1140–1149. | ||
Wu XN, Miao LY, Yao YF, et al. Electrospun fibrous scaffolds combined with nanoscale hydroxyapatite induce osteogenic differentiation of human periodontal ligament cells. Int J Nanomed. 2014;9:4135–4143. | ||
Thomas V, Derrick RD, Jose MV, Mathew BN, Chowdhury S, Vohra YK. Nanostructured Biocomposite Scaffolds Based on Collagen Coelectrospun with Nanohydroxyapatite. Biomacromolecules. 2007;8(2):631–637. | ||
Stanishevsky A, Chowdhury S, Chinoda P, Thomas V. Hydroxyapatite nanoparticle loaded collagen fiber composites: microarchitecture and nanoindentation study. J Biomed Mater Res A. 2008;86(4):873–882. | ||
Song JH, Kim HE, Kim HW. Electrospun fibrous web of collagen-apatite precipitated nanocomposite for bone regeneration. J Mater Sci Mater Med. 2008;19(8):2925–2932. | ||
Li MY, Mondrinos MJ, Gandhi MR, Ko FK, Weiss AS, Lelkes PI. Electrospun protein fibers as matrices for tissue engineering. Biomaterials. 2005;26(30):5999–6008. | ||
Rho KS, Jeong L, Lee G, et al. Electrospinning of collagen nanofibers: Effects on the behavior of normal human keratinocytes and early-stage wound healing. Biomaterials. 2006;27(8):1452–1461. | ||
Yang L, Fitié CC, Werf KO, Bennink ML, Dijkstra PJ, Feijen J. Mechanical properties of single electrospun collagen type I fibers. Biomaterials. 2008;29(8):955–962. | ||
Shih YV, Chen CN, Tsai SW, Wang YJ, Lee OK. Growth of Mesenchymal Stem Cells on Electrospun Type I Collagen Nanofibers. Stem Cells. 2006;24(11):2391–2397. | ||
Ki CS, Baek DH, Gang KD, Lee KH, Um IC, Park YH. Characterization of gelatin nanofiber prepared from gelatin–formic acid solution. Polymer. 2005;46(14):5094–50102. | ||
Kapllani A, Tran C, Kalra V. Self-assembly of fully conjugated rod-rod diblock copolymers within nanofibers. Soft Matter. 2013;9:11014–11020. | ||
Gu SY, Wang ZM, Ren J, Zhang CY. Electrospinning of gelatin and gelatin/poly(l-lactide) blend and its characteristics for wound dressing. Mater Sci Eng C Mater Biol Appl. 2009;29:1822–1828. | ||
Song JH, Kim HE, Kim HW. Production of electrospun gelatin nanofiber by water-based co-solvent approach. J Mater Sci Mater Med. 2008;19(1):95–102. | ||
Chen HC, Jao WC, Yang MC. Characterization of gelatin nanofibers electrospun using ethanol/formic acid/water as a solvent. Polymers for Advanced Technologies. 2009;20:98–103. | ||
Dong B, Arnoult O, Smith ME, Wnek GE. Electrospinning of Collagen Nanofiber Scaffolds from Benign Solvents. Macromol Rapid Commun. 2009;30(7):539–542. | ||
Dong ZX, Wu YQ, Clark RL. Thermodynamic Modeling and Investigation of the Formation of Electrospun Collagen Fibers. Langmuir. 2011;27(20):12417–12422. | ||
Mantz RA, Fox DM, Green JM, Fylstra PA, DeLong HC, Trulove PC. Dissolution of Biopolymers Using Ionic Liquids. Z Naturforsch. 2007;62A:275–280. | ||
Li LM, Miao JJ, Dordick JS, Robert JL. Electrospinning from room temperature ionic liquids for biopolymer fiber formation. Green Chemistry. 2010;12(11):1883–1892. | ||
Teng SH, Lee EJ, Wang Peng, Kim HE. Collagen/hydroxyapatite composite nanofibers by electrospinning. Materials Letters. 2008;62(17–18):3055–3058. | ||
Ruzin E. Plant Microtechnique and Microscopy. Oxford: Oxford University Press; 1999. | ||
Zhang Y, Reddy VJ, Wong SY, et al. Enhanced biomineralization in osteoblasts on a novel electrospun biocomposite nanofibrous substrate of hydroxyapatite/collagen/chitosan. Tissue Eng Part A. 2010;16(6):1949–1960. | ||
Feldkamp LA, Goldstein SA, Parfitt AM. The direct examination of three dimensional bone architecture in vitro by computed tomography. J Bone Miner Res. 1989;4(1):3–11. | ||
Chen F, Tang QL, Zhu YJ, et al. Hydroxyapatite nanorods/poly(vinyl pyrolidone) composite nanofibers, arrays and three-dimensional fabrics: Electrospun preparation and transformation to hydroxyapatite nanostructures. Acta Biomaterialia. 2010;6:3013–3020. | ||
Asran AS, Henning S, Michler GH. Polyvinyl alcohol-collagen-hydroxyapatite biocomposite nanofibrous scaffold: Mimicking the key features of natural bone at the nanoscale level. Polymer. 2010;51(4):868–876. | ||
Areias AC, Ribeiro C, Sencadas V, et al. Influence of crystallinity and fiber orientation on hydrophobicity and biological response of poly(L-lactide) electrospun mats. Soft Matter. 2012;8:5818–5825. | ||
Zhang XJ, Zhang YM, Wang ZQ, Li QJ, Li BX. The effect of non-growth factors on chondrogenic differentiation of mesenchymal stem cells. Cell Tissue Bank. 2014;15(3):319–327. | ||
Simmons DM, Kearney JN. Evaluation of collagen cross-linking techniques for the stabilization of tissue matrices. Biotechnol Appl Biochem. 1993;17(Pt 1):23–29. | ||
Olde DH, Dijkstra PJ, Vanluyn MJ, Wachem PB, Nieuwenhuis P, Feijen J. In vitro degradation of dermal sheep collagen cross-linked using a water-soluble carbodiimide. Biomaterials. 1996;17(7):679–684. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.