Back to Journals » Cancer Management and Research » Volume 14
GPR27 Regulates Hepatocellular Carcinoma Progression via MAPK/ERK Pathway
Authors Wang H ![]() , Du D, Huang J, Wang S, He X, Yuan S, Xiao J
, Du D, Huang J, Wang S, He X, Yuan S, Xiao J
Received 24 August 2021
Accepted for publication 8 February 2022
Published 17 March 2022 Volume 2022:14 Pages 1165—1177
DOI https://doi.org/10.2147/CMAR.S335749
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Ahmet Emre Eşkazan
Hongxv Wang,1,* Danyu Du,2,* Jianwen Huang,3,* Shuai Wang,2 Xv He,3 Shengtao Yuan,2 Jing Xiao1
1Zhuhai Precision Medical Center, Zhuhai People’s Hospital, Zhuhai Hospital Affiliated with Jinan University, Jinan University, Zhuhai, Guangdong, 519000, People’s Republic of China; 2Jiangsu Key Laboratory of Drug Screening, China Pharmaceutical University, Nanjing, Jiangsu, People’s Republic of China; 3Zhuhai Interventional Medical Center, Zhuhai Precision Medical Center, Zhuhai People’s Hospital, Zhuhai Hospital Affiliated with Jinan University, Zhuhai, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jing Xiao; Shengtao Yuan, Tel +86 15118802570 ; +86 13914798635, Email [email protected]; [email protected]
Purpose: Orphan GPCRs (GPRs) play important roles in the malignant progression of cancer and have the potential to develop into anti-tumor drug targets. However, the biological processes and molecular mechanisms of GPR27 have not been properly assessed in cancer. Our objective was to reveal the effect of GPR27 on the progression of hepatocellular carcinoma (HCC).
Methods: GPR27 levels were detected in HCC cell lines using quantitative reverse transcriptase-polymerase chain reaction and Western blot analysis. Next, the changes of phenotypes after GPR27 knockdown or overexpression were evaluated using in vitro methods. Finally, the mechanism of GPR27 in HCC was tested using RNA-seq and in vivo mouse xenograft model.
Results: In the present study, we reported that suppression of GPR27 expression inhibited proliferation, colony formation, cell viability, and induced cell S phase arrest of HCC cells, whereas GPR27 overexpression led to the opposite outcomes. Moreover, suppression of GPR27 expression resulted in blocking MAPK/ERK signal pathway which indicated the inhibition of HCC cells proliferation. Further study in vivo confirmed that GPR27 can affect the proliferation of HCC cells through the MAPK/ERK pathway.
Conclusion: Taken together, the findings of the present study uncover biological functions of GPR27 in HCC cells, and delineate preliminary molecular mechanisms of GPR27 in modulating HCC development and progression.
Keywords: GPR27, hepatocellular carcinoma, proliferation, MAPK/ERK pathway
Graphical Abstract:

Introduction
Primary hepatic carcinoma is the fourth leading cause of cancer-related death, accounting for 8.2% of global cancer deaths.1 Among them, the vast majority (>80%) are hepatocellular carcinoma (HCC), and about 85% of HCC patients are in East Asia and sub-Saharan Africa.2,3 According to the statistics of China Cancer Annual Report in 2015, there are about 370,000 new cases of liver cancer in China each year and 326,000 patients lose their lives. Globally, 782,000 liver cancer patients died in 2018 alone. It can be seen that the situation of HCC prevention and control in China and even in the world is extremely severe.4 For far, there are primarily two targeted small-molecule tyrosine kinase inhibitors (TKIs), sorafenib and lenvatinib,5,6 are approved by FDA as the first-line choice for HCC treatment, they are far from meeting the needs of HCC patients yet.7
G-protein-coupled receptors (GPCRs) are composed of more conservative seven-transmembrane structural proteins which are divided into an extracellular domain, a transmembrane domain, and an intracellular domain.8 The extracellular region is responsible for receiving extracellular signals, such as ions, amines, peptides, lipids, nucleotides, organic odorants, and so on,9 and the transmembrane region binds ligands and transduces signals to the intracellular region, then the intracellular region activates intracellular signal transduction by combining Gs, Gq/11, Gi, G12/13 and other different forms of G proteins,10 thus participates in the regulation of immune system activities, autonomic nervous system transmission, nutrient metabolism and water balance.11–14 At present, there are more than 830 types of GPCRs have been identified. The classic classification method divides them into A-F types. Among them, type A GPCRs account for nearly 85%,15 and more than one-third to one-half of the marketed drugs work through GPCRs.16 Therefore, the research on GPCRs has always been a hot spot in the field of medicine.
Orphan GPCRs (GPRs), as a class of GPCR receptors whose endogenous ligands are not yet known, currently have more than 140, accounting for 15% of GPCRs.17 Among them, GPR87, GPR81, GPR124, etc. have been reported to affect the growth of tumor cells and have the potential to develop into anti-tumor drug targets,18–20 but most of the remaining GPRs have not been extensively studied. GPR27 is a highly conserved orphan G protein-coupled receptor, belonging to the super conserved receptor in brain family. GPR27 is expressed in a variety of tissues, with high distribution in the brain, endocrine tissues, bone marrow, and lymphatic tissues according to the database of THE HUMAN PROTEIN ATLAS (https://www.proteinatlas.org/). Previous study reveals that GPR27 is related to the methylation of the 3p11-p14 promoter region, which may promote the occurrence of cancers such as epithelial cancer and cervical cancer.21 In addition, knocking down GPR27 in vitro can downregulate the activity of the insulin promoter and the secretion of glucose-stimulated insulin, thereby affecting the occurrence and progression of type 2 diabetes.22 Moreover, knockdown of GPR27 in the zebrafish model can increase the level of medium-chain acylcarnitines and reduce the sensitivity to insulin, thereby enhancing the increase in glucose levels caused by pharmacological or nutritional interference.23 However, the biological processes and molecular mechanisms of GPR27 in hepatocellular carcinoma remain unexplored. A better understanding of whether GPR27 functions in HCC is essential.
Here, we used a series of techniques such as gene overexpression, RNAi knockdown, RNA-seq for in vitro and in vivo studies to illustrate the promotive role of GPR27 in HCC. It was found that inhibiting the expression of GPR27 could lead to the S phase arrest and down-regulate the MAPK/ERK pathway, thus inhibiting the proliferation of HCC cells. Our research revealed the important role of GPR27 in the proliferation of HCC cells and will provide a new train of thought for the diagnosis and treatment of HCC.
Materials and Methods
Cell Culture
Human liver cancer cell lines SMMC-7721 and BEL-7402 were obtained from BeNa Culture Collection (Beijing, China); and human liver cancer cell lines Huh-7, SNU387, HCCLM3, HepG2, SK-Hep-1, and human embryonic kidney cell line HEK293T were obtained from National Infrastructure of Cell Line Resource (Shanghai, China). These cells were cultured according to the supplier’s instructions. All cell lines were cultured in a humidified incubator at 37°C with 5% CO2.
siRNA and Plasmids
GPR27 siRNAs and non-targeting siRNA (siNC) were purchased from Shanghai GenePharma Co., Ltd (Shanghai, China). The sequences of siGPR27#1 and siGPR27#2 are 5′-UCCUCGUGCUGGAAGAAUUTT-3′ and 5′-GCUGUGCAAGAUGUUCUACTT-3′. The sequence of siNC is 5′-UUCUCCGAACGUGUCACGUTT-3′. After being treated with GPR27 siRNAs or siNC, the samples were collected to detect the mRNA expression of GPR27 after 48 hours and detect the protein expression of GPR27 after 72 hours. The plasmids of pLenti-CMV-SREB1-GFP-Puro (PPL01344-4b) and pLenti-CMV-GFP-Puro (PE064) were purchased from Public Protein/Plasmid Library.
Western Blot Analysis
Cells were lysed using RIPA lysis buffer (Beyotime Biotech, Shanghai, China). Protein samples mixed with loading buffer and were separated using 10% gradient SDS-PAGE, and then transferred onto a polyvinylidene fluoride membrane. The membrane was incubated with a primary antibody at 4°C overnight followed by the secondary antibody for 2 hours at room temperature. Enhanced chemiluminescence (ECL) reagents were used to visualize the targeted protein brands. The following primary antibodies were used: Anti-GPR27 (PA5-110977) was purchased from Invitrogen (Cambridge, MA, USA), Anti-Ras (#6704), anti-c-Raf (#9665), anti-phospho-c-Raf (#2933), anti-ERK1/2 (#4585), anti-phospho-ERK (#4370), anti-MEK (#2774), anti-phospho-MEK (#4661), anti-β-actin (#2146) were obtained from Cell Signaling Technology (Danvers, MA, USA). Anti-CDK2 (10993-1-AP) was purchased from Proteintech. Anti-Cyclin E (AC026) and anti-Cyclin A2 (AC001) were obtained from Abclonal Technology (Shanghai, China).
Quantitative Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR)
RNA was extracted from tumor cells or tumor specimens using Trizol reagent (Vazyme Biotech, Nanjing, China). RNase free materials and reagents were used to avoid RNase contamination and all processes were conducted on the ice to avoid RNA degradation. The quality and quantity for the RNA extracted from cells and tumor specimens were detected by Nanodrop (Thermo scientificTM, US), and the ratio of A260/A280 is generally between 1.8–2.0, and the ratio of A260/A230 is generally between 1.8–2.2. The cDNA was synthesized using PrimeScript™ RT reagent Kit (Vazyme Biotech, Nanjing, China). qRT-PCR was performed with the AceQ® qPCR SYBR Green Master Mix (High ROX Premixed) kit (Vazyme Biotech, Nanjing, China). Conditions for qRT-PCR were as follows: 95°C for 5mins, 40 cycles of 95°C for 10s, 60°C for 30s, and a melting curve of 15s mins at 95°C, the 60s at 60°C and 15s at 95°C. The primers for amplification were as follows: GPR27-Forward: 5ʹ-GCCTCCGTGTGGCTGACCTTC-3ʹ, GPR27-Reverse: 5ʹ- ACCAATGCCTTTCAGGTCGCAG-3ʹ, 18s rRNA-Forward: 5ʹ- ACCCGTTGAACCCCATTCGTGA-3ʹ, 18s rRNA -Reverse: 5ʹ-GCCTCACTAAACCATCCAATCGG-3ʹ.
Establishment of GPR27 Overexpressed HCC Cell Lines
Lentiviruses were generated with either pLVX or pLVX-GFP co-transfecting the 293T cells, together with the packaging plasmids pMD2.G and psPAX2. After 48 hours, the supernatant containing the viral particles was collected, filtered, and then used to infect targeted cells for 72 hours. The infected cells were cultured in fresh growth medium containing 10 μg/mL puromycin (Beyotime, Shanghai, China) as described previously.24
In vitro Functional Assays
The real-time cell analyzer (xCelligence RTCA SP; Roche Diagnostics GmbH, Penzberg, Germany) was used to assess cell proliferation. Briefly, 5 × 103 cells were seeded onto the E plate-16 plates. After 30 mins, the plate was transport to analyzer and electrode resistance was recorded. 16 hours later, cells were treated with control siRNA or GPR27 siRNAs and cultured for 8 hours. After that, the media were changed and cell growth was monitored continuously up to another 144 hours. Growth curve and colony formation assays were performed as previously described.25 Apoptosis, cell cycle and cell viability assay were measured using Annexin V-FITC apoptosis detection kit (Vazyme Biotech, Nanjing, China), Cell cycle analysis kit (Beyotime Biotech, Shanghai, China) and Cell Counting-Lite® 2.0 Luminescent Cell Viability Assay kit (Vazyme Biotech, Nanjing, China) according to the manufacturer’s instructions.
Nude Mice Xenograft Model
4 to 6-week-old male Balb/c nude mice (Hangzhou ZiYuan Laboratory Animal Technology Co., Ltd., China) were housed under specific pathogen-free conditions and cared for in accordance with protocols approved by the Experimental Animal Care Commission in China Pharmaceutical University. SMMC-7721 cells (2.0 × 106) were injected subcutaneously into the right flank of mice. When tumor volume reached about 100 mm3, mice were randomly allocated (six mice per group) and treated with an intratumoral injection of control siRNA or GPR27 siRNA (10 μg per tumor) complexed with EntransterTM-in vivo transfection reagent (10 μL per tumor) (Engreen Biosystem, Beijing, China) every other day.26 After siRNAs and in vivo transfection reagent were solved and diluted in 5% glucose solution individually, they were mixed gently and placed at room temperature for 30 minutes. Then, the mixture were injected slowly for twice or three times into the tumor xenografts per mouse. Tumor volumes and body weight of mice were monitored throughout the experiment. Nude mice were sacrificed after 2 weeks of treatment, xenograft tumors were harvested, photographed, and processed for hematoxylin-eosin (H&E), immunohistochemical (IHC), qRT-PCR and Western blot analysis.
Tumor volume (TV) = ab2/2(a, the longest diameter of tumor; b, the shortest diameter of tumor).
Statistical Analysis
Statistical analyses were performed using SPSS 19.0 (SPSS, Chicago, IL, USA) and Prism 5.0 (GraphPad Software, La Jolla, CA, USA) software. Data are presented as the mean ± SD of at least three independent experiments. Quantitative data were evaluated by the Student’s t-test. The p-values < 0.05 were considered statistically significant for all tests.
Results
Knockdown of GPR27 Inhibits the Proliferation of HCC Cells in vitro
To investigate the potential roles of GPR27 in HCC, we first determined the expression levels of GPR27 in seven well-characterized HCC cells. As shown in Figure 1A, SMMC-7721 and SK-Hep-1 cells expressed relatively high mRNA and protein levels of GPR27 as compared with other HCC cells. Besides, these two cells are convenient for us to conduct knockdown experiments, and are easier to form xenografts to assess the biological function of GPR27 in vivo. Therefore, SMMC-7721 and SK-Hep-1 cells were selected to explore the impact of GPR27 on malignant phenotypes of HCC cells after GPR27 knockdown. Both short interference RNA fragments of GPR27 (siGRP27#1 and siGPR27#2) showed a marked reduction in mRNA and protein expression of GPR27 compared with negative control in SMMC-7721 and SK-Hep-1 cell lines (about 70% decrease; Figure 1B and C). Colony formation assay revealed that knockdown of GPR27 significantly decreased cell clonogenicity in SMMC-7721 and SK-Hep-1 cells (Figure 1D). To further confirm the effect of GPR27 on the proliferation of HCC cells, cell viability of SMMC-7721 and SK-Hep-1 cells after GPR27 knockdown was examined by measuring the intracellular ATP content.27 Results showed that ATP levels were significantly reduced compared with the negative control (Figure 1E). Besides, the Real Time Cellular Analysis showed that the cell proliferation in the siGPR27#1 and siGPR27#2 groups was significant slower compared with negative control (Figure 1F). Taken together, the above results indicate that knockdown of GPR27 inhibits the proliferation of HCC cells in vitro.
 |
Figure 1 Knockdown of GPR27 inhibits HCC cells proliferation in vitro. (A) The mRNA and protein expression of GPR27 were examined in HCC cell lines. (B and C) GPR27 knockdown efficiency of siRNAs was determined by qRT-PCR and Western blot analysis. (D) Colony formation assays were performed to determine the cell clonogenicity of SMMC-7721 and SK-Hep-1 cells treated with control or GPR27 siRNAs. (E) The effects of GPR27 on cell proliferation of SMMC-7721 and SK-Hep-1 cells were detected by measuring the intracellular ATP content treated with control or GPR27 siRNAs. (F) Real Time Cellular Analysis (RTCA) assays were performed to evaluate the effect of GPR27 on HCC cell proliferation treated with control or GPR27 siRNAs. Data are represented as the mean ± SD of three independent experiments. The p-values < 0.05 were considered statistically significant for all tests. |
Overexpression of GPR27 Promotes the Proliferation of HCC Cells in vitro
To further examine the effect of GPR27 on the proliferation of HCC cells, we stably expressed GPR27 in SMMC-7721 and SK-Hep-1 cells through lentiviral infection. As shown in Figure 2A, GPR27 mRNA levels significantly increased over 200–400 folds in GPR27-overexpressed (GPR27-OE) SMMC-7721 and SK-Hep-1 cells compared with the vector control cells (P<0.001; Figure 2A). Meantime, GPR27 protein levels also increased significantly in GPR27-OE SMMC-7721 and SK-Hep-1 cells (Figure 2A). Colony formation assay showed that overexpression of GPR27 significantly increased cell clonogenicity in GPR27-OE SMMC-7721 and SK-Hep-1 cells (Figure 2B). Growth curve analysis revealed that overexpression of GPR27 significantly promoted the proliferation of GPR27-OE SMMC-7721 and SK-Hep-1 cells (Figure 2C). In summary, these results indicate that GPR27 significantly promotes HCC cells proliferation in vitro.
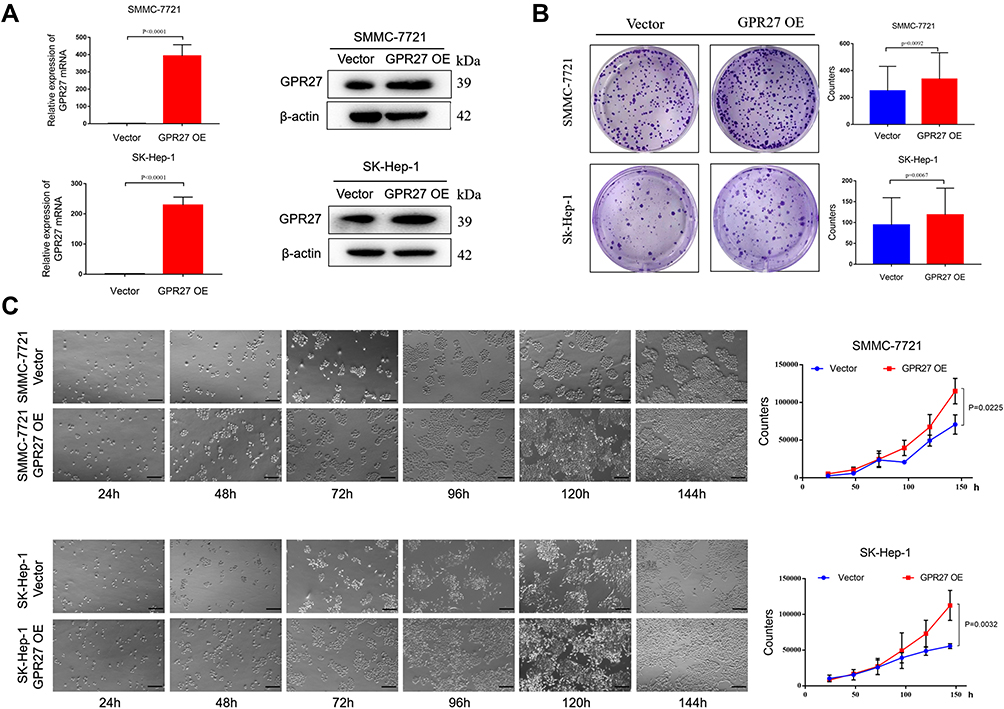 |
Figure 2 Overexpression of GPR27 promotes HCC cells proliferation in vitro. (A) GPR27 overexpression efficiency was determined by qRT-PCR and Western blot analysis after lentiviral infection in SMMC-7721 and SK-Hep-1 cells. (B) Colony formation assays were performed to determine the cell clonogenicity of GPR27-OE SMMC-7721 and SK-Hep-1 cells. (C) Growth curve analysis was conducted to examine the cell proliferation in GPR27-OE SMMC-7721 and SK-Hep-1 cells. Data are represented as the mean ± SD of three independent experiments. The p-values < 0.05 were considered statistically significant for all tests. |
Knockdown of GPR27 Induces S-Phase Arrest in HCC Cells
Abnormal activity of the cell-cycle engine is present in essentially all tumor types including HCC and functions as a driving force of tumorigenesis.28 To further investigate the mechanisms for GPR27 boosting the proliferation of HCC cells, cell cycle distribution in HCC cell lines SMMC-7721 and SK-Hep-1 was analyzed by flow cytometry after GPR27 knockdown. As shown in Figure 3A, siGPR27#1 and siGPR27#2 triggered an increased percentage in S phase in SMMC-7721 and SK-Hep-1 cells compared with the negative control group. Accumulating evidence suggests that cyclin A and E were responsible for G1/S transition and S phase progression, respectively. They combined with CDK2 and activated CDK2 to facilitate S phase entry and progression.29–31 In our studies, the expression of cyclin A-CDK2 complex was significantly downregulated in SMMC-7721 and SK-Hep-1 cells after GPR27 knockdown and the expression of cyclin E was slightly increased but had no significant change (Figure 3B). Moreover, deregulation of apoptosis is one of the important characteristics of cancer and is closely related to tumor cell proliferation.32 Hence, we analyzed cell apoptosis by Annexin V-FITC and propidium iodide (PI) staining after GPR27 knockdown. However, knockdown of GPR27 could not significantly affect cell apoptosis in SMMC-7721 and SK-Hep-1 cells (Figure S1). Together, above results suggest that GPR27 knockdown induced S phase arrest in HCC cells.
 |
Figure 3 Knockdown of GPR27 induces S phase arrest in HCC cells. (A) Cell cycle distribution was measured by flow cytometry using PI staining in SMMC-7721 and SK-Hep-1 cells treated with control or GPR27 siRNAs. (B) The cell cycle related proteins were analyzed through Western blot analysis after GPR27 knockdown. Data are represented as the mean ± SD of three independent experiments. The p-values < 0.05 were considered statistically significant for all tests (*P < 0.05, **P < 0.01, ***P < 0.001 vs SiNC). |
GPR27 Accelerates the Proliferation of HCC Cells Through the MAPK/ERK Pathway
Above results reveal that knockdown of GPR27 induces S phase arrest and inhibits the proliferation of HCC cells. To further explore the mechanism of action of GPR27 in HCC cells, we performed RNA-seq to characterize the gene expression profile in SMMC-7721 cell treated with control or siGPR27s. As shown in Figure 4A, a total of 519 differentially expressed genes (|fold change|>2 and p<0.05) were detected in the GPR27 knockdown group compared with control group, of which 257 genes were upregulated and 262 genes were downregulated. By Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis, the most enriched top 30 pathways were displayed in Figure 4B, including cancer-related pathways and the MAPK signaling pathway (Figure 4B). Moreover, a previous study found that knockdown of GPR27 inhibits phosphorylation of ERK caused by plasmalogens.33 Therefore, we hypothesized that abnormalities in the MAPK signaling pathway are associated with the impact of GPR27 on malignant phenotypes of HCC cells. To test this, we detected the expression of key proteins and their phosphorylation state in this signaling pathway in GPR27 knockdown group and control group. As shown in Figure 4C, both the phosphorylation levels of ERK1/2 and MEK were significantly downregulated in SMMC-7721 and SK-Hep-1 cells after GPR27 siRNAs treatment. Ras protein functions as an essential upstream regulatory signaling molecule of the ERK1/2 signaling pathway.34 The protein expression level of Ras in SMMC-7721 and SK-Hep-1 cells was significantly downregulated following GPR27 knockdown by siRNAs. Besides, the phosphorylation level of c-Raf was also significantly downregulated. In contrast, the protein levels of Ras, p-c-Raf, p-ERK1/2 and p-MEK were significantly upregulated in GPR27-OE SMMC-7721 and SK-Hep-1 cells (Figure 4D). Based on these results, we reasoned that GPR27 may promote the proliferation of HCC cells through MAPK/ERK pathway.
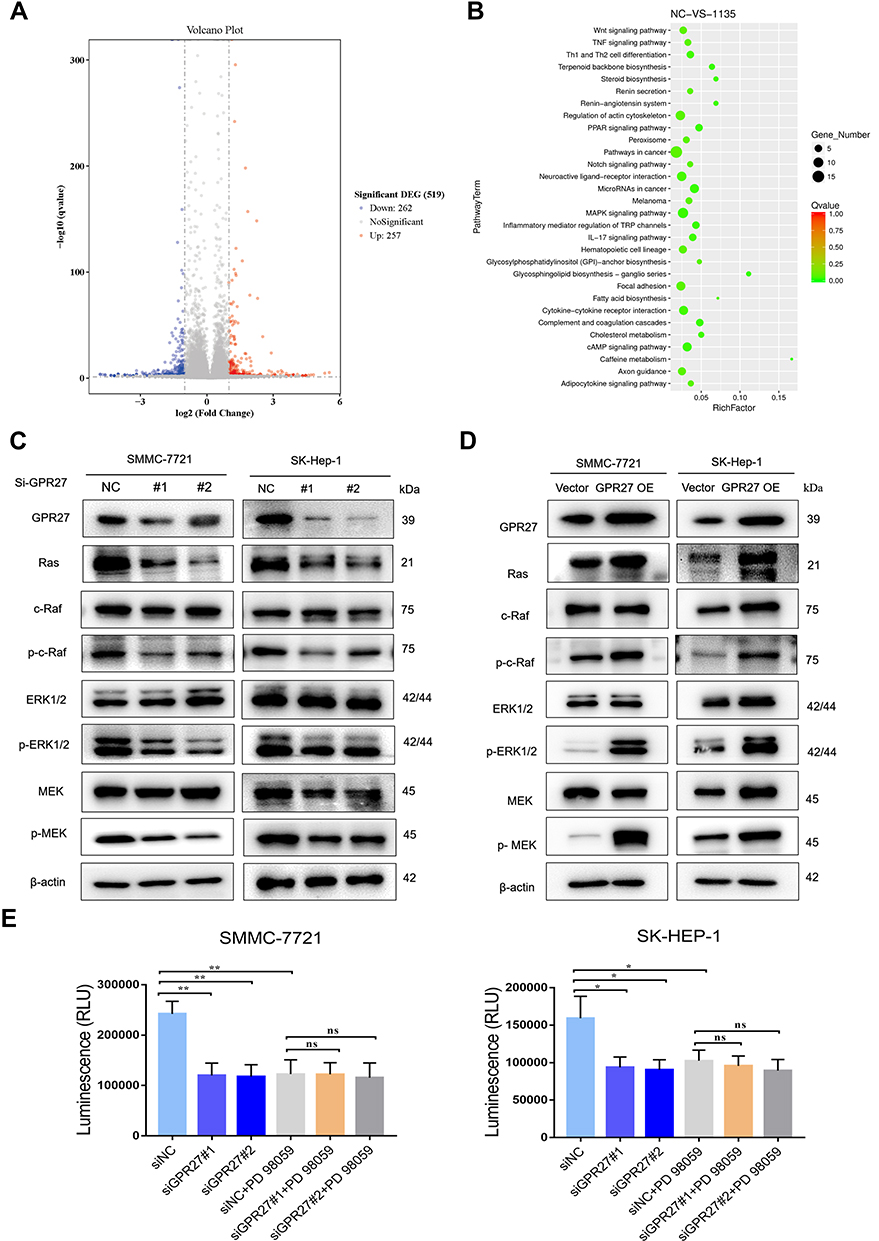 |
Figure 4 GPR27 accelerates the proliferation of HCC cells through the MAPK/ERK signaling pathway. (A) Volcano map of differential genes in SMMC-7721 cells treated with control or GPR27 siRNAs. (B) Annotated map of KEGG pathway of differential genes in SMMC-7721 cells treated with control or GPR27 siRNAs. (C and D) The protein expression levels of Ras, c-Raf, p-c-Raf, ERK1/2, P-ERK1/2, MEK and p-MEK in SMMC-7721 and SK-Hep-1 cells treated with control or GPR27 siRNAs (C) or stably overexpressed GPR27 (D). (E) Cell viability was determined by measuring the intracellular ATP content in the absence and presence of MEK inhibitor PD98059 (10μM) after GPR27 knockdown. Data are represented as the mean ± SD of three independent experiments. The p-values < 0.05 were considered statistically significant for all tests (*P < 0.05, **P < 0.01, vs SiNC). |
In support of this notion, we used a well-characterized MEK pharmacological inhibitor PD98059 to block the MAPK pathway. As shown in Figure 4E, PD98059 (10 μM) or siGPR27 alone could effectively inhibit the cell proliferation with a similar inhibitory effect, and PD98059 alone may not inhibit GPR27 expression in both mRNA and protein levels in SMMC-7721 and SK-Hep-1 cell lines (Figure S4). Nevertheless, there was no significant difference in the inhibitory activity between GPR27 siRNAs combined treatment with PD98059 and PD98059 alone. These results suggest that GPR27 accelerates the proliferation of HCC cells, at least in part, through the MAPK/ERK pathway.
Knockdown of GPR27 Inhibits HCC Tumor Growth in vivo
To investigate whether GPR27 could promote tumorigenic capacity of HCC cells in vivo, SMMC-7721 cells were subcutaneously inoculated into the right flank of BALB/c nude mice. After the tumor volume reached ~100 mm3, control or GPR27 siRNAs were injected into the HCC tumor xenografts. Consistent with in vitro results, xenograft tumors after GPR27 knockdown grew slower than the control group (Figure 5A, B and D) without any significant change in body weight (Figure 5C). Nude mice were sacrificed on day 21, the H&E pictures showed that the size and morphology of cancer cells are different, and the nucleo-plasmic ratio of cancer cells is significantly higher than that of normal cells, and megakaryon, dikaryon or multikaryon may be present, these are the characteristics of tumor tissue (Figure 5F). And, the size and weight of the tumors were significantly smaller compared with the control group (Figure 5E). Besides, the GPR27 knockdown efficiency in tumors were confirmed by qRT-PCR (about 45% decrease, P<0.010) (Figure 5G) and Western blot analysis (Figure 5H, Figure S5). Consistent with retarded tumor growth, Ki67 immunohistochemical staining showed there is 75–80% nuclear positivity in siNC group, while about 20% nuclear positivity in siGPR27 group (Figure 5F).
 |
Figure 5 GPR27 knockdown inhibits HCC growth in vivo. (A–E). SMMC-7721 cells (2 × 106 cells per mouse) were injected into the axilla of 6-week-old male BALB/c nude mice (n=6). When tumor volume reaches at 100 mm3, the control siRNA or GPR27 siRNA (10 μg per tumor) was injected into tumors every other day. The size (D) of the tumors and body weight (C) of mice were measured every 2 or 3 days. Nude mice were sacrificed on day 25 after inoculation, xenograft tumors were harvested. Tumor growth curves (D), photographs of mice (A) and harvested tumors (B), and tumor weight (E) are shown. (F) Representative images of H&E-stained sections of tumors and Ki67 positive cells (F) are shown. Scale bar: 50μm. (G and H) GPR27 knockdown efficiency in vivo was validated by qRT-PCR (G) and Western blot analysis (H). And the protein expression levels of c-Raf, p-c-Raf, ERK1/2, p-ERK1/2, MEK and p-MEK in tumor tissues were determined by Western blot. The p-values < 0.05 were considered statistically significant. |
Consistent with in vitro findings, the protein expression levels of p-c-Raf, p-ERK1/2 and p-MEK in the tumor tissues of the GPR27 knockdown group were significantly downregulated compared with control group. Together, these results suggest that knockdown of GPR27 could inhibit HCC tumor growth in vivo, partly through the MAPK/ERK signaling pathway.
Discussion
Previous study shows that GPR27, an orphan GPCR that belongs to the super conserved receptors expressed in the brain family and conserves highly among vertebrates. Currently, studies have shown that GPR27 affects fatty acid and glucose metabolism. In particular, loss of GPR27 can lead to an increase in the expression of Carnitine O-palmitoyl transferase 1, CPT1 in the liver and thus affect fatty acid metabolism.35 Also, the whole-body knockout of GPR27 in mouse can reduce the expression of Insulin and Pdx1 mRNA by 30% at the pancreatic islets, thereby leading to a defect in insulin secretion.36 Since aberrantly deregulated glucose metabolism and fatty acid metabolism are closely linked to HCC progression,37 thus, whether GPR27 plays a promotive role in HCC progression were examined. In this paper, we uncovered an important role of GPR27 in the progression of HCC. We demonstrated that the expression of GPR27 was significantly upregulated in HCC cells. However, GPR27 mRNA expression in human HCC tumor tissues was not significantly higher than in human normal liver tissues, according to the RNA sequencing data from The Cancer Genome Atlas (TCGA) database which analyzed through UALCAN online website (http://ualcan.path.uab.edu/index.html).38 Meanwhile, the GPR27 mRNA expression had no significant correlation with the survival time of HCC patients through the survival curve of patients with HCC. One possible reason for this discrepancy is that GPR27 mRNA expression is relatively low and cannot better reflect the correlation with the progression of HCC, and the data on its protein expression should be considered. Besides, a tissue microarray containing liver cancer tissues and adjacent liver tissues, or clinical samples obtained from pathologically confirmed HCC patients should be conducted to identify the role of GPR27 in HCC further.
GPR27 knockdown significantly inhibited HCC cells proliferation in vitro and retarded HCC tumor growth in vivo. In contrast, GPR27 overexpression accelerates HCC cell proliferation in vitro. Whether GPR27 contributes to glucose and fatty acid homeostasis in HCC cells deserves further exploration, although expression levels of several key enzymes and transporters were significantly changed (our unpublished data).
Accumulating evidence shows that aberrant activity of the cell-cycle machinery is present in essentially all tumor types including HCC and functions as a driving force of tumor progression.28 Indeed, GPR27 deficiency leads to S phase arrest in HCC cells, and then testing the expression of cycle-related proteins, such as CDK2, Cyclin A2 and Cyclin E, and found that inhibiting the expression of GPR27 can down-regulate the protein expression of CDK2 and Cyclin A2.39 Although tumor cells need continuous input of survival signals to suppress apoptosis and promote proliferation,40 significantly apoptotic effects were not observed after knocking down of GPR27. Therefore, it is concluded that GPR27 deficiency induces S phase arrest and GPR27 overexpression plays an important role in the proliferation of HCC cells.
By using RNA-seq transcriptomics and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis, cancer-related pathways and the MAPK signaling pathway were enriched among those related pathways. Previous evidence suggests that knockdown of GPR27 inhibits phosphorylation of ERK caused by plasmalogens.33 Moreover, the activation of the MAPK pathway initiates from a conformational change of GDP-bound RAS (inactive) switches to GTP-bound RAS (active), thereby triggering the recruitment and activation of RAF. Activated RAF further phosphorylates and actives MEK, thus directly leads to the phosphorylation of ERK.41 In addition, the MAPK/ERK signaling pathway is activated in more than 50% of human HCC cases.34
Here, we found that GPR27 knockdown downregulates Ras, p-c-Raf, p-MEK and p-ERK significantly in HCC cells and xenograft tumors, whereas GPR27 overexpression exerts the opposite effects. To further verify the MAPK/ERK pathway functions in the promotive effect of HCC progression, we used a MEK pharmacological inhibitor, PD98059, to block the MAPK pathway. Results showed that there was no significant difference in the suppressive effect of GPR27 siRNAs combined treatment with PD98059 compared with PD98059 alone in HCC cells. The p38 MAPK, the Jun N-terminal kinases (JNK) and the ERK are the main MAPK signaling pathways that affect the proliferation, differentiation and survival of various tumors.42 In addition to MAPK/ERK signaling pathway, we have also evaluated the P38 and JNKs pathway after GPR27 knockdown, and found that P38 expression did not change significantly. Besides, the expression levels of p-JNK in the two cell lines were inconsistent, p-JNK expression was down-regulated after GPR27 knockdown in SK-Hep-1 cells while not changed in SMMC-7721 cells (Figure S2). In addition, signal transducer and activator of transcription 3 (STAT3) has recently become a potential therapeutic target for HCC due to its key role in tumorigenesis,43 and STAT3 supports Ras-induced malignant transformation of tumor cell.44 Therefore, we wondered whether the phosphorylation level of STAT3 was regulated by GPR27, and it was an interesting founding that pSTAT3 expression was significantly downregulated after GPR27 disruption in both SK-Hep-1 and SMMC-7721 cells (Figure S3). The underlying mechanism of GPR27 regulating STAT3 is still under research. Taken together, GPR27 may accelerate the proliferation of HCC cells partly through the MAPK/ERK pathway.
Conclusion
We carried out a relatively comprehensive study on GPR27 in HCC development and progression in vitro and in vivo (Figure 6). The findings demonstrate that GPR27 exerts a promotive effect on the proliferation of HCC cells through regulation of S phase of cell cycle and MAPK/ERK signaling pathway. This study provides valuable insights into the underlying mechanisms of GPR27 in modulating proliferation of HCC, and points GPR27 as a promising target for HCC treatment.
 |
Figure 6 Schematic representation of GPR27 inhibits HCC cells proliferation based on this study. GPR27 regulates proliferation of HCC cells via modulation of S phase entry and the MAPK/ERK signaling pathway . |
Animal Ethics Statement
The procedures for care and use of animals were performed in accordance with the regulations of the principles of the 3Rs (Replacement, Reduction and Refinement), and all animal experiments were approved by the Ethics Committee of the China Pharmaceutical University and the animal trial registration number is 2021-08-001.
Funding
This study is supported by the National Key Research and Development Program of China (Grant No. 2017YFA0205200) and the National Natural Science Foundation of China (Grant No.81801811, No. 81773766 and 81872892). Thanks to Dr. Qipeng Wu for guiding and advising in this work.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi:10.3322/caac.21492
2. McGuire S. World cancer report 2014. Geneva, Switzerland: world health organization, international agency for research on cancer, who press, 2015. Adv Nutr. 2016;7:418–419. doi:10.3945/an.116.012211
3. Yang JD, Hainaut P, Gores GJ, et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16:589–604. doi:10.1038/s41575-019-0186-y
4. Vos T, Allen C, Arora M, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the global burden of disease study 2015. Lancet. 2016;388:1545–1602. doi:10.1016/S0140-6736(16)31678-6
5. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised Phase 3 non-inferiority trial. Lancet. 2018;391:1163–1173. doi:10.1016/S0140-6736(18)30207-1
6. Keating GM. Sorafenib: a review in hepatocellular carcinoma. Target Oncol. 2017;12:243–253. doi:10.1007/s11523-017-0484-7
7. Huang A, Yang X-R, Chung W-Y, et al. Targeted therapy for hepatocellular carcinoma. Signal Transduct Target Ther. 2020;5:146. doi:10.1038/s41392-020-00264-x
8. Venkatakrishnan A, Deupi X, Lebon G, et al. Molecular signatures of g-protein-coupled receptors. Nature. 2013;494:185–194. doi:10.1038/nature11896
9. Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of g-protein-coupled receptors. Nature. 2009;459:356–363. doi:10.1038/nature08144
10. Ritter SL, Hall RA. Fine-tuning of gpcr activity by receptor-interacting proteins. Nat Rev Mol Cell Biol. 2009;10:819–830. doi:10.1038/nrm2803
11. Wang D. The essential role of g protein-coupled receptor (gpcr) signaling in regulating t cell immunity. Immunopharmacol Immunotoxicol. 2018;40:187–192. doi:10.1080/08923973.2018.1434792
12. Li S, Yang C, Zhang L, et al. Promoting axon regeneration in the adult cns by modulation of the melanopsin/gpcr signaling. Proc Nat Acad Sci. 2016;113:1937–1942. doi:10.1073/pnas.1523645113
13. Husted AS, Trauelsen M, Rudenko O, et al. Gpcr-mediated signaling of metabolites. Cell Metab. 2017;25:777–796. doi:10.1016/j.cmet.2017.03.008
14. Hazell GG, Hindmarch CC, Pope GR, et al. G protein-coupled receptors in the hypothalamic paraventricular and supraoptic nuclei–serpentine gateways to neuroendocrine homeostasis. Front Neuroendocrinol. 2012;33:45–66. doi:10.1016/j.yfrne.2011.07.002
15. Joost P, Methner A. Phylogenetic analysis of 277 human g-protein-coupled receptors as a tool for the prediction of orphan receptor ligands. Genome Biol. 2002;3:RESEARCH0063–RESEARCH0063. doi:10.1186/gb-2002-3-11-research0063
16. Hu G-M, Mai T-L, Chen C-M. Visualizing the gpcr network: classification and evolution. Sci Rep. 2017;7:15495. doi:10.1038/s41598-017-15707-9
17. Odoemelam CS, Percival B, Wallis H, et al. G-protein coupled receptors: structure and function in drug discovery. RSC Adv. 2020;10:36337–36348. doi:10.1039/D0RA08003A
18. Niss Arfelt K, Fares S, Sparre-Ulrich AH. Signaling via g proteins mediates tumorigenic effects of gpr87. Cell Signal. 2017;30:9–18. doi:10.1016/j.cellsig.2016.11.009
19. Brown TP, Bhattacharjee P, Ramachandran S. The lactate receptor gpr81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene. 2020;39:3292–3304. doi:10.1038/s41388-020-1216-5
20. Cherry AE, Vicente JJ, Xu C. Gpr124 regulates microtubule assembly, mitotic progression, and glioblastoma cell proliferation. Glia. 2019;67:1558–1570. doi:10.1002/glia.23628
21. Lando M, Fjeldbo CS, Wilting SM, et al. Interplay between promoter methylation and chromosomal loss in gene silencing at 3p11-p14 in cervical cancer. Epigenetics. 2015;10:970–980. doi:10.1080/15592294.2015.1085140
22. Ku GM, Pappalardo Z, Luo CC, German MS, McManus MT. An sirna screen in pancreatic beta cells reveals a role for gpr27 in insulin production. PLoS Genet. 2012;8:e1002449. doi:10.1371/journal.pgen.1002449
23. Nath AK, Ma J, Chen -Z-Z. Genetic deletion of gpr27 alters acylcarnitine metabolism, insulin sensitivity, and glucose homeostasis in zebrafish. FASEB J. 2020;34:1546–1557. doi:10.1096/fj.201901466R.
24. Wei X, Li S, He J, et al. Tumor-secreted pai-1 promotes breast cancer metastasis via the induction of adipocyte-derived collagen remodeling. Cell Commun Signal. 2019;17:58. doi:10.1186/s12964-019-0373-z
25. Li H, Sun L, Li H, et al. Dt-13 synergistically enhanced vinorelbine-mediated mitotic arrest through inhibition of foxm1-bicd2 axis in non-small-cell lung cancer cells. Cell Death Dis. 2017;8:e2810. doi:10.1038/cddis.2017.218
26. Jin C, Shi L, Li K, et al. Mechanism of tumor‑derived extracellular vesicles in regulating renal cell carcinoma progression by the delivery of malat1. Oncol Rep. 2021;46:187. doi:10.3892/or.2021.8138
27. Riss T, Niles A, Moravec R, et al. Cytotoxicity assays: in vitro methods to measure dead cells. In: Markossian S, Grossman A, Brimacombe K, Arkin M, Auld D, Austin CP, editors. Assay Guidance Manual. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2004.
28. Suski JM, Braun M, Strmiska V, et al. Targeting cell-cycle machinery in cancer. Cancer Cell. 2021;39:759–778. doi:10.1016/j.ccell.2021.03.010
29. Matsui T, Chiyo T, Kobara H, et al. Telmisartan inhibits cell proliferation and tumor growth of esophageal squamous cell carcinoma by inducing s-phase arrest in vitro and in vivo. Int J Mol Sci. 2019;20:3197. doi:10.3390/ijms20133197
30. Sharma GP, Gurung SK, Inam A, et al. Cid-6033590 inhibits p38mapk pathway and induces s-phase cell cycle arrest and apoptosis in du145 and pc-3 cells. Toxicol in Vitro. 2019;60:420–436. doi:10.1016/j.tiv.2019.06.003
31. Gopinathan L, Tan SLW, Padmakumar VC, et al. Loss of cdk2 and cyclin a2 impairs cell proliferation and tumorigenesis. Cancer Res. 2014;74:3870. doi:10.1158/0008-5472.CAN-13-3440.
32. Pistritto G, Trisciuoglio D, Ceci C, et al. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging. 2016;8:603–619. doi:10.18632/aging.100934
33. Hossain MS, Mineno K, Katafuchi T. Neuronal orphan g-protein coupled receptor proteins mediate plasmalogens-induced activation of erk and akt signaling. PLoS One. 2016;11:e0150846. doi:10.1371/journal.pone.0150846
34. Moon H, Ro SW. Mapk/erk signaling pathway in hepatocellular carcinoma. Cancers. 2021;13:3026. doi:10.3390/cancers13123026
35. Nath AK, Ma J, Chen ZZ, et al. Genetic deletion of gpr27 alters acylcarnitine metabolism, insulin sensitivity, and glucose homeostasis in zebrafish. FASEB j. 2020;34:1546–1557. doi:10.1096/fj.201901466R
36. Chopra DG, Yiv N, Hennings TG, Zhang Y, Ku GM. Deletion of gpr27 in vivo reduces insulin mrna but does not result in diabetes. Sci Rep. 2020;10:5629. doi:10.1038/s41598-020-62358-4
37. Satriano L, Lewinska M, Rodrigues PM, et al. Metabolic rearrangements in primary liver cancers: cause and consequences. Nat Rev Gastro Hepat. 2019;16:748–766. doi:10.1038/s41575-019-0217-8
38. Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. Ualcan: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–658. doi:10.1016/j.neo.2017.05.002
39. Gopinathan L, Tan SL, Padmakumar VC, et al. Loss of cdk2 and cyclin a2 impairs cell proliferation and tumorigenesis. Cancer Res. 2014;74:3870–3879. doi:10.1158/0008-5472.CAN-13-3440
40. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi:10.1038/35077213
41. Liu F, Yang X, Geng M, et al. Targeting erk, an achilles’ heel of the mapk pathway, in cancer therapy. Acta Pharm Sin B. 2018;8:552–562. doi:10.1016/j.apsb.2018.01.008
42. Lee S, Rauch J, Kolch W. Targeting mapk signaling in cancer: mechanisms of drug resistance and sensitivity. Int J Mol Sci. 2020;21. doi:10.7150/ijms.39074
43. Lee C, Cheung ST. Stat3: an emerging therapeutic target for hepatocellular carcinoma. Cancers. 2019;11:1646. doi:10.3390/cancers11111646
44. Gough DJ, Corlett A, Schlessinger K, et al. Mitochondrial stat3 supports ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi:10.1126/science.1171721
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.