Back to Journals » Journal of Inflammation Research » Volume 18
Glycochenodeoxycholic acid induces the release of IL-1beta from L02 cells via the NLRP3/caspase-1/GSDMD pathway to activate LX2 cells
Authors Li J, Han L, Feng S, Lin G, Zou G, Huang T, Ran T, Zhao X ![]()
Received 2 April 2025
Accepted for publication 24 September 2025
Published 5 November 2025 Volume 2025:18 Pages 15429—15440
DOI https://doi.org/10.2147/JIR.S532042
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Fatih Türker
Jianchao Li,1,2,* Lu Han,1,* Shu Feng,1,* Guoyuan Lin,1 Gaoliang Zou,1 Tao Huang,1 Tao Ran,1 Xueke Zhao1,2
1Department of Infectious Diseases, Affiliated Hospital of Guizhou Medical University, Guiyang, 550004, People’s Republic of China; 2Clinical Medical College, Guizhou Medical University, Guiyang, 550004, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xueke Zhao, Email [email protected]
Purpose: Cholestatic liver diseases, characterized by hepatic bile acid accumulation, often progress to fibrosis through incompletely understood mechanisms. While hydrophobic bile acids like glycochenodeoxycholic acid (GCDCA) are known to induce hepatocyte apoptosis or necrosis and their role in pyroptosis—a pro-inflammatory programmed cell death—remains unclear. This study investigates whether GCDCA drives hepatic stellate cell (HSC) activation via NLRP3 inflammasome-mediated pyroptosis in hepatocytes.
Patients and Methods: Using L02 hepatocytes and LX2 HSCs, we evaluated the effects of GCDCA on NLRP3 inflammasome activation, gasdermin D (GSDMD)-dependent pyroptosis, and interleukin-1beta (IL-1β) secretion. Cells were treated with GCDCA (25– 400 μM) ± lipopolysaccharide (LPS) to simulate cholestatic injury. Caspase-1 inhibition (Ac-YVAD-cmk), GSDMD knockdown (shRNA), and IL-1 receptor antagonist (IL-1RA) were employed to dissect mechanistic pathways. Pyroptosis (LDH release, Annexin V/PI staining), inflammasome components (NLRP3, ASC, caspase-1), GSDMD cleavage, IL-1β secretion (ELISA), and HSC activation (α-SMA, Collagen-I, proliferation/migration assays) were analyzed.
Results: GCDCA alone upregulated NLRP3, pro-caspase-1, and pro-IL-1β in L02 cells but required LPS co-stimulation to trigger caspase-1 activation and GSDMD cleavage, indicating a dual-signal mechanism (priming + activation). This led to pyroptotic cell death (dose/time-dependent LDH release, Annexin V⁺/PI⁺ cells) and IL-1β secretion. GSDMD knockdown abolished pyroptosis and IL-1β release, while caspase-1 inhibition suppressed GSDMD-N-terminal (GSDMD-NT) generation. Conditioned media from pyroptotic hepatocytes activated LX2 HSCs, upregulating α-SMA and Collagen-I, and enhancing proliferation/migration—effects reversed by IL-1RA.
Conclusion: GCDCA promotes HSC activation via NLRP3 inflammasome-mediated pyroptosis in hepatocytes, driven by caspase-1-dependent GSDMD cleavage and IL-1β paracrine signaling. This mechanism bridges bile acid toxicity to fibrogenesis, highlighting therapeutic potential for targeting NLRP3-GSDMD-IL-1β signaling in cholestatic liver diseases.
Plain Language Summary: Cholestatic liver diseases, marked by toxic bile acid buildup, often progress to liver scarring (fibrosis), but the exact mechanisms are unclear. This study explored how a bile acid (GCDCA) triggers inflammatory cell death (pyroptosis) and drives fibrosis. Using lab-grown liver and scar-forming cells, the team exposed liver cells to GCDCA and bacterial toxins (LPS) to mimic disease conditions. They blocked key molecules to identify critical pathways. GCDCA activates the NLRP3 inflammasome, priming liver cells for pyroptosis. Combined with LPS, GCDCA triggers pyroptosis, releasing IL-1β, a protein that activates scar-forming cells. Blocking IL-1β reversed scar cell activity. This reveals how bile acids like GCDCA cause liver scarring through pyroptosis and IL-1β signaling. Targeting this pathway—NLRP3, GSDMD (a cell death protein), or IL-1β—could lead to new treatments for cholestatic liver diseases.
Keywords: glycochenodeoxycholic acid, NLRP3 inflammasome, pyroptosis, gasdermin D, interleukin-1beta, hepatic stellate cell
Introduction
Liver fibrosis, a common pathological endpoint of chronic liver diseases (eg, cholestasis, viral hepatitis, metabolic dysfunction-associated steatotic liver disease (MASLD)), is characterized by the excessive extracellular matrix (ECM) deposition and impaired repair following hepatic injury.1,2 Cholestatic disorders, such as intrahepatic cholestasis of pregnancy (ICP), biliary atresia and primary biliary cholangitis (PBC), act as critical drivers of fibrosis. Globally, cirrhosis due to advanced fibrosis affects over 2.4 million people annually, with cholestatic diseases accounting for 15–20% of cases. Primary Biliary Cholangitis (PBC) has a prevalence rate of 1.9–4.5 per 100,000 in Western populations, while Intrahepatic Cholestasis of Pregnancy (ICP) exhibits regional variations in incidence—Europe (0.3–1.5%) and South America (up to 5.6%)—which can induce preterm birth and fetal distress.3 These conditions are marked by persistent bile duct injury and chronic portal inflammation, progressing to fibrosis, cirrhosis and ultimately liver failure or transplantation.4–6
Although the precise mechanisms remain incompletely elucidated, the systemic and intrahepatic accumulation of hydrophobic bile salts has been recognized as a key unifying pathogenic hallmark.7 Bile acid accumulation directly inflicts hepatocyte injury by inducing mitochondrial dysfunction and excessive generation of reactive oxygen species (ROS), depleting antioxidant defense systems including glutathione and activating oxidative stress-sensing pathways such as KEAP1-Nrf2. Impaired Nrf2 signaling further exacerbates intracellular redox imbalance.8 The ensuing oxidative stress triggers inflammatory responses: ROS activate NF-κB and MAPK signaling pathways, promoting the expression of pro-inflammatory factors such as TNF-α and IL-6 and recruiting/activating immune cells.9 Concurrently, ROS can activate the NLRP3 inflammasome by inducing mitochondrial DNA release and K⁺ efflux.5,10 Sustained inflammatory microenvironments ultimately drive hepatic fibrogenesis, primarily through the activation and transdifferentiation of hepatic stellate cells (HSCs) into collagen-secreting myofibroblasts. Activated HSCs further secrete TIMP-1, inhibiting the degradation of the extracellular matrix (ECM).2
Central to linking bile acid toxicity with inflammation is NLRP3 inflammasome-mediated pyroptosis. During the priming phase, hydrophobic bile acids (eg, GCDCA) and gut dysbiosis-derived LPS activate TLR4/NF-κB signaling to upregulate NLRP3, pro-caspase-1 and pro-IL-1β.11 In the activation phase, bile acid-induced ROS and lysosomal rupture provide the second signal for NLRP3 oligomerization, recruiting ASC adaptor protein to activate caspase-1. Activated caspase-1 cleaves gasdermin D (GSDMD) to form plasma membrane pores that induce pyroptotic cell death, while concurrently processing pro-IL-1β into mature IL-1β for extracellular release through GSDMD pores.12 This pyroptotic cascade significantly amplifies fibrogenesis: Hepatocyte-derived IL-1β activates PI3K/AKT and JAK/STAT signaling in HSCs via IL-1 receptor binding, inducing α-smooth muscle actin (α-SMA) and type I collagen expression to establish a self-perpetuating pyroptosis-fibrosis cycle.13
Ursodeoxycholic acid (UDCA) and obeticholic acid (OCA), first-line therapeutics for cholestatic disorders, exhibit constrained clinical efficacy: approximately 40% of ICP patients demonstrate refractoriness to UDCA, while pharmacologic FXR agonism (OCA) paradoxically exacerbates fibrogenesis via hepatic stellate cell (HSC) activation in decompensated cirrhosis.14 Consequently, delineating the dynamic interactome of hydrophobic bile salt metabolism and its crosstalk with the immune microenvironment holds transformative potential for developing precision antifibrotic therapies.
Glycochenodeoxycholic acid (GCDCA), a core hydrophobic bile acid, serves as a sensitive diagnostic biomarker for ICP, with elevated concentrations strongly correlating with disease severity and adverse fetal outcomes.15 However, the mechanistic role of GCDCA in hepatic injury and fibrogenesis remains incompletely elucidated. This study aims to delineate the molecular mechanisms by which GCDCA activates hepatocyte pyroptosis to drive HSC activation. By unraveling the bile acid toxicity-pyroptosis-fibrosis regulatory cascade network, this study provides novel therapeutic targets to overcome the clinical challenge of currently irreversible fibrosis in cholestasis treatment.
Materials and Methods
Cell Culture
L02 cells (Cellverse) and LX2 cells (Procell) were cultured in RPMI 1640 medium (Gibco) and DMEM (Gibco), respectively, both supplemented with 10% FBS (VivaCell). Cells were cultured under standard conditions (37°C, 5% CO2 humidified atmosphere) and restricted to ≤20 passages to preserve genomic integrity.
L02 and LX2 cells were synchronized in serum-free medium for 12 h. L02 cells were exposed to graded GCDCA concentrations (25–400 μM, HY-N2334, MCE) for 24 h or 200 μM GCDCA for 6–72 h to establish dose/time-response profiles.3 For LPS (L8880, Solarbio) co-stimulation, cells were pre-treated with 1 μg/mL LPS (4 h) followed by GCDCA (50–400 μM, 24 h) or combined LPS+200 μM GCDCA (LG200) for 12–72 h. To investigate NLRP3-specific mechanisms, cells were pretreated with LPS (1 μg/mL) for 4 h, followed by incubation with the selective NLRP3 inhibitor MCC950 (10 μM, HY-12815, MCE) for 2 h prior to GCDCA exposure (200 μM, 24 h). Caspase-1 was pharmacologically inhibited by pre-incubating with Ac-YVAD-cmk (50 μM, HY-16990; MCE) for 2 h prior to stimulation, with DMSO and LPS serving as negative and positive controls, respectively.11 Conditioned media from treated L02 cells were co-cultured with LX2 cells in the presence/absence of Interleukin-1 receptor antagonist (IL-1RA) (200 ng/mL, HY-P7029A, MCE).
For lentivirus-mediated GSDMD knockdown, recombinant lentiviruses expressing short hairpin RNA (shRNA) targeting human GSDMD or non-targeting control (Genechem) were generated using a three-plasmid packaging system (psPAX2/pMD2.G/shRNA plasmid). Viral supernatants were concentrated to titers ≥1×108 TU/mL (System Biosciences). L02 cells at 40% confluence were transduced with lentivirus at a multiplicity of infection (MOI) of 10 in the presence of 8 μg/mL polybrene and transduction enhancer A for 20 h. After replacing virus-containing medium with fresh complete medium, stable cell lines were selected with 1 μg/mL puromycin for 7 days. Knockdown efficiency was confirmed by Western blot prior to experiments.
LDH Assay
L02 cells were seeded in 96-well plates and stratified into experimental groups. LDH release reagent was introduced 1 h prior to analysis. Supernatants (120 μL) were combined with LDH working solution (60 μL) and subjected to dark-phase incubation (30 min, 25°C). Absorbance was quantified via dual-wavelength detection (490 nm, 600 nm) using a microplate reader. LDH release rates were calculated using standardized calibration curves.
Flow Cytometry
L02 cells were co-stained with Annexin V-FITC/PI (5 μL and 10 μL, respectively) and analyzed via NAVIOS™ (Beckman Coulter), ≥10,000 events collected per sample. FlowJo v10.7 was used for quadrant analysis, distinguishing viable (Annexin V−/PI−), early pyroptotic (Annexin V⁺/PI−) and late pyroptotic/necrotic (Annexin V⁺/PI⁺) populations.
Caspase-1/4 Activity Analysis
Caspase-1/4 activity was quantified via enzymatic kinetic assays using a commercial Caspase-1/4 Activity Kit (Beyotime). Whole-cell lysates were prepared and incubated with caspase-specific substrates, which release chromogenic products upon cleavage by activated caspases. Absorbance at 405 nm was measured to determine enzymatic activity.
ELISA
Supernatants were subjected to IL-1β quantification using a sandwich ELISA kit. Absorbance at 450 nm was measured to determine cytokine concentration.
Western Blot
Cellular proteins were extracted using RIPA lysis buffer (R0010, Solarbio) supplemented with 1 mM PMSF, with 150–250 μL of lysis buffer added per 106 cells. The mixture was incubated on ice for 30 minutes, followed by centrifugation at 12,000 × g for 15 minutes at 4°C. The middle clear supernatant was carefully aspirated and quantified via NanoDrop 3000 (Thermo Scientific), with samples adjusted to a concentration of 25 μg protein per 15 μL loading volume. Following blocking with 5% nonfat milk, membranes were incubated overnight at 4°C with primary antibodies targeting specific proteins (listed in Table 1). HRP-conjugated secondary antibodies (1:7,000) were applied at room temperature for 1 h. Protein bands were visualized with ECL substrate. Pre-stained protein molecular weight markers (Yeasen, 20352ES) were used as references.
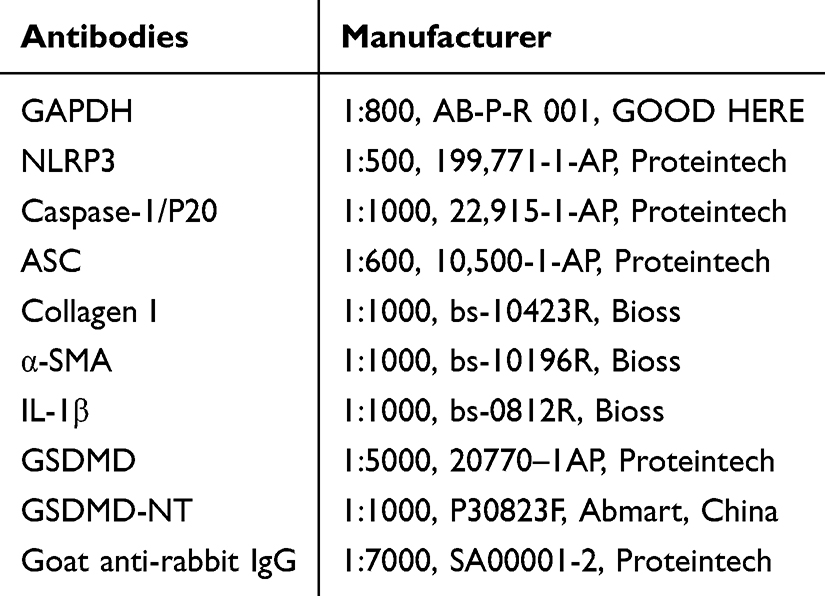 |
Table 1 Antibodies List |
Wound Healing Assay
LX2 cells were plated in 6-well plates and cultured to full confluency. Uniform scratches were generated using a 200 μL pipette tip, followed by gentle PBS washing to remove debris. Conditioned medium from L02 hepatocytes (with or without IL-1RA) was mixed with fresh complete medium at a 1:1 volume ratio for co-culture with LX2 cell for 48 h and wound closure was documented at fixed positions (0, 12, 24, 48 h) under standardized imaging conditions.
CCK-8 Assay
LX2 cells were plated in 96-well plates and co-cultured with L02 cell supernatant (1:1volume ratio in 10% FBS medium) for 1–7 days. Post-incubation, 20 µL CCK-8 reagent was added per well, followed by a standardized incubation. Absorbance at 450 nm was quantified using a microplate reader.
Statistical Analysis
All experimental data were analyzed using GraphPad Prism 9.0. Group comparisons were performed via Student’s t-test (two-group analysis), or one-way ANOVA followed by Tukey’s post hoc test (multi-group analysis). All experiments were independently repeated three times (n = 3), with data expressed as mean ± standard deviation (SD). Statistical significance was defined as P < 0.05.
Results
GCDCA Activates the NLRP3 Inflammasome in Hepatocytes
To investigate the effect of GCDCA on hepatocyte pyroptosis, we examined the NLRP3 inflammasome signaling pathway. The results showed that treatment of L02 cells with varying concentrations of GCDCA significantly upregulated the protein expression of NLRP3, pro-caspase-1 and pro-IL-1β in a dose-dependent manner (Figure 1A). When L02 cells were treated with a fixed concentration of GCDCA for different durations, the expression of these proteins increased over time (Figure 1B). However, the levels of the adaptor protein ASC and activated cleaved caspase-1 remained unchanged, suggesting that GCDCA alone is insufficient to trigger inflammasome assembly or caspase-1 activation.
 |
Figure 1 GCDCA activates the NLRP3 inflammasome in hepatocytes. (A) Dose-dependent effects of GCDCA (25–400 μM, 24 h) on protein levels effects of NLRP3, ASC, pro-caspase-1, caspase-1 P20, pro-IL-1β and GAPDH. (B) Time-dependent effects of these proteins in L02 cells treated with 200 μM GCDCA. (C and D) Synergistic priming by LPS (1 μg/mL, 4 h pre-treatment): GCDCA (50–400 μM, 24 h) (C) or time-dependent (12–72 h) (D) enhancement of NLRP3, pro-caspase-1, and pro-IL-1β expression in LPS-primed cells. (E) Comparative analysis of NLRP3 inflammasome activationin L02 cells treated with GCDCA (200 μM, 24 h) with/without LPS co-stimulation. (F) Essential role of NLRP3 in GCDCA-induced pyroptosis. L02 cells were pretreated with/without LPS (1 μg/mL, 4 h) and MCC950 (10 μM, 2 h) prior to GCDCA (200 μM, 24 h) exposure. Whole cell lysates were examined by Western Blot analysis. Molecular weight markers (kDa) are indicated. n ≥ 3 for each group. (LG200: LPS+200 μM GCDCA; G200: GCDCA 200 μM). |
To simulate the canonical activation pathway of the NLRP3 inflammasome, L02 cells were pretreated with LPS (a “Signal 1” priming stimulus) before exposure to GCDCA. Combined treatment synergistically enhanced the expression of NLRP3, pro caspase-1 and IL-1β in both time- and dose-dependent manners (Figures 1C and D). Furthermore, co-treatment with GCDCA and LPS further amplified NLRP3 inflammasome signaling, indicating a bile acid-specific potentiation of LPS-driven inflammasome activation (Figures 1E).
Critically, to confirm the essential role of NLRP3 in this process, we employed the selective NLRP3 inhibitor MCC950. As shown in Figure 1F, compared to the GCDCA+LPS co-treatment group, additional MCC950 treatment significantly suppressed the expression of NLRP3 and completely abolished the downstream activation events, specifically caspase-1 cleavage (p20 subunit) and GSDMD processing (GSDMD-NT formation). Concurrently, MCC950 treatment led to the accumulation of the precursor forms pro-caspase-1 and full-length GSDMD, indicating effective blockade of proteolytic activation. This demonstrates that NLRP3 inhibition not only affects upstream expression but also completely blocks downstream execution, confirming that GCDCA-induced pyroptosis is specifically mediated through the NLRP3 inflammasome pathway.
GCDCA Induces GSDMD-Mediated Pyroptosis in Hepatocytes
As the terminal executor of NLRP3 inflammasome signaling, GSDMD facilitates pyroptosis by forming transmembrane pores upon caspase-1/4-mediated cleavage.11 To validate GSDMD-dependent pyroptosis in GCDCA/LPS-treated L02 hepatocytes, we assessed LDH release and GSDMD proteolytic activation. GCDCA induced time- and dose-dependent LDH release, indicative of progressive plasma membrane rupture (Figure 2A and B). Concurrently, Western blot analysis revealed kinetic GSDMD-N-terminal (GSDMD-NT) accumulation (activated pore-forming N-terminal fragment) with full-length GSDMD depletion, confirming caspase-1-dependent GSDMD cleavage (Figure 2C and D).
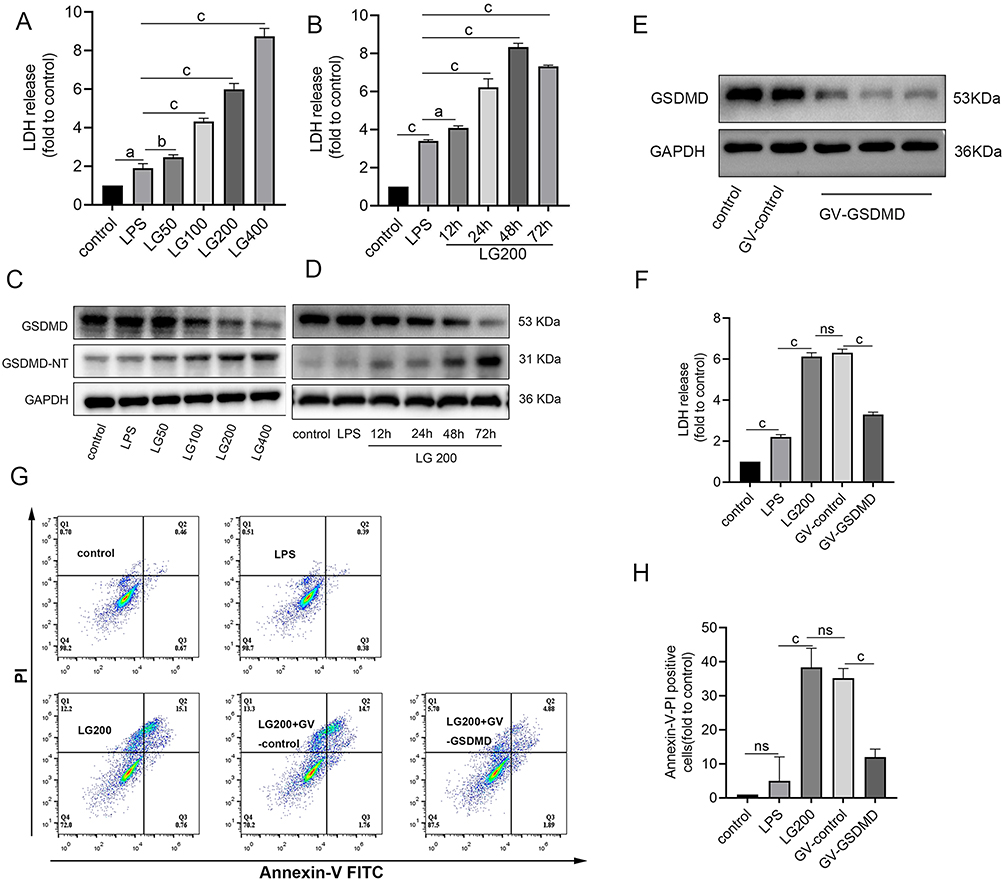 |
Figure 2 GCDCA induces GSDMD-mediated pyroptosis in hepatocytes. (A and B) Time- and dose-dependent LDH release in LPS-primed L02 cells treated with GCDCA: (A) Dose-response (50–400 μM GCDCA, 24 h); (B) Time-course (200 μM GCDCA, 12–72 h). (C) Dose-dependent GSDMD proteolysis in LPS-primed cells treated with GCDCA (50–400 μM, 24 h); (D) Time-dependent GSDMD proteolysis (200 μM GCDCA, 12–72 h). GAPDH serves as loading control. (E) Validation of GSDMD knockdown efficiency in L02 cells. (F) LDH release in lentiviruses-transfected L02 cells treated with GCDCA (200 μM, 24 h). (G) Representative flow plots showing Annexin V/PI staining in lentiviruses-transfected L02 cells. (H) Flow cytometry quantification of pyroptotic cells (Annexin V⁺/PI⁺). n ≥ 3 for each group. aP < 0.005, bP < 0.05, cP < 0.0001. Abbreviation: GV, Empty Vector Control. |
GSDMD knockdown attenuated GCDCA-induced cytotoxicity, with reduction in LDH release and decrease in pyroptotic cell ratio (Annexin V⁺/PI⁺ subpopulation) compared to scrambled controls (Figure 2E–H). These data conclusively demonstrate that GCDCA triggers GSDMD-mediated pyroptosis in hepatocytes through NLRP3 inflammasome activation.
GCDCA Triggers Caspase-1-Dependent Pyroptosis in Hepatocytes
GSDMD mediates pyroptosis through two distinct pathways: the canonical caspase-1-dependent pathway16 and the non-canonical caspase-4/5/11-dependent pathway.17 To elucidate the mechanism of GCDCA-induced pyroptosis in hepatocytes, we examined caspase-1 and caspase-4 activity in L02 cells. The results demonstrated significant upregulation of caspase-1 activity with no notable change in caspase-4 activity, confirming caspase-1 as the dominant pathway in this context (Figure 3A and B).
 |
Figure 3 GCDCA triggers caspase-1-dependent pyroptosis in hepatocytes. (A and B) Caspase-1/4 activity in LPS-primed L02 cells treated with GCDCA. (C and D) Flow cytometry quantification of pyroptotic cells (Annexin V⁺/PI⁺) in L02 cells with caspase-1 inhibitor Ac-YVAD-cmk (AC, 50 μM) followed by LPS + GCDCA (200 μM, 24 h). (E) LDH release in L02 cells with AC pretreatment. (F)Western blot analysis of pyroptosis-related proteins in L02 cells with AC pretreatment. n ≥ 3 for each group. aP < 0.005, bP < 0.0001. |
Subsequently, we investigated whether the caspase-1 inhibitor Ac-YVAD-CMK (AC) could reverse GCDCA-mediated pyroptosis. The results demonstrated a significant reduction in LDH release and pyroptotic cell ratio in AC-pretreated L02 cells compared to GCDCA-alone groups (Figure 3C–E). Western blot analysis further corroborated these findings, revealing suppressed pro-caspase-1, diminished caspase-1 P20 subunit activation and attenuated GSDMD-NT accumulation, alongside restored full-length GSDMD expression (Figure 3F). These data mechanistically validate caspase-1 as the dominant executor of GCDCA-induced pyroptosis, excluding contributions from non-canonical pathways.
GCDCA Drives IL-1β Secretion Through GSDMD-Dependent Activation
Pro-IL-1β, a downstream substrate of caspase-1, undergoes proteolytic cleavage to generate the mature IL-1β P17 subunit, which is subsequently secreted extracellularly to exert its biological functions.18 To investigate this process, we quantified mature IL-1β levels in the supernatant of L02 hepatocytes. ELISA results demonstrated a significant elevation of extracellular IL-1β in GCDCA-treated cells, exhibiting both time-dependent and dose-dependent dynamics (Figure 4A and B).
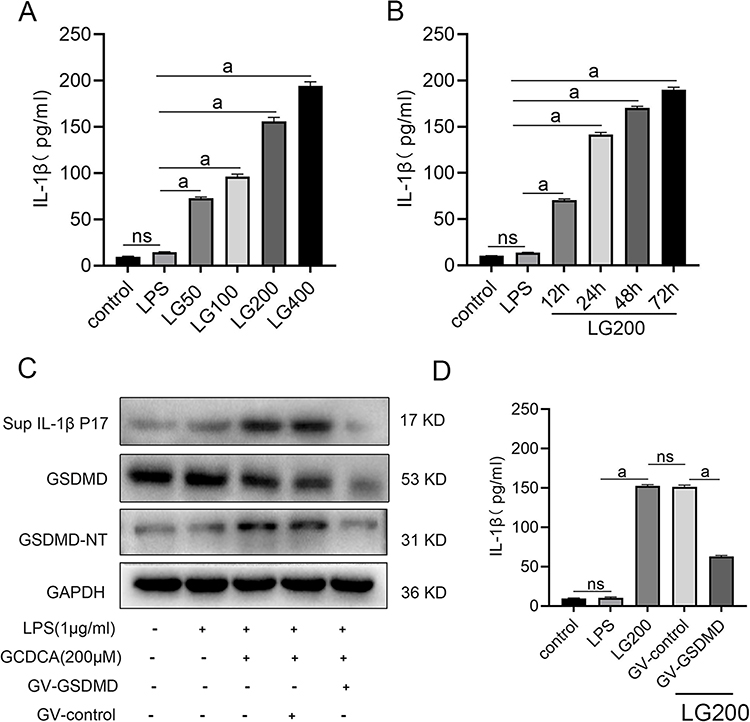 |
Figure 4 GCDCA drives IL-1β secretion through GSDMD-dependent activation. (A) Dose-dependent IL-1β secretion in LPS-primed L02 cells treated with GCDCA (50–400 μM, 24 h). (B)Time-dependent IL-1β secretion (200 μM GCDCA, 12–72 h). (C) Western blot analysis of GSDMD-NT and GSDMD in L02 cells and mature IL-1β in cell supernatant. (D) ELISA quantification of extracellular IL-1β. n ≥ 3 for each group. aP < 0.01. |
To establish GSDMD’s essential role in IL-1β secretion, we genetically silenced GSDMD in L02 cells. Western blot analysis revealed marked reduction of full-length GSDMD and its activated GSDMD-NT, concomitant with diminished mature IL-1β levels in the supernatant (Figure 4C). This observation was corroborated by ELISA, which showed decrease in extracellular IL-1β in GSDMD-knockdown cells compared to controls (Figure 4D).
GCDCA-Mediated Hepatocyte IL-1β Secretion Activates HSCs
To delineate the profibrotic effects of IL-1β secreted during hepatocyte pyroptosis, conditioned media from pyroptotic L02 hepatocytes were applied to LX2 HSCs. This intervention markedly upregulated the expression of α-smooth muscle actin (α-SMA) and collagen type I (Collagen-I) in LX2 cells (Figure 5A), consistent with their activation into myofibroblasts-a hallmark of fibrogenesis driven by hepatocyte-derived inflammatory mediators.
 |
Figure 5 GCDCA-mediated hepatocyte IL-1β secretion activates HSCs. (A) Western blot analysis ofα-SMA and Collagen-I in LX2 cells treated with conditioned medium (CM) from GCDCA/LPS-stimulated L02 hepatocytes. (B) The protein levels of α-SMA and Collagen-I in LX2 cells with treatment of IL-1 receptor antagonist (IL-1RA, 200 ng/mL) upon CM exposure. (C and D) Representative images and quantification of LX2 migration upon CM exposure detected by wound healing assay. (E) CCK-8 assay of LX2 proliferation by CM treatment. n ≥ 3 for each group. aP < 0.05. |
Subsequent co-treatment with the IL-1 receptor antagonist (IL-1RA) significantly suppressed α-SMA and Collagen-I expression (Figure 5B), unequivocally establishing IL-1β as a critical mediator in orchestrating HSC activation and extracellular matrix (ECM) deposition. Functional assays further revealed that IL-1RA attenuated LX2 cell proliferation and impaired migratory capacity (Figure 5C–E).
Discussion
This study reveals that GCDCA, a core cytotoxic bile acid in cholestatic diseases, activates the NLRP3 inflammasome in hepatocytes, thereby inducing GSDMD-dependent pyroptosis and subsequent IL-1β release, which drives HSC activation and fibrogenesis. These findings unveil a novel molecular bridge linking bile acid metabolic dysregulation to hepatic fibrogenesis, offering groundbreaking insights into the pathological progression of cholestatic liver diseases.
While hydrophobic bile acids like GCDCA are traditionally recognized for inducing hepatocyte apoptosis or necrosis via Fas-dependent pathways and mitochondrial permeability transition pore opening,19–21 their role in pyroptosis remains underexplored. This study unveils GCDCA’s dual regulatory role in pyroptosis: it primes the NLRP3 inflammasome by upregulating NLRP3, pro-caspase-1 and pro-IL-1β expression, yet requires a secondary signal (eg, LPS, mimicking gut-derived endotoxin translocation in cholestasis) to trigger caspase-1 activation, GSDMD cleavage and IL-1β maturation. The dual regulatory role of GCDCA aligns with the classical “two-signal” model of NLRP3 inflammasome activation. Given documented evidence that bile acids induce mitochondrial stress,22 we propose that GCDCA likely promotes NLRP3 oligomerization by triggering “Signal 2”-associated events (eg, ROS generation or ion channel perturbation). However, GCDCA inherently lacks the capacity to initiate TLR4/NF-κB-mediated “Signal 1” – the priming signal essential for transcriptional upregulation of inflammasome components.This mechanistic framework explains the clinical observation that cholestatic patients exhibit gut dysbiosis and systemic inflammation,23 where GCDCA accumulation synergizes with LPS to amplify hepatic injury. Therapeutically, targeting the “Signal 2” pathway may specifically block bile acid-induced inflammasome activation while preserving host defense functions. This study establishes GCDCA as a critical driver of pyroptosis-dependent inflammation, offering a mechanistic rationale for targeting NLRP3-GSDMD-IL-1β signaling in cholestatic liver disease.
GSDMD, as the central executor of pyroptosis, exhibits dual functionalities in cholestatic liver injury.24 On one hand, GSDMD-NT forms transmembrane pores on hepatocyte membranes, leading to LDH release and cell lysis, a process strictly dependent on caspase-1-mediated cleavage.25 On the other hand, sublytic GSDMD pores allow selective release of inflammatory cytokines such as IL-1β while transiently preserving membrane integrity during early disease stages.26 This dynamic regulation explains the spatiotemporal progression from localized inflammation to widespread hepatocyte necrosis in advanced cholestasis. Our experiments confirmed that GCDCA-induced hepatocyte pyroptosis promotes hepatocyte injury and fibrosis through the dual function of GSDMD. Importantly, GSDMD knockdown not only abolishes pyroptosis but also completely blocks IL-1β secretion, highlighting its pivotal role in amplifying bile acid-induced inflammatory signals. This finding aligns with recent therapeutic breakthroughs, such as paeoniflorin, a bioactive compound derived from Paeonia species, which directly targets GSDMD to inhibit pyroptosis and significantly alleviates cholestatic liver injury.27 These results validate GSDMD as a therapeutic linchpin for cholestatic disorders.
IL-1β, a key effector of pyroptosis, was identified as a central driver of HSCs activation in this study.27 Hepatocyte-derived IL-1β upregulating α-SMA and type I collagen expression while enhancing HSC proliferation and migration. These findings align with clinical evidence showing a significant positive correlation between serum IL-1β levels and liver fibrosis staging in patients with PBC and ICP.3,28 Notably, pharmacological blockade of IL-1β signaling using an IL-1 receptor antagonist effectively reversed the pro-fibrotic phenotype of activated HSCs, suggesting therapeutic potential for targeting this axis in fibrotic liver diseases.
While this study systematically elucidates the pro-fibrotic mechanisms of GCDCA in cellular models, clinical translation faces challenges: (1) the regulatory effects of the NLRP3/GSDMD pathway in cholestatic liver disease models (eg, bile duct ligation or DDC-induced models) require validation; (2) the involvement of other hepatic cell types (eg, Kupffer cells, cholangiocytes) via similar pyroptosis-driven mechanisms remains unclarified; (3) long-term safety profiles of pyroptosis inhibitors (eg, MCC950, disulfiram) in chronic liver diseases need rigorous evaluation. Future studies should integrate single cell sequencing and spatial transcriptomics to map spatiotemporal pyroptosis signaling patterns across hepatic zones and develop personalized therapeutic strategies targeting this axis.
Conclusion
This study reveals that GCDCA drives hepatic fibrosis through the NLRP3/GSDMD/IL-1β axis, challenging the conventional view that bile acids solely induce liver injury via apoptosis or necrosis. These findings not only redefine GCDCA’s pathogenic role but also provide a mechanistic foundation for developing precision therapies targeting pyroptosis-dependent fibrotic progression
Acknowledgments
Jianchao Li, Lu Han and Shu Feng contributed equally to the work and should be regarded as co-first authors for this study. We would like to express our sincere gratitude to all those who have contributed to this study with their support and assistance.
Funding
The study received funding from Guizhou Provincial Science and Technology Program (QKH JC-ZK [2023]-214 and No. [2021] 094) and National Natural Science Foundation of China (No. 82060116 and No. 82260129).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hammerich L, Tacke F. Hepatic inflammatory responses in liver fibrosis. Nat Rev Gastroenterol Hepatol. 2023;20(10):633–646. doi:10.1038/s41575-023-00807-x
2. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18(3):151–166. doi:10.1038/s41575-020-00372-7
3. Ovadia C, Seed PT, Sklavounos A, et al. Association of adverse perinatal outcomes of intrahepatic cholestasis of pregnancy with biochemical markers: results of aggregate and individual patient data meta-analyses. Lancet. 2019;393(10174):899–909. doi:10.1016/S0140-6736(18)31877-4
4. Goldstein J, Levy C. Novel and emerging therapies for cholestatic liver diseases. Liver Int. 2018;38(9):1520–1535. doi:10.1111/liv.13880
5. Fuchs CD, Trauner M. Role of bile acids and their receptors in gastrointestinal and hepatic pathophysiology. Nat Rev Gastroenterol Hepatol. 2022;19(7):432–450. doi:10.1038/s41575-021-00566-7
6. Zhang Y, Hou L, Guo T, Lu H, Zhang X, Xing M. An in-depth analysis of the effects of excessive acetochlor exposure on chicken liver health. Pestic Biochem Physiol. 2025;208:106280. doi:10.1016/j.pestbp.2024.106280
7. Hohenester S, Gates A, Wimmer R, et al. Phosphatidylinositol-3-kinase p110gamma contributes to bile salt-induced apoptosis in primary rat hepatocytes and human hepatoma cells. J Hepatol. 2010;53(5):918–926. doi:10.1016/j.jhep.2010.05.015
8. Gao R, Huang Q, Zeng Y, et al. Pueraria lobata-prunus mume complex alleviates alcoholic liver disease by regulating lipid metabolism and inhibiting inflammation: a transcriptome and gut microbiota analysis. Foods. 2024;13(15):2431. doi:10.3390/foods13152431
9. Zhang Y, Lu H, Hou L, et al. GPR120 exacerbates the immune-inflammatory response in chicken liver by mediating acetochlor induced macrophage M1 polarization. J Hazard Mater. 2025;485:136928. doi:10.1016/j.jhazmat.2024.136928
10. Lu H, Guo T, Zhang Y, et al. 内质网应激诱导的NLRP3炎性体激活是聚苯乙烯微塑料 (PS-MPs) 诱导的鸡肺部炎症的新机制. [Endoplasmic reticulum stress-induced NLRP3 inflammasome activation as a novel mechanism of polystyrene microplastics (PS-MPs)-induced pulmonary inflammation in chickens]. J Zhejiang Univ Sci B. 2024;25(3):233–243. doi:10.1631/jzus.B2300409
11. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489. doi:10.1038/s41577-019-0165-0
12. Liu Z, Wang C, Yang J, et al. Caspase-1 engages full-length gasdermin d through two distinct interfaces that mediate caspase recruitment and substrate cleavage. Immunity. 2020;53(1):106–114e5. doi:10.1016/j.immuni.2020.06.007
13. Gaul S, Leszczynska A, Alegre F, et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol. 2021;74(1):156–167. doi:10.1016/j.jhep.2020.07.041
14. Chascsa DMH, Lindor KD. Emerging therapies for PBC. J Gastroenterol. 2020;55(3):261–272. doi:10.1007/s00535-020-01664-0
15. Han X, Wang J, Gu H, et al. Predictive value of serum bile acids as metabolite biomarkers for liver cirrhosis: a systematic review and meta-analysis. Metabolomics. 2022;18(7):43. doi:10.1007/s11306-022-01890-y
16. Fink SL, Cookson BT. Pillars Article: caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006. 8: 1812-1825. J Immunol. 2019;202(7):1913–1926. doi:10.1093/jimmunol/202.7.1913
17. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341(6151):1250–1253. doi:10.1126/science.1240988
18. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677–687. doi:10.1038/nm.3893
19. Sasaki M, Nakanuma Y. Bile acids and deregulated cholangiocyte autophagy in primary biliary cholangitis. Dig Dis. 2017;35(3):210–216. doi:10.1159/000450913
20. Woolbright BL, Jaeschke H. Novel insight into mechanisms of cholestatic liver injury. World J Gastroenterol. 2012;18(36):4985–4993. doi:10.3748/wjg.v18.i36.4985
21. Ashby K, Navarro Almario EE, Tong W, Borlak J, Mehta R, Chen M. Review article: therapeutic bile acids and the risks for hepatotoxicity. Aliment Pharmacol Ther. 2018;47(12):1623–1638. doi:10.1111/apt.14678
22. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11(1):55–67. doi:10.1038/nrgastro.2013.151
23. Sun D, Xie C, Zhao Y, et al. The gut microbiota-bile acid axis in cholestatic liver disease. Mol Med. 2024;30(1):104. doi:10.1186/s10020-024-00830-x
24. Li Y, Guo B. GSDMD-mediated pyroptosis: molecular mechanisms, diseases and therapeutic targets. Mol Biomed. 2025;6(1):11. doi:10.1186/s43556-025-00249-8
25. Zhao T, Chi Z, Wang D. Versatility of gasdermin D beyond pyroptosis. Trends Cell Biol. 2025. doi:10.1016/j.tcb.2025.02.011
26. Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in pyroptosis. Acta Pharm Sin B. 2021;11(9):2768–2782. doi:10.1016/j.apsb.2021.02.006
27. Ma X, Zhang W, Chen Y, et al. Paeoniflorin inhibited GSDMD to alleviate ANIT-induced cholestasis via pyroptosis signaling pathway. Phytomedicine. 2024;134:156021. doi:10.1016/j.phymed.2024.156021
28. Colapietro F, Gershwin ME, Lleo A. PPAR agonists for the treatment of primary biliary cholangitis: old and new tales. J Transl Autoimmun. 2023;6:100188. doi:10.1016/j.jtauto.2023.100188
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Pyroptosis: A Novel Intervention Target in the Progression of Osteoarthritis
Chang X, Kang Y, Yang Y, Chen Y, Shen Y, Jiang C, Shen Y
Journal of Inflammation Research 2022, 15:3859-3871
Published Date: 11 July 2022
Characterization of Cathepsin B in Mediating Silica Nanoparticle-Induced Macrophage Pyroptosis via an NLRP3-Dependent Manner
Ma L, Han Z, Yin H, Tian J, Zhang J, Li N, Ding C, Zhang L
Journal of Inflammation Research 2022, 15:4537-4545
Published Date: 8 August 2022
Inhibition of Dectin-1 Alleviates Neuroinflammatory Injury by Attenuating NLRP3 Inflammasome-Mediated Pyroptosis After Intracerebral Hemorrhage in Mice: Preliminary Study Results
Ding Z, Zhong Z, Wang J, Zhang R, Shao J, Li Y, Wu G, Tu H, Yuan W, Sun H, Wang Q
Journal of Inflammation Research 2022, 15:5917-5933
Published Date: 19 October 2022
Lactobacillus Plantarum Promotes Wound Healing by Inhibiting the NLRP3 Inflammasome and Pyroptosis Activation in Diabetic Foot Wounds
Wang X, Li X, Liu J, Tao Y, Wang T, Li L
Journal of Inflammation Research 2024, 17:1707-1720
Published Date: 16 March 2024
The Impact of NLRP3 Inflammasome on Osteoblasts and Osteogenic Differentiation: A Literature Review
Yang Z, Xu J, Kang T, Chen X, Zhou C
Journal of Inflammation Research 2024, 17:2639-2653
Published Date: 29 April 2024