")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Glucagon-like peptide-1 receptor (GLP-1R) signaling ameliorates dysfunctional immunity in COPD patients
Authors Huang J , Yi H , Zhao C, Zhang Y, Zhu L, Liu B, He P , Zhou M
Received 23 May 2018
Accepted for publication 21 August 2018
Published 9 October 2018 Volume 2018:13 Pages 3191—3202
DOI https://doi.org/10.2147/COPD.S175145
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Jingwen Huang,1,* Huahua Yi,1,* Chunliu Zhao,2 Yifan Zhang,3 Liying Zhu,3 Bing Liu,1 Ping He,4 Min Zhou1
1Department of Pulmonary and Critical Care Medicine, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China; 2Department of Respiratory Medicine, Luwan Branch, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China; 3Department of Nuclear Medicine, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China; 4Department of Microbiology and Immunology, Institutes of Medical Science, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Background: The glucagon-like peptide-1 receptor (GLP-1R) agonist – liraglutide (LIR) – is an insulin secretagogue for the treatment of diabetes and has been proven to have therapeutic potential in the treatment of COPD. Evidence suggested that activating GLP-1R signaling might have immunomodulating and anti-inflammatory effects. COPD is characterized by dysregulation of immunity, oxidative stress, and excessive inflammation responses. The aim of this study was to investigate whether GLP-1R signaling had a regulatory role in COPD immunity.
Patients and methods: Fifty-seven COPD patients in a stable condition and 51 age-, sex-, and smoking history-matched non-COPD subjects provided blood samples for isolation of peripheral blood mononuclear cells (PBMCs). GLP-1R expression was measured, and its association with clinical parameters and plasma cytokines was analyzed. T cell function was assessed at baseline and after regulating GLP-1R expression.
Results: GLP-1R expression decreased in circulating PBMCs of COPD patients, which was associated with decreased interferon (IFN)-γ expression. Reduced IFN-γ production stimulated by phytohemagglutinin (PHA) and increased programmed cell death protein 1 (PD-1) expression on T cells were observed in COPD patients compared with non-COPD subjects. Treatment with LIR could upregulate the GLP-1R expression, and this was observed to restore the antigen-stimulated IFN-γ production and downregulate PD-1 expression in T cells.
Conclusion: PBMCs of COPD patients showed defective GLP-1R signaling and functional T-lymphocyte abnormalities that could be rescued by LIR treatment.
Keywords: COPD, glucagon-like peptide-1 receptor, liraglutide, interferon-γ, T cell
Introduction
COPD is a major cause of morbidity and mortality worldwide. According to the GOLD 2018, COPD is the fourth leading cause of death in the world and is projected to be the third by 2020.1 There is no proven therapy to reverse pulmonary injury due to COPD, emphasizing the urgent need to explore novel targets to prevent disease progression.
Lung inflammation caused by recurrent pulmonary infection often leads to acute exacerbation of COPD (AECOPD).2,3 Innate immunity is the first line of defense against invading pathogens, and lymphocytes play pivotal roles in the regulation of immune response via either direct cell-to-cell interactions or secretion of cytokines, including tumor necrosis factor (TNF)-α, IL-12 p40, and interferon (IFN)-γ.4–6 Currently, dysregulation of immunity is thought to be an important disease mechanism in COPD locally and systematically. Chronic infection or antigen presentation would cause abnormalities in the distribution and function of T-lymphocytes, resulting in the dysfunction of cell-mediated immune responses.7,8 Functional exhausted effector T cells expressing programmed cell death protein 1 (PD-1), a negative costimulatory molecule that is associated with defective immune response, increase in COPD patients.9
Glucagon-like peptide-1 (GLP-1) is a hormone secreted by L cells of the distal ileum in response to food intake.10 GLP-1 receptor (GLP-1R) is a G-protein-coupled membrane receptor detected not only in the cells of the pancreatic islets but also in several other tissues such as the central nervous system, kidney, heart, blood vessels, and lung.11,12 Liraglutide (LIR), a GLP-1 analog, is a clinically used drug for metabolic syndromes with a wide knowledge in improving insulin sensitivity and anti-inflammatory function.13,14 In COPD, it has been found that activation of GLP-1R signaling by LIR could decrease the severity of AECOPD and airway remodeling,15,16 but the mechanism is not well known.
Recent studies suggested that the exacerbations of obstructive pulmonary disease and airway remodeling are closely associated with T-lymphocyte immune influences.17,18 It is also reported that GLP-1 can inhibit chemokine-induced differentiation of CD4-positive lymphocyte into TH1 cells.19 It is reasonable to hypothesize that GLP-1R signaling is associated with the function of T-lymphocytes in COPD patients. Our results may provide a possible therapeutic approach for COPD via modulation of GLP-1R signaling.
Patients and methods
Subjects
We obtained peripheral blood from the patients of respiratory department in Ruijin Hospital affiliated to the School of Medicine, Shanghai Jiao Tong University. All COPD patients met the criteria proposed by GOLD 2018 and were in a stable condition at the time of the study. The non-COPD group comprised age-, sex-, and smoking history-matched non-COPD subjects randomly selected from Ruijin Physical Examination Center. These non-COPD subjects had normal pulmonary images, routine blood screening tests, and biochemical examination without any history of asthma or chronic bronchitis. Subject characteristics are summarized in Table 1. No patients were diagnosed with metabolic diseases or recent (8 weeks) respiratory tract infection or any exacerbations requiring oral steroid and nonsteroidal anti-inflammatory agents. All the subjects underwent clinical evaluation and pulmonary function test and provided written informed consent to participate in the study. The study was reviewed and approved by Ruijin Hospital Ethics Committee, Shanghai Jiao Tong University School of Medicine.
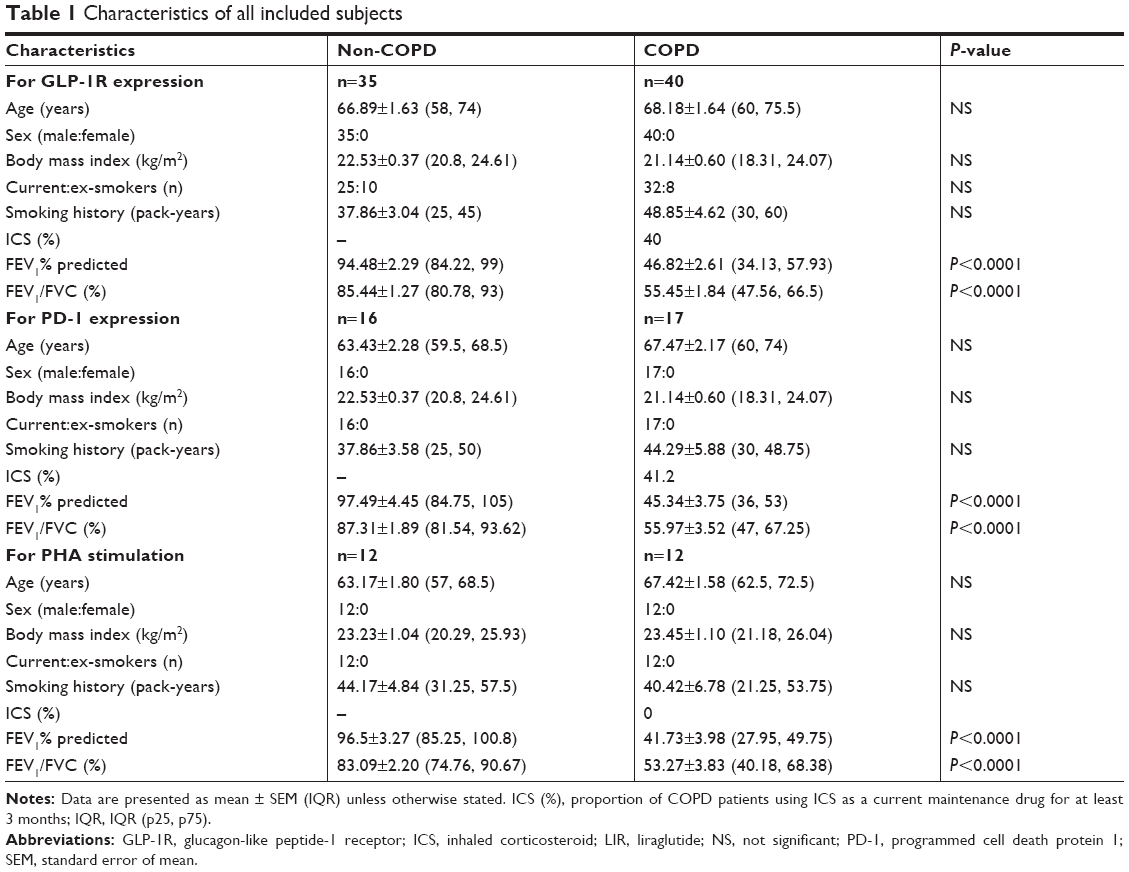 | Table 1 Characteristics of all included subjects |
Isolation of peripheral blood mononuclear cells (PBMCs)
Blood samples were collected in 5 mL EDTA evacuated test tubes (BD Vacutainer Systems, Plymouth, UK) from all subjects. After the plasma was isolated, PBMCs were separated from the peripheral blood using Lymphoprep™ (density gradient: 1.077±0.001 g/mL; Axis-Shield, Oslo, Norway) for 30 minutes at 800 g. The mononuclear layer was harvested and transferred to new tubes. PBS was added to the PBMCs, and the cells were washed twice by centrifugation for 10 minutes at 500 g. This study was conducted in two phases: 1) detection of GLP-1R expression was carried out in a cohort of COPD patients (n=40) and non-COPD (n=35) subjects and 2) fluorescence-activated cell sorter (FACS) analysis of PD-1 expression was evaluated in a different cohort of COPD patients (n=17) and non-COPD (n=16) subjects. LIR treatment was studied in a subset of patients who did not use inhaled corticosteroids (ICSs) as a current maintenance drug during the past 3 months and were current smokers in COPD (n=12) patients and non-COPD (n=12) subjects, depending on the availability of cells.
RNA extraction and real-time PCR
RNA extraction and cDNA synthesis from PBMCs were performed using TRIzol reagents (Thermo Fisher Scientific, Waltham, MA, USA) and RevertAid™ First Strand cDNA Synthesis Kit (Fermentas, Waltham, MA, USA), according to the manufacturer’s protocol. Total RNA (500 ng) was amplified with a 7500 Fast Real-Time PCR System (Thermo Fisher Scientific) and performed with FastStart Universal SYBR Green (Hoffman-La Roche Ltd., Basel, Switzerland) for real-time PCR after cDNA synthesis. The standard curve for quantification was created with a previously described modified procedure.20 The sequences of all primers used are as follows: hGLP-1R: sense 5′-ACCTGTCGGAGTGTGAAGAG-3′ and antisense 5′-GCGGAGAAAGAAGTGCGTA-3′; hIFN-γ: sense 5′-TCAACTTCTTTGGCTTAATTCTCTC-3′ and antisense 5′-ATATGGGTCCTGGCAGTAACA-3′.
Fold change of the gene expression was calculated by 2−ΔCt relative to the internal reference gene (glyceraldehyde-3-phosphate dehydrogenase, GAPDH).
Western blotting
Briefly, 106 PBMCs isolated from peripheral blood were lysed in 100 μL lysis buffer (P0013B; Beyotime, Suzhou, China) containing protease inhibitors. Equal amounts of proteins (30 μL) were subjected to a 10% SDS-PAGE and then transferred onto a polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA, USA). After blocking with Tris-buffered saline and Tween 20 containing 5% nonfat milk for 2 hours at room temperature, the membrane was incubated overnight at 4°C with mouse monoclonal anti-hGLP-1R antibody (1:500; sc-390774; Santa Cruz Biotechnology Inc., Dallas, TX, USA) or anti-rabbit GAPDH antibody (1:1,000; Cell Signaling, Danvers, MA, USA). The membrane was washed for 10 minutes with Tris-buffered saline and Tween 20 three times and then incubated with horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit IgG antibody (1:1,000; Cell Signaling) for 1 hour at room temperature. After the membrane was washed, chemiluminescent substrate (Thermo Fisher Scientific) was applied. The signal was detected and quantified with an enhanced chemiluminescence system (ImageQuant LAS-4000 MINI; GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA).
PBMCs: in vitro treatment
Purified PBMCs (1×106/mL) were resuspended in Roswell Park Memorial Institute (RPMI)-1640 complete medium containing 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin and then cultured with or without LIR (Victoza, Novo Nordisk, Denmark) at concentrations of 100 nM overnight (5% CO2 atmosphere at 37°C). After incubation, the cells were harvested for analysis of GLP-1R expression using quantitative real-time PCR (qRT-PCR) and Western blotting. The cells were also collected and assessed for PD-1 expression using flow cytometry. Mitogen stimulation of PBMCs was performed as described earlier.21 Triplicate cultures of 105 cells/well were stimulated with 10 μg/mL of phytohemagglutinin (PHA; Sigma-Aldrich Co., St Louis, MO, USA) in complete medium in 96-well, flat-bottomed microtiter plates (Corning Incorporated, Corning, NY, USA). Cells were either untreated or treated with LIR. After 3 days of stimulation, the culture supernatants were analyzed using an IFN-γ ELISA kit (R&D Systems, Inc., Minneapolis, MN, USA).
Cytokine ELISAs
The concentrations of IFN-γ in the plasma and the cell supernatant were detected using DuoSet ELISA kits (R&D Systems, Inc.) according to the manufacturer’s instructions.
Flow cytometry staining and analysis
Samples were immunostained with mAbs including CD3-FITC, CD4-PE, CD8-BV521, PD-1-BV421, and relevant isotype controls for 30 minutes at room temperature. All the antibodies were from BD Biosciences (San Jose, CA, USA). Cells were then resuspended in 1% paraformaldehyde (PFA) and stored at 4°C until flow cytometry could be conducted.
Statistical analyses
Results were presented as mean ± standard error of the mean if not stated otherwise. Analysis of two groups was performed using Wilcoxon’s signed-rank test for paired data and the Mann–Whitney test for unpaired data. The chi-squared test was used for categorical data. Spearman’s rank correlation coefficients were evaluated to access the relationships between GLP-1R mRNA levels, IFN-γ expression levels, and clinical lung function parameters. P-values of <0.05 were considered statistically significant. Statistical analyses were performed as shown in Figures 1–5, using GraphPad Prism version 7.0 (GraphPad Software, Inc., La Jolla, CA, USA).
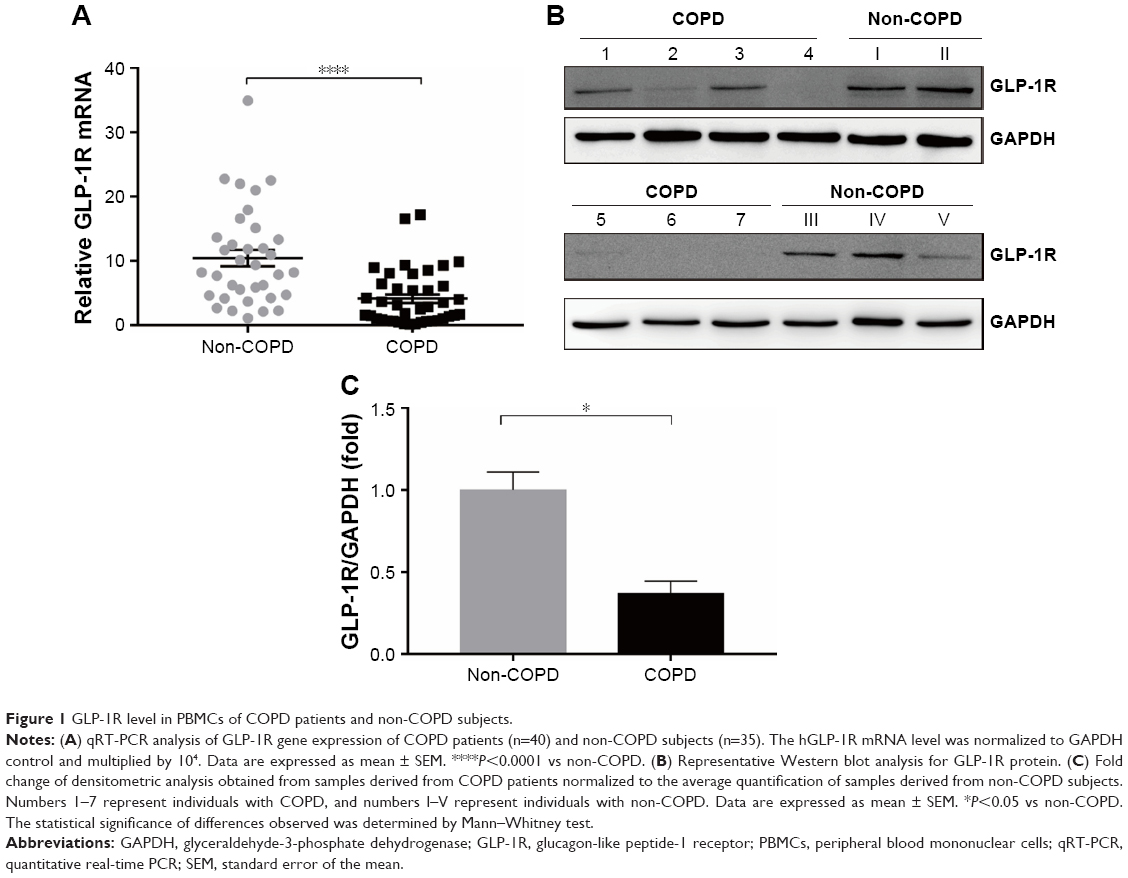 | Figure 1 GLP-1R level in PBMCs of COPD patients and non-COPD subjects. |
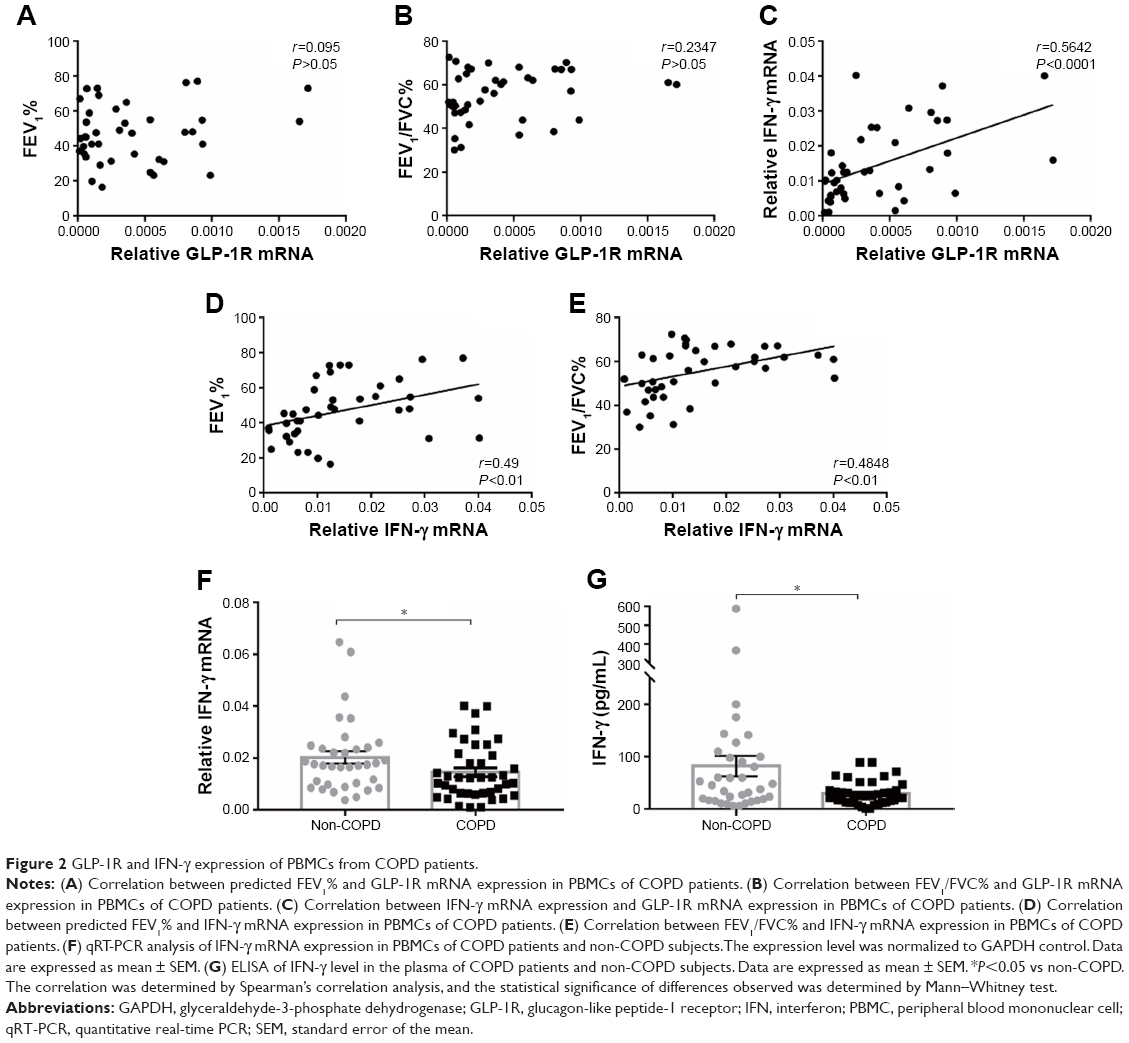 | Figure 2 GLP-1R and IFN-γ expression of PBMCs from COPD patients. |
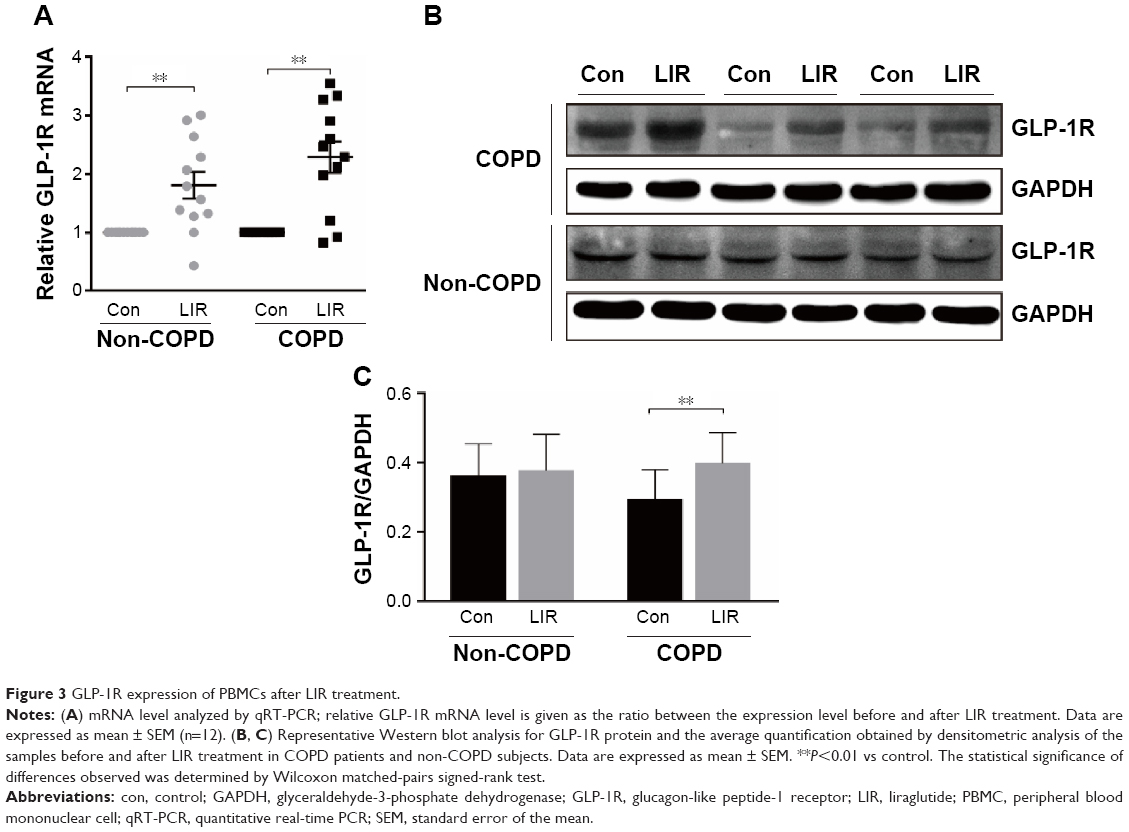 | Figure 3 GLP-1R expression of PBMCs after LIR treatment. |
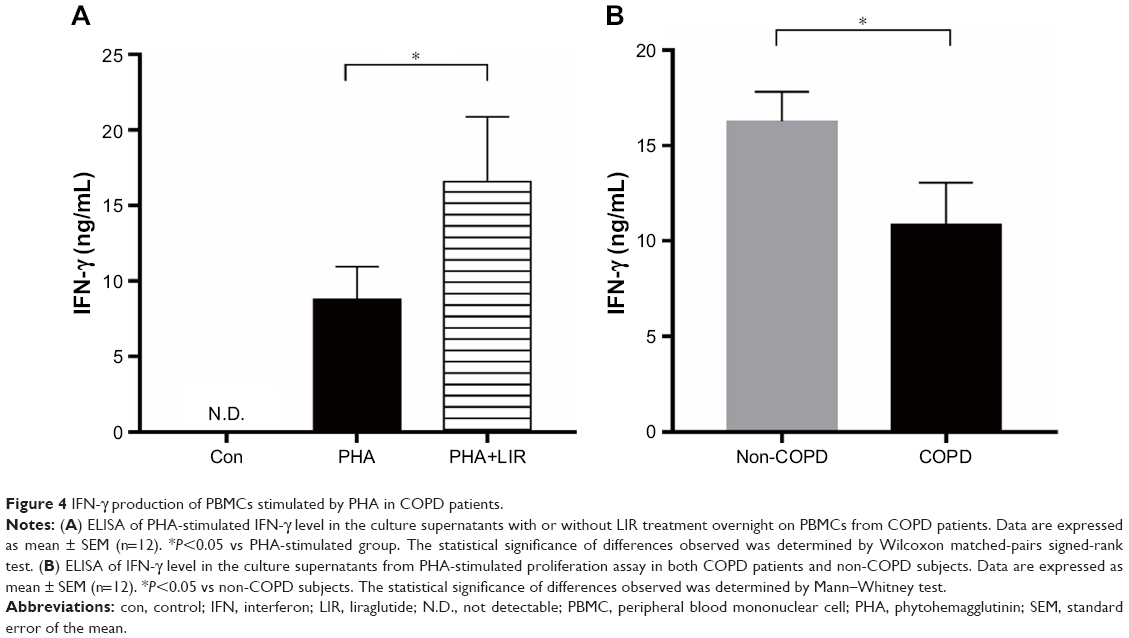 | Figure 4 IFN-γ production of PBMCs stimulated by PHA in COPD patients. |
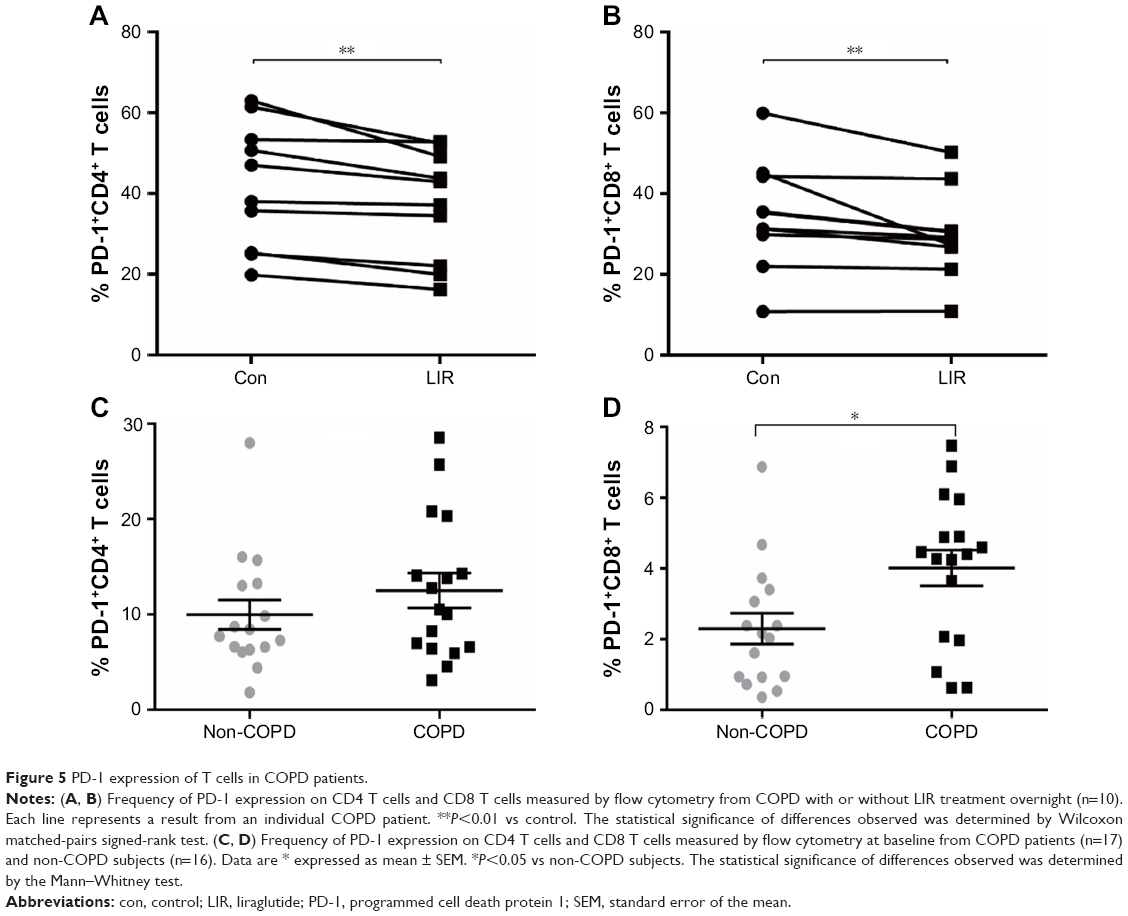 | Figure 5 PD-1 expression of T cells in COPD patients. |
Results
GLP-1R level in PBMCs in COPD patients and non-COPD subjects
We profiled the expression of GLP-1R in PBMCs at both the mRNA and protein levels. The expression level of GLP-1R in PBMCs was significantly lower in COPD patients than non-COPD subjects (P<0.0001; Figure 1). To ensure that the difference was not mixed by the effect of ICS, we also compared the expression level of GLP-1R in PBMCs of COPD patients based on ICS use and found no significant intergroup differences (Figure S1A).
Correlation between IFN production and GLP-1R expression
We found that relative GLP-1R mRNA level of PBMCs had no correlation with lung function parameters of COPD patients, including FEV1/FVC% and predicted FEV1% (Figure 2A and B). However, a positive correlation existed between GLP-1R mRNA level and IFN-γ mRNA level in COPD patients (r=0.5642, P<0.001; Figure 2C). Meanwhile, both FEV1/FVC% (r=0.49, P<0.01) and predicted FEV1% (r=0.4848, P<0.01) had moderate correlation with IFN-γ mRNA levels (Figure 2D and E). Furthermore, IFN-γ production decreased in COPD patients when compared with non-COPD subjects (Figure 2F and G). These results indicated that GLP-1R expression was associated with the functionality of lymphocytes.
Functionality of T cells after LIR treatment
Previous studies reported that although LIR had therapeutic potential in COPD by decreasing the severity of acute exacerbations, it had had no modulation on pulmonary inflammation and surfactant production.16 To ensure that GLP-1R signaling plays a critical role in enhancing effector T cell functionality of COPD patients, cytokine production of antigen-stimulated effector T cells was examined in the presence or absence of LIR. We first treated the COPD patients’ PBMCs with LIR; GLP-1R expression in PBMCs was first examined to ensure a potential direct effect, as GLP-1R expression was proven to be upregulated after GLP-1R agonist or dipeptidyl peptidase 4 (DPP-4) inhibitor treatment in mice.12,22 As shown in Figure 3, LIR administration promoted a 2.3- and 1.8-fold increase in the GLP-1R mRNA level of both COPD patients and non-COPD subjects as well as a significant increase in GLP-1R protein level of COPD patients. Meanwhile, LIR treatment resulted in an enhancement of IFN-γ production of PBMCs from COPD patients (Figure 4A), suggesting that the activation of GLP-1R signaling could contribute to enhanced antibacterial immunity in COPD patients. The reduced secretion of IFN-γ by PBMCs from COPD patients was also confirmed by analyzing its production after PHA stimulation (Figure 4B).
Downregulated PD-1 expression in COPD patients after LIR treatment
To further assess whether GLP-1R signaling has potential effects on ameliorating immunosuppression, we treated COPD patients’ PBMCs with LIR and found that the addition of LIR downregulated PD-1 expression in both CD4 and CD8 T cells of COPD patients (Figures 5A and B and S2). Meanwhile, the proportion of both PD-1+ CD4+ T cells (12.49±1.82 vs 9.97±1.55) and PD-1+ CD8+ T cells (4.01±0.51 vs 2.29±0.44) was greater in COPD patients than non-COPD subjects, although the difference in CD4+ T cell percentage did not reach statistical significance (P=0.40; Figure 5C and D). No significant difference of PD-1 expression was found on lymphocytes from COPD patients based on ICS use (Figure S1B and C), which was consistent with the results conducted by the McKendry’s group.23
Discussion
Our study revealed that GLP-1R expression decreased in circulating PBMCs from COPD patients, which was associated with IFN-γ expression. The treatment with LIR could restore the defective IFN-γ production with PHA stimulation and downregulated the PD-1 expression in COPD T cells.
Prior study has documented the association between inflammation and GLP-1R signaling. Islets were less frequently found to be GLP-1R positive or expressed receptors in low density in chronic pancreatitis than in the normal pancreas.24 COPD is widely recognized as a chronic inflammatory disease.3–5,25,26 The principal pathogenesis of COPD airways is characterized by the presence of an inflammatory cellular infiltrate and remodeling of the airway wall, thereby resulting in the airway obstruction.27,28 As per the current concepts, COPD is increasingly being recognized as a whole systemic inflammatory disorder rather than an airway-located disease. The presence of airway and systemic inflammation in COPD has been suggested by multiple studies showing increased oxidative stress and circulating levels of inflammatory mediators.29,30 Sun et al31 proved that GLP-1R expression levels were decreased in airway smooth muscle (ASM) cells from COPD patients compared with those from healthy subjects and upregulation of GLP-1R inhibited cell proliferation, migration, and inflammatory cytokine expression in ASM cells. In our study, we found that the expression level of GLP-1R was lower in PBMCs of COPD patients than non-COPD subjects, indicating that the impairment of GLP-1R signaling also occurred in circulating PBMCs of COPD patients.
GLP-1 and its analogs have been verified to play an obvious anti-inflammatory role in various diseases, including type 2 diabetes, asthma, and atherogenesis.13,14,32 However, the therapeutic potential of the specific GLP-1R agonist – LIR – in COPD was only confirmed in a mice model of obstructive pulmonary disease induced by ovalbumin (OVA) and lipopolysaccharide (LPS).16 We found that there was a positive correlation between IFN-γ production and GLP-1R expression. Meanwhile, PD-1-expressing lymphocyte populations were deceased and IFN-γ production was increased after LIR treatment, which suggests that restoring GLP-1R signaling might have regulatory effects on innate and adaptive immunity of COPD. Our finding is reminiscent of the GLP-1R signaling modulation in controlling innate immune responses and host microbial responses, as observed by Yusta et al33 in a mouse model.
Emerging data suggest the immune deficiency of T cells in COPD.9,34,35 Sustaining signaling in COPD can lead to progressive loss of T cell function. T cells from diseased lungs express the key receptor PD-1 and demonstrate loss of cytotoxicity.23,36 PBMCs from COPD patients show impaired responses to antigen, such as lipoprotein P6 and PHA stimulation.9,35,37 COPD patients with more severe stage have lower proportions of IFN-γ CD8+ T cells than those with mild stage.38 IFN-γ and its specific receptors have long been recognized as a critical component of innate immune defense to microorganisms and viruses.5,6,39,40 In keeping with these findings, we also found that PBMCs from COPD patients expressed lower IFN-γ mRNA than non-COPD subjects and showed greater occurrence of reduced IFN-γ production with the progression of COPD severity. A greater proportion of PD-1-expressing lymphocytes was seen in COPD patients, and impaired response of T cells toward PHA stimulation was found.
GLP-1R expression was reported to be downregulated in response to dexamethasone in islets,22 and COPD patients in stable condition often use ICS combined with long-acting β2 agonists (LABAs) as the maintenance treatment. To rule out the possibility of ICS-induced differences in GLP-1R expression between COPD patients and non-COPD subjects, we evaluated the GLP-1R expression in COPD patients based on ICS use and found that there were no significant intergroup differences. We acknowledge that the observational nature of our study and the relatively small cohort sizes are significant limitations of our study. Another limitation is the heterogeneity of GOLD stages of the COPD patients. However, all the patients included in the study were in stable state and proportions of lymphocyte subpopulations did not significantly differ. Our data provided a plausible theory that restoration of GLP-1R signaling could ameliorate functional defect of T cells in COPD patients. As metabolic syndrome and diabetes are common comorbidities of COPD41 and GLP-1 analogs have been clinically used in obesity and diabetes, more preclinical and clinical studies are required to be carried out to clarify the regulatory mechanism of GLP-1R signaling in COPD.
Conclusion
We found that PBMCs from COPD patients underwent GLP-1R signaling defect, which was associated with decreased IFN-γ production. Treatment with LIR significantly restored the IFN-γ production of PBMCs and downregulated the PD-1-expressing T cell proportions in these patients. These findings indicated that the restoration of GLP-1R signaling might ameliorate T cell dysfunction in COPD patients. It remains to be seen if GLP-1R signaling and GLP-1 analogs are likely to serve as possible immunomodulatory targets for COPD therapy.
Acknowledgments
We are grateful to Dr Xianwen Sun and Qiurui Zhang for their assistance with the recruitment of patients. We also thank all the members of flow cytometry platform, Shanghai Jiao Tong University School of Medicine for their generous technical assistance in the flow cytometric characterization of PBMCs. This study was funded by grants from National Natural Science Foundation of China (award number: 81570029) and Ministry of Science and Technology of the People’s Republic of China (No. 2017YFC1309700) and Shanghai Key Discipline for Respiratory Diseases (2017ZZ02014).
Author contributions
Yifan Zhang and Liying Zhu gave the initial concept. Jingwen Huang, Chunliu Zhao, and Bing Liu collected the samples of COPD patients. Jingwen Huang and Huahua Yi performed the experiments. Jingwen Huang and Min Zhou drafted the work and revised it. Min Zhou and Ping He provided guarantee for the data presented in this manuscript. All the authors read and approved the final manuscript. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Diaz-Guzman E, Mannino DM. Epidemiology and prevalence of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):7–16. | ||
Chapman KR, Mannino DM, Soriano JB, et al. Epidemiology and costs of chronic obstructive pulmonary disease. Eur Respir J. 2006;27(1):188–207. | ||
Hilleman DE, Dewan N, Malesker M, Friedman M. Pharmacoeconomic evaluation of COPD. Chest. 2000;118(5):1278–1285. | ||
Alonso JM. Immunity and pathophysiology of respiratory tract infections. Med Mal Infect. 2008;38(8):433–437. | ||
Reynolds HY. Modulating airway defenses against microbes. Curr Opin Pulm Med. 2002;8(3):154–165. | ||
Reid PT, Sallenave JM. Cytokines in the pathogenesis of chronic obstructive pulmonary disease. Curr Pharm Des. 2003;9(1):25–38. | ||
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | ||
Prieto A, Reyes E, Bernstein ED, et al. Defective natural killer and phagocytic activities in chronic obstructive pulmonary disease are restored by glycophosphopeptical (inmunoferón). Am J Respir Crit Care Med. 2001;163(7):1578–1583. | ||
Kalathil SG, Lugade AA, Pradhan V, et al. T-regulatory cells and programmed death 1+ T cells contribute to effector T-cell dysfunction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;190(1):40–50. | ||
Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet. 1987;2(8571):1300–1304. | ||
Bullock BP, Heller RS, Habener JF. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology. 1996;137(7):2968–2978. | ||
Romaní-Pérez M, Outeiriño-Iglesias V, Moya CM, et al. Activation of the GLP-1 Receptor by Liraglutide Increases ACE2 Expression, Reversing Right Ventricle Hypertrophy, and Improving the Production of SP-A and SP-B in the Lungs of Type 1 Diabetes Rats. Endocrinology. 2015;156(10):3559–3569. | ||
Lee YS, Park MS, Choung JS, et al. Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia. 2012;55(9):2456–2468. | ||
Arakawa M, Mita T, Azuma K, et al. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes. 2010;59(4):1030–1037. | ||
Viby NE, Isidor MS, Buggeskov KB, et al. Glucagon-like peptide-1 (GLP-1) reduces mortality and improves lung function in a model of experimental obstructive lung disease in female mice. Endocrinology. 2013;154(12):4503–4511. | ||
Gou S, Zhu T, Wang W, et al. Glucagon like peptide-1 attenuates bleomycin-induced pulmonary fibrosis, involving the inactivation of NF-κB in mice. Int Immunopharmacol. 2014;22(2):498–504. | ||
Damera G, Tliba O, Panettieri RA. Airway smooth muscle as an immunomodulatory cell. Pulm Pharmacol Ther. 2009;22(5):353–359. | ||
Al Heialy S, Risse PA, Zeroual MA, et al. T cell-induced airway smooth muscle cell proliferation via the epidermal growth factor receptor. Am J Respir Cell Mol Biol. 2013;49(4):563–570. | ||
Marx N, Burgmaier M, Heinz P, et al. Glucagon-like peptide-1(1-37) inhibits chemokine-induced migration of human CD4-positive lymphocytes. Cell Mol Life Sci. 2010;67(20):3549–3555. | ||
Zhang Y, Lou XL, Yang HL, et al. Establishment of a leptospirosis model in guinea pigs using an epicutaneous inoculations route. BMC Infect Dis. 2012;12:20. | ||
Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y. Higher frequencies of GARP(+)CTLA-4(+)Foxp3(+) T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res. 2013;73(8):2435–2444. | ||
Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132(6):2131–2157. | ||
McKendry RT, Spalluto CM, Burke H, et al. Dysregulation of Antiviral Function of CD8(+) T Cells in the Chronic Obstructive Pulmonary Disease Lung. Role of the PD-1-PD-L1 Axis. Am J Respir Crit Care Med. 2016;193(6):642–651. | ||
Körner M, Stöckli M, Waser B, Reubi JC. GLP-1 receptor expression in human tumors and human normal tissues: potential for in vivo targeting. J Nucl Med. 2007;48(5):736–743. | ||
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8(3):183–192. | ||
Costa C, Traves SL, Tudhope SJ, et al. Enhanced monocyte migration to CXCR3 and CCR5 chemokines in COPD. Eur Respir J. 2016;47(4):1093–1102. | ||
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–2454. | ||
Agustí AG, Noguera A, Sauleda J, Sala E, Pons J, Busquets X. Systemic effects of chronic obstructive pulmonary disease. Eur Respir J. 2003;21(2):347–360. | ||
Vernooy JH, Küçükaycan M, Jacobs JA, et al. Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. Am J Respir Crit Care Med. 2002;166(9):1218–1224. | ||
Sun YH, He L, Yan MY, et al. Overexpression of GLP-1 receptors suppresses proliferation and cytokine release by airway smooth muscle cells of patients with chronic obstructive pulmonary disease via activation of ABCA1. Mol Med Rep. 2017;16(1):929–936. | ||
Zhu T, Wu XL, Zhang W, Xiao M. Glucagon Like Peptide-1 (GLP-1) Modulates OVA-Induced Airway Inflammation and Mucus Secretion Involving a Protein Kinase A (PKA)-Dependent Nuclear Factor-κB (NF-κB) Signaling Pathway in Mice. Int J Mol Sci. 2015;16(9):20195–20211. | ||
Yusta B, Baggio LL, Koehler J, et al. GLP-1R Agonists Modulate Enteric Immune Responses Through the Intestinal Intraepithelial Lymphocyte GLP-1R. Diabetes. 2015;64(7):2537–2549. | ||
Kim WD, Kim WS, Koh Y, et al. Abnormal peripheral blood T-lymphocyte subsets in a subgroup of patients with COPD. Chest. 2002;122(2):437–444. | ||
Abe Y, Murphy TF, Sethi S, et al. Lymphocyte proliferative response to P6 of Haemophilus influenzae is associated with relative protection from exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;165(7):967–971. | ||
Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–687. | ||
Reyes E, Prieto A, de La Hera A, et al. Treatment with AM3 restores defective T-cell function in COPD patients. Chest. 2006;129(3):527–535. | ||
Paats MS, Bergen IM, Hoogsteden HC, van der Eerden MM, Hendriks RW. Systemic CD4+ and CD8+ T-cell cytokine profiles correlate with GOLD stage in stable COPD. Eur Respir J. 2012;40(2):330–337. | ||
Nalos M, Santner-Nanan B, Parnell G, Tang B, McLean AS, Nanan R. Immune effects of interferon gamma in persistent staphylococcal sepsis. Am J Respir Crit Care Med. 2012;185(1):110–112. | ||
Jarvis JN, Meintjes G, Rebe K, et al. Adjunctive interferon-γ immunotherapy for the treatment of HIV-associated cryptococcal meningitis: a randomized controlled trial. AIDS. 2012;26(9):1105–1113. | ||
Cavaillès A, Brinchault-Rabin G, Dixmier A, et al. Comorbidities of COPD. Eur Respir Rev. 2013;22(130):454–475. |
Supplementary materials
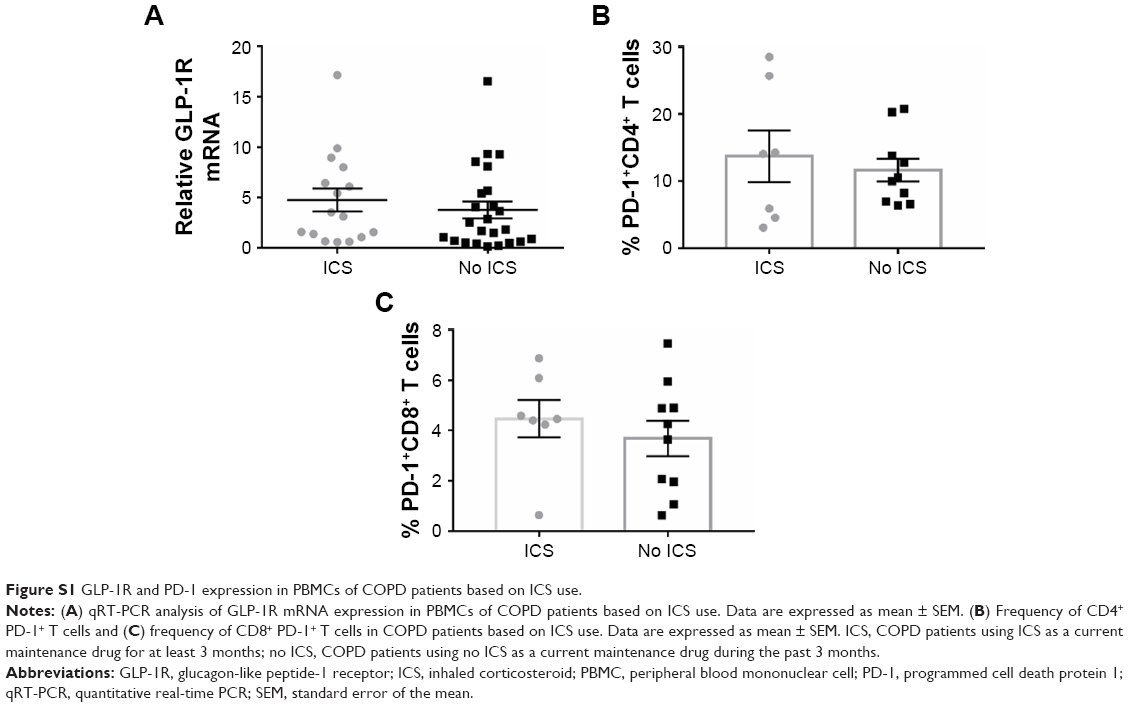 | Figure S1 GLP-1R and PD-1 expression in PBMCs of COPD patients based on ICS use. |
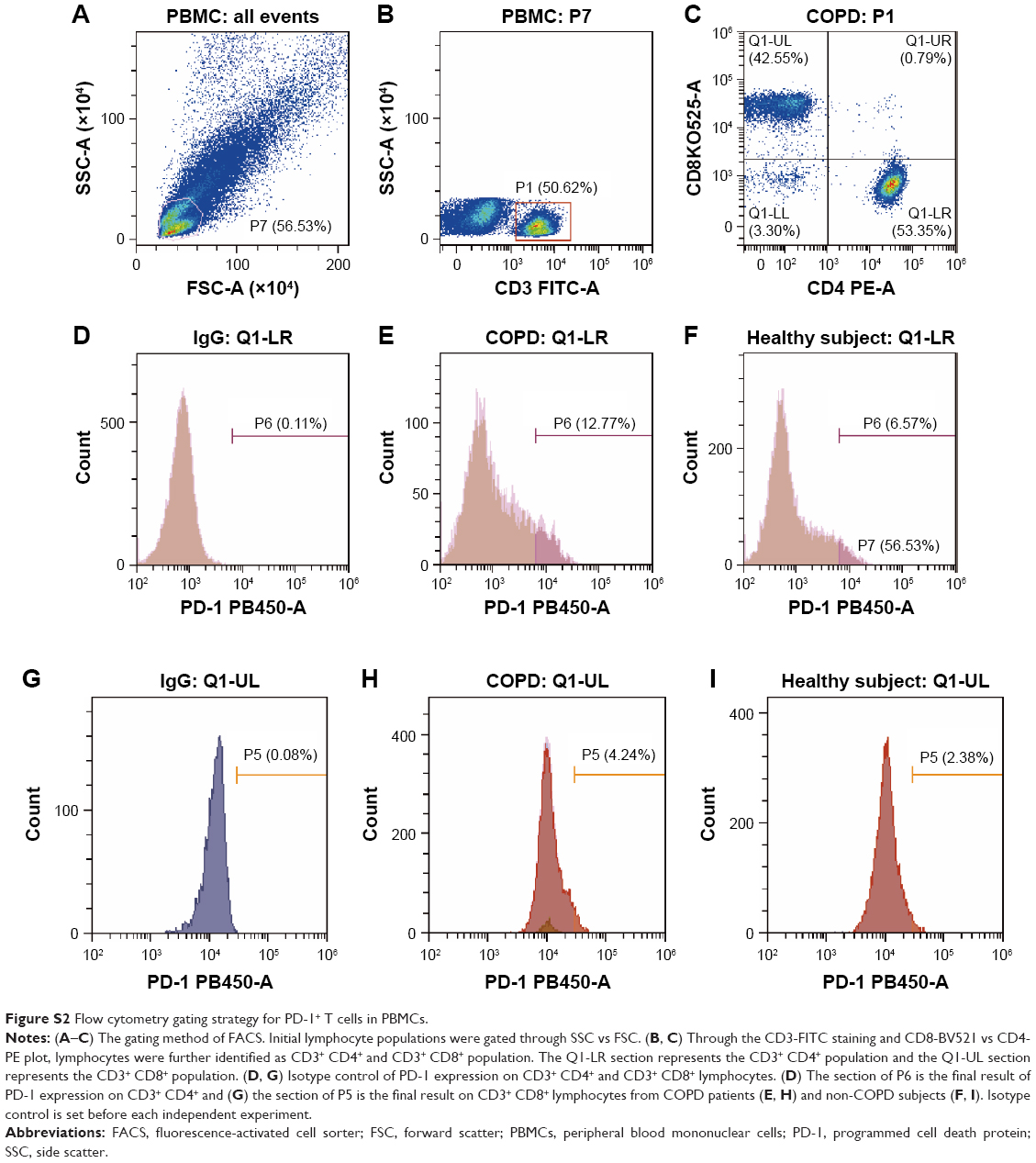 | Figure S2 Flow cytometry gating strategy for PD-1+ T cells in PBMCs. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.