Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Glia maturation factor-β: a potential therapeutic target in neurodegeneration and neuroinflammation
Authors Fan J ![]() , Fong T
, Fong T ![]() , Chen X, Chen C
, Chen X, Chen C ![]() , Luo P
, Luo P ![]() , Xie H
, Xie H
Received 15 November 2017
Accepted for publication 14 December 2017
Published 9 February 2018 Volume 2018:14 Pages 495—504
DOI https://doi.org/10.2147/NDT.S157099
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Wai Kwong Tang
Junsheng Fan,1,2,* Tszhei Fong,1,* Xinjie Chen,3,* Chuyun Chen,1 Peng Luo,1 Haiting Xie1
1Zhujiang Hospital of Southern Medical University, Guangzhou, China; 2Shanghai Tenth People’s Hospital, Tongji University, Shanghai, China; 3Second School of Clinic Medicine, Guangzhou University of Chinese Medicine, Guangzhou, China
*These authors contributed equally to this work
Abstract: Glia maturation factor-β (GMFB) is considered to be a growth and differentiation factor for both glia and neurons. GMFB has been found to be upregulated in several neuroinflammation and neurodegeneration conditions. It may function by mediating apoptosis and by modulating the expression of superoxide dismutase, granulocyte-macrophage colony-stimulating factor, and neurotrophin. In this review, we mainly discussed the role of GMFB in several neuroinflammatory and neurodegenerative diseases. On review of the literature, we propose that GMFB may be a promising therapeutic target for neuroinflammatory and neurodegenerative diseases.
Keywords: glia maturation factor-β, neurodegeneration, neuroinflammation
Introduction
Glia maturation factor (GMF), as a growth and differentiation factor, was first isolated from bovine brain in 1972.1 The protein is actually a mixture of two compounds: glia maturation factor-β (GMFB) and GMF-γ. GMFB was further purified from crude GMF.2
GMFB is expressed predominantly in the central nervous system (CNS),3 while its expression has also been detected in various other tissues including colon, thymus, and kidney.4,5 It is a highly evolutionary conserved protein and is expressed in a wide range of vertebrates;6 however, the whole-body knockout (KO) of GMF using homologous recombination appears to be nonlethal to C57BL/6 mice in some studies.7–9
Neurodegenerative diseases, which include Alzheimer’s disease (AD) and Parkinson’s disease (PD), are a group of disorders characterized by the loss of neurons.10 During neurodegeneration, astrogliosis is often observed.11 On one hand, reactive astrocytes can demarcate lesions and play a protective role by secreting neurotrophic factors.12 On the other hand, reactive astrocytes are implicated in the upregulation of various cytokines, chemokines, as well as reactive oxygen species (ROS) and reactive nitrogen species (RNS),11 which may lead to oxidative stress.
Neuroinflammation is the inflammatory response in the nervous system. Uncontrolled neuroinflammation would lead to the overproduction of neurotoxic factors including pro-inflammatory cytokines/chemokines and ROS/RNS, which would impair the normal structure and function of neurons. Chronic microglia activation is a common feature of all neuroinflammatory responses. Activated microglia, characterized by the release of various inflammatory mediators and phagocytosis, could have both protective and deleterious effects on the nervous system.13
Neuroinflammation is often identified in many neurodegenerative diseases.13 Neurodegenerative diseases are often driven by the innate immune system while neuroinflammatory diseases are more commonly driven by the adaptive immune system.14 The close relationship between neuroinflammation and neurodegeneration cannot be overlooked.
In this review, we mainly focus on the structure, function, and possible signaling pathways of GMFB in several neurodegenerative diseases, and touch upon the progress of research on GMFB beyond CNS. As a result, we propose that GMFB may be a potential therapeutic target for neurodegenerative and neuroinflammatory diseases.
Gene structure and function of GMFB
Human GMFB is located on the long arm of human chromosome 14.15 With six introns and seven exons, it is 7 kb in length.16 The gene encodes an acidic protein with an isoelectric point of pH 4.9.2 The purified GMFB protein has been identified to have 142 amino acid residues and it has a molecular weight of ~17 kDa.2
Lacking a signal peptide sequence, GMFB is primarily an intracellular protein which is implicated in various signaling pathways.17 Considering that some proteins can also be secreted even in the absence of a signal sequence,18 GMFB may also be a secretory protein. In fact, GMFB can be secreted under certain conditions.19,20 Also, it has been reported that GMFB is expressed on the cell surface of some cells including astrocytes and thymus epithelial cell line.21,22 An in vitro study has demonstrated that the introduction of GMFB to the culture media can elicit some signaling and metabolic alterations in glioblastoma cells.23 These evidence indicate that GMFB may act as a signaling molecule both intracellularly and extracellularly and can influence the signaling transduction as well as cell communication via autocrine or paracrine fashion.
GMFB is predominantly expressed in the CNS although the relative expression quantity in different parts of the CNS remains controversial.3,5,6,24 This may result from the difference of experimental specimens and methods. The expression of GMFB has also been reported in various other tissues including thymus, testis, ovary, colon, and renal proximal tubule.3,5,6 The major cells that express GMFB in the nervous system are glial cells, including astrocytes, Bergmann glia in the cerebellum, Schwann cells in the peripheral nervous system (PNS), as well as Müller cells in the retina.6,25,26 However, its expression in some neurons has also been detected.24 Likewise, GMFB is highly expressed in some cell lines which have a glia or neuron origin, such as glioma, schwannoma, and neuroblastoma cell lines.3
GMFB expression pattern is development dependent. In rat brain, its mRNA was detectable as early as embryonic day 10 and there is no significant change throughout the rats’ life.3 However, the protein was first detected at embryonic day 14 (E14), and its expression level increases steadily and reached its peak at 1 week after birth. Also, the expression of the protein did not change significantly in the rats’ adult life.3 As mentioned earlier, GMFB can be secreted although it has no signal sequence. An immunoassay has been carried out and confirmed that the serum concentration of GMFB is relatively stable throughout the life of both humans and rats with an unknown secretion mechanism.5 Furthermore, its expression can also be upregulated during some pathological conditions.16,27,28
Additionally, as a member of the actin-depolymerizing factor/cofilin family, GMFB can interact with Arp2/3 complex. Therefore, it is implicated in the regulation of actin debranching, lamellipodial dynamics, and consequently the migration of cells.29,30 Although the whole-body KO of GMF appeared to be nonlethal to C57BL/6 mice, the absence of GMFB may lead to compromised motor learning and performance comparing with their wild-type (WT) counterparts.8 These evidence indicate that GMFB plays an important, although not indispensable, role in the development of CNS.
GMFB-related signaling pathway: friend or foe?
In an in vitro assay, GMFB can be phosphorylated by protein kinase A (PKA), protein kinase C, casein kinase II, and p90 ribosomal S6 kinase.17 PKA-phosphorylated GMFB is an enhancer of p38 mitogen-activated protein kinase (MAPK) and an inhibitor of extracellular signal-regulated kinase 1 and 2 (ERK1/ERK2).31,32 The overexpression of GMFB using an adenovirus vector carrying GMFB cDNA in rat primary astrocytes led to a slight increase in the activity of ERK MAPK.33 This distinct outcome may be explained by the fact that GMFB may affect the activity of ERK MAPK through multiple pathways. It is also shown that the overexpression of GMFB using an adenovirus vector carrying GMFB cDNA in mouse primary astrocytes led to the upregulation of nuclear factor-κB (NF-κB) and a significant increase of granulocyte-macrophage colony-stimulating factor (GM-CSF) secretion (the concentration of GM-CSF in the culture media reached a level of 78, 150, and 320 pg/mL at 12, 24, and 48 hours after GMFB transfection; in comparison, the concentration of GM-CSF remained at a low level [5–8 pg/mL] in the mock or control lacZ infection groups at all incubation times). These effects can be blocked by p38 MAPK inhibitor and NF-κB inhibitor.34 These evidence indicate that GMFB can induce the expression and secretion of GM-CSF through the p38 MAPK pathway (Figure 1A).
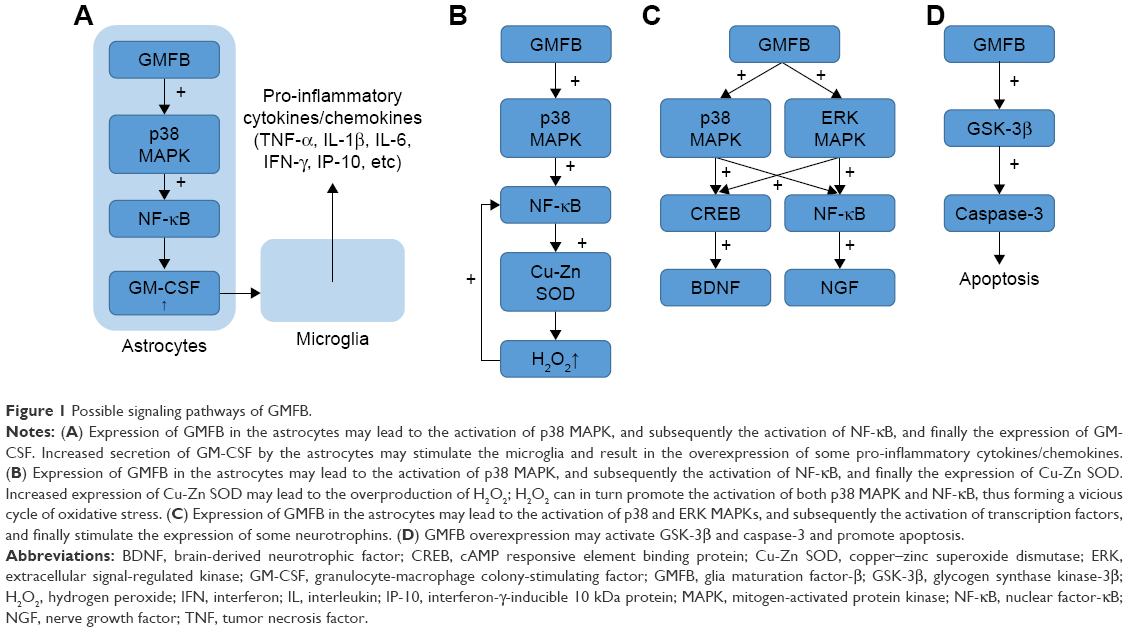 | Figure 1 Possible signaling pathways of GMFB. |
In the CNS, GM-CSF is a pro-inflammatory cytokine and a potent mitogen for microglia.35 Consequently, the increase in GM-CSF level may lead to the activation and proliferation of microglia. It has been reported that the expression and secretion of various pro-inflammatory cytokines/chemokines, including tumor necrosis factor (TNF)-α and interleukin (IL)-1β, are upregulated in activated microglia. These pro-inflammatory factors are cytotoxic to both neurons and oligodendrocytes.34 On the other hand, the presence of GM-CSF receptors on rat oligodendrocytes and the mitogenic effect of GM-CSF on oligodendrocytes indicate that GM-CSF might promote remyelination of neurons after demyelination caused by trauma or multiple sclerosis (MS).36 Taken together, the expression of GMFB and the subsequent secretion of GM-CSF may exacerbate neuroinflammation or promote glia regeneration in different conditions.
Another clue to GMFB signaling is oxidative stress. Lim et al have reported that overexpression of GMFB using an adenovirus vector carrying GMFB cDNA in C6 rat glioma cells led to the activation of NF-κB as well as an increase in copper–zinc superoxide dismutase (Cu-Zn SOD) expression. This effect can be blocked by p38 inhibitor and NF-κB nuclear translocation blocker.37
Cu-Zn SOD is a cytosolic enzyme which can convert superoxide radical (O2−) to oxygen and hydrogen peroxide (H2O2).38 H2O2 can be further reduced to water and oxygen by catalase or glutathione peroxidase (GPX).39 As part of the cellular antioxidant system, Cu-Zn SOD is initially protective since it can scavenge ROS in the presence of catalase and/or GPX. Nevertheless, an in vitro study carried out on renal proximal tubular cell line mProx24 and NIH 3T3 showed that the overexpression of GMFB using liposome transfection led to the downregulation of both catalase and GPX although Cu-Zn SOD was upregulated.4 This was corroborated by experiments carried out on primary astrocytes derived from GMFB-KO mice: both catalase and GPX were significantly upregulated while the activity of Cu-Zn SOD decreased significantly compared with WT controls.9 Based on these results, it seems that GMFB overexpression is responsible for the upregulation of Cu-Zn SOD, as well as the downregulation of catalase and GPX. Considering that biological membranes are permeable to H2O2 but not to superoxide, accumulated H2O2 could diffuse into the extracellular fluid, thus impairing other cells.40 Additionally, H2O2 can in turn promote the activation of both p38 MAPK and NF-κB,39 thus forming a vicious cycle under some pathological conditions. In summary, the GMFB-dependent upregulation of Cu-Zn SOD may account for the increased oxidative stress during some neuroinflammatory and neurodegenerative diseases (Figure 1B).
When it comes to oxidative stress, the role of RNS cannot be overlooked. Nitric oxide (NO) has been reported to be implicated in several neurodegenerative and neuroinflammatory diseases including MS and PD.41 During pathological conditions, NO is mainly synthesized by inducible nitric oxide synthase (iNOS) and secreted by reactive astrocytes as well as microglia.41 It was found that iNOS was downregulated in GMFB-KO mice compared with WT mice; the iNOS mRNA level of GMFB-KO brain was ~1/18 of that of its WT counterparts at the peak of experimental autoimmune/allergic encephalomyelitis (EAE) onset.42 A significant decrease in NO production was also observed in primary GMFB-KO neuron-glia cells after 20 μM MPTP treatment (the NO levels were 11.31±0.19 and 46.21±5.88 μmol/L in GMFB-KO and WT cells, respectively; no significant difference in NO production has been observed at the basal level between the KO and WT groups).43 These evidence indicate that GMFB is closely related to the production of NO during some pathological conditions. The precise mechanism of how GMFB induces the overexpression of iNOS, and subsequently the overproduction of NO, remains largely elusive. It has been shown that the upregulation of iNOS in endotoxin-stimulated glial cells was ERK and p38 dependent.41 Moreover, p38 MAPK may induce the transcription of iNOS possibly by activating NF-κB.44 Recently, cross talk between the p38 MAPK pathway and Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway has also been reported.45,46 The relationship between GMFB, MAPK, JAK-STAT, and NF-κB needs further investigation.
Another clue to GMFB signaling lies in neurotrophins (NTs), including nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF). An in vivo study has reported a 10-fold increase in BDNF and a 1.7-fold increase in NGF mRNA expression in the cerebellum of WT mice following exercise, while no increase in BDNF and NGF expression was observed in the cerebellum of GMFB-KO mice (homologous recombination).7 This finding was corroborated by in vitro studies carried out on C6 glioma cells and primary astrocytes in which overexpression of GMFB led to the upregulation of both BDNF and NGF.33,47,48 (For example, overexpression of GMFB [transient transfection] in rat C6 glioma cells led to an 8 to 10-fold increase in both NGF and BDNF mRNA expression.47) Moreover, Zaheer et al reported the activation of p38 MAPK, and to a less extent, ERK MAPK, as well as the activation of transcription factors cAMP responsive element binding protein (CREB) and NF-κB after adenovirus vector induced GMFB overexpression in primary astrocytes derived from embryonic rat brains. Moreover, it has been demonstrated that the upregulation of NGF is NF-κB dependent, while the upregulation of BDNF is CREB dependent33 (Figure 1C).
As members of the NT family, it has been reported that both BDNF and NGF have neuroprotective function while BDNF also has a neurotrophic effect.47 The neuroprotective effect of NGF may be partly due to its ability to upregulate catalase, which may counteract the detrimental effect of the upregulation of Cu-Zn SOD caused by GMFB overexpression.49 Nevertheless, as mentioned earlier, catalase was significantly downregulated in GMFB overexpressing models. There must be other ways in which GMFB can act either directly or indirectly on catalase.
Last but not least, GMFB is associated with apoptosis. The activation of glycogen synthase kinase-3β (GSK-3β) and caspase-3 in GMFB-overexpressed neuroblastoma cells (transient transfection with adenoviral vector) has been observed, while the upregulation of caspase-3 can be blocked by GSK-3β inhibitors.50 This indicates that GMFB may play a role in neurodegenerative diseases by mediating apoptosis (Figure 1D).
In addition, experiments carried out on PC-12 pheochromocytoma demonstrated that the activation of p38 and c-jun N-terminal kinase (JNK) MAPK as well as the inhibition of ERK MAPK contribute to apoptosis after NGF retraction.51 Concerning the interaction between GMFB and MAPK, GMFB may also regulate apoptosis via MAPK pathways. Although the precise mechanism remains to be elucidated, present research studies have paved the way for it.
GMFB in the nervous system
GMFB in the retina
As a special part of the CNS, the retina is considered to share many common features with the brain.52 Several types of glial cells in the brain including microglia and astrocytes are also found to reside in the retina.53 Another type of macroglia, the Müller cells, which spans the entire thickness of the neural retina and ensheathes all retinal neurons, is characteristic of the retina.54
Both GMFB mRNA and protein could be detected in rat retina as early as rat E14, while mRNA reached its highest level at postnatal day 14. The expression of GMFB is primarily localized to the Müller cells in the rat retina.25 The expression pattern of GMFB in the retina highlighted its role in the development and differentiation of Müller cells in the retina. Moreover, it has been reported that GMFB causes the upregulation of BDNF in the Müller cells, thus indicating that it may have a neuroprotective function in the retina.25 However, the role of GMFB in retinal neurodegeneration is hardly investigated.
Age-related macular degeneration (AMD), characterized by drusens in the macula lutea and the progressive loss of central vision, can be considered as a neurodegenerative disease.55 Several molecular factors such as the overproduction of pro-inflammatory cytokines/chemokines and the activation of the complement system, along with some cellular factors including chronic astrogliosis, Müller cell gliosis and the activation of microglia, and oxidative stress are thought to contribute to the development of AMD.53,56,57
Recent work showed that compared with adjacent non-lesioned areas, IL-33 immunoreactivity was higher in Müller cells of AMD lesioned areas in the human retina.57 IL-33 level in the vitreous from AMD patients was also higher than age-matched controls. It is believed that the release of IL-33 is increased under stressed retinas, and bioactive IL-33 can in turn elicit the upregulation of various pro-inflammatory cytokines/chemokines in the Müller cells.57 In the CNS, it has been reported that GMFB can enhance the expression of IL-33 in mouse astrocytes, and IL-33 can in turn augment the release of TNF-α as well as other pro-inflammatory cytokines/chemokines from astrocytes. Moreover, IL-33 together with other pro-inflammatory mediators might in turn induce the expression of GMFB in the CNS.10 GMFB and IL-33 constitute a vicious cycle. Considering that astrocytes and Müller cells are both macroglial cells in the retina, Müller cells can be perceived as radial astroglia.52,53 We propose that IL-33 might be a promising link between GMFB and neuroinflammatory and neurodegenerative diseases in the retina. It seems reasonable to postulate that GMFB may be implicated in the pathogenesis of neuroinflammation and neurodegeneration in the retina in a similar fashion to that in the brain. Notwithstanding, these hypotheses need to be corroborated by further investigation.
GMFB in PNS
In PNS, the glial cells which myelinate the axons are Schwann cells. Actually, there are two types of Schwann cells: the myelin-forming Schwann cell, which is similar to oligodendrocytes in the CNS, and non-myelin-forming Schwann cell, which resembles the astrocytes in the CNS.26
The expression of GMFB has been detected in cultured rat Schwann cells.26 GMFB immunoreactivity is upregulated in the Schwann cells after axotomy of rat sciatic nerve. However, GMFB was not detected in Schwann cells in an intact nerve and it seems that the expression of GMFB was suppressed by the regeneration of axons.58 These evidence suggest that the expression of GMFB may be induced by the loss of cell contact between Schwann cells and axons in the PNS.
Axotomy would lead to the degeneration of axons, and this degeneration can occur in nerve segments distal to the severed site. This is called Wallerian degeneration (WD). Axotomy-induced WD can be considered as a neurodegenerative disorder while Wallerian-like degeneration was also observed in other neurodegenerative diseases including amyotrophic lateral sclerosis. It is believed that WD is an essential procedure before axon regeneration.59
Considering that JNK MAPK is involved in the signal transduction of axotomy-induced WD,60 GMFB may probably play a role in WD via the MAPK pathways.
GMFB in CNS
AD
AD is one of the major causes of dementia that mainly affects the elderly. As a typical neurodegenerative disease, AD is pathologically characterized by amyloid plaques (APs), which are mainly formed by aggregated amyloid-beta peptide (Aβ), and neurofibrillary tangles (NFTs), which are composed of hyperphosphorylated tau.61 Evidence collected from human brain samples has shown a significant upregulation of GMFB in AD-affected brain, especially in the hippocampus and entorhinal cortex.19,62,63 The expression of GMFB was co-localized with APs and NFTs in the AD-affected brain. Furthermore, GMFB expression was detected in both the vicinity and interior of APs.16,19,62,63 Co-localization of GMFB-immunoreactive astrocytes with glial fibrillary acidic protein-labeled reactive astrocytes as well as ionized calcium binding adaptor molecule 1-labeled activated microglia has also been reported.19 In AD mouse models induced either by triple transgene or Aβ infusion, a significant increase in GMFB expression as well as pro-inflammatory cytokines/chemokines such as TNF-α, IL-1β, interferon-γ, and interferon-γ-inducible 10 kDa protein has been observed.64,65 However, compared with WT mice, the expression of pro-inflammatory cytokines/chemokines such as IL-1β and TNF-α was much lower in the hippocampal extracts of GMFB-KO mice (homologous recombination) after Aβ injection. Likewise, ameliorated neuronal loss and suppressed astrocyte/microglia activation were reported in GMFB-KO mice. Behavioral experiments also demonstrated better cognitive performance in GMFB-KO mice after Aβ treatment.65 Collectively, these evidence support the hypothesis that GMFB is involved in the pathogenesis of AD, though the exact mechanism remains elusive. It has been reported that overexpression of GMFB using an adenovirus vector carrying GMFB cDNA in neuroblastoma cells led to significant activation of GSK-3β.50 Intriguingly, the activation of GSK-3β can not only upregulate the expression of caspase-3 but also lead to the hyperphosphorylation of Tau as well.50 This suggests that GMFB may probably mediate the neuronal loss and the formation of NFTs in AD. Considering that GM-CSF was detected in reactive astrocytes in AD lesions, both serum and cerebrospinal fluid levels of GM-CSF were much higher in AD patients compared with healthy people.35,36 Meanwhile, overexpression of GMFB in astrocytes leads to increased secretion of GM-CSF, and subsequently the activation of microglia.66 It seems reasonable to propose that astrocytes may be responsible for the initiation of GMFB–GM-CSF–cytokines/chemokines cascade in AD (Figure 1). Additionally, since oxidative stress plays an important role in the pathogenesis and development of AD, GMFB-KO astrocytes showed decreased Cu-Zn SOD expression.9 GMFB may probably mediate oxidative stress in AD via upregulation of the Cu-Zn SOD pathway.
MS
MS is an autoimmune-mediated chronic inflammatory demyelinating disease which affects the CNS. It can be further divided into several subtypes including relapse-remitting MS, secondary progressive MS, and primary progressive MS.40,67 MS is characterized by multiple demyelinating plaques, while chronic inflammation, progressive neurodegeneration, as well as the impairment of blood–brain barrier are also its pathologic features.68 Hitherto, although the etiology and the pathogenesis of MS remain largely unknown, several hypotheses have been proposed. Some believe that the disease starts from the infiltration of autoimmune reactive T-cells from the peripheral immune system (the outside-in hypothesis), while others insist that the primary cause is the disturbance within the CNS, and inflammation is secondary to it (the inside–out hypothesis).68 What is clear is that autoimmune reactive CD4+ T-cells and activated microglia play a pivotal role in the immune response of MS.69 Oxidative stress and inflammation are also important factors that contribute to demyelination and neurodegeneration.40 Over the past few decades, several animal models were used to mimic the pathologic changes of MS, among which EAE of mouse/rat is the most widely used.70
A significant increase in the expression of GMFB has been reported in the CNS of EAE-affected mice compared with healthy controls.71 After infiltration of myelin oligodendrocyte glycoprotein, GMFB-KO mice (homologous recombination) showed a significant decreased incidence (20% in the GMFB-KO group vs 100% in the WT control group) and alleviated symptoms (maximal clinical score of 0.65±0.35 in the GMFB-KO group vs 3.5±0.4 in the WT control group) compared with the WT counterparts. Also, mRNA expression levels of some pro-inflammatory cytokines/chemokines such as TNF-α and GM-CSF are reported to be lower in GMFB-KO mice after injection (TNF-α and GM-CSF mRNA expression in the spinal cord of GMFB-KO mice were ~1/26 and 1/21 of that of the WT counterparts).71 However, reintroduction of GMFB to KO mice by either adenovirus infection or intraperitoneal injection led to the restoration of symptoms and increased pro-inflammatory mediators during onset of EAE.72,73 Intriguingly, intravenous injection of anti-GMFB antibody led to significantly ameliorated symptoms and a clear-cut decrease in pro-inflammatory cytokine/chemokine production.74 These results suggest that GMFB may be closely involved in the pathogenesis and development of EAE and mainly plays a detrimental role.
Some previous research studies may be helpful to understand its possible mechanism. It has been reported that overexpression of GMFB using an adenovirus vector in primary astrocytes isolated from embryonic rat brain led to significantly increased secretion of GM-CSF. The GM-CSF level in the culture media was determined with enzyme-linked immunosorbent assay: GM-CSF level was 380.0±20.6 pg/mL in the GMFB-transfected group while the mock control, virus control, and LacZ control groups remained at 15.0±1.2, 10.0±0.3, and 8.0±0.1 pg/mL, respectively. Interestingly, GMFB-transfected astrocyte conditioned medium with increased GM-CSF level induced the expression of class II major histocompatibility complex II and IL-1β in microglia.66 Moreover, adding GMFB into the culture media (0.02–0.4 μg/mL) led to an increased secretion of IL-1 by primary mouse glial cells, and IL-1 is known to play an important role in activating T-cells.75 Taken together, these evidence suggest that GMFB may probably play an immune modulatory role in EAE. Recently, it has been reported that the activation of STATs in EAE mice is GMFB dependent,45 and the JAK/STAT pathway is essential for the upregulation of iNOS after TNF-γ stimulation.46 Considering that GMFB can also mediate the expression of Cu-Zn SOD, it seems reasonable to assume that GMFB may be responsible for the overproduction of ROS and RNS and exacerbate oxidative stress during EAE. Additionally, GMFB may also be responsible for the overexpression of pro-inflammatory cytokines/chemokines during EAE via the GM-CSF pathway.
In summary, it is quite clear that GMFB is implicated in the pathogenesis of MS, as well as its animal model, EAE, although limitations cannot be overlooked in that animal models cannot well represent all the features of MS.70 As a result, potential therapies such as GMFB-antibody infusion need to be tested by clinical trials. Further investigation is warranted to elucidate the exact mechanism of GMFB’s involvement in MS/EAE.
PD
PD is a progressive neurodegenerative disease clinically featured by the impairment of motor functions as well as other nonmotor symptoms such as depression and cognitive disorders.76 The two major pathological changes of PD are the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) and the formation of Lewy bodies, whose major component is misfolded α-synuclein.77 Up till now, the exact mechanism of the pathogenesis of PD remains largely elusive, although several risk factors including age, genetic background, and environmental factors such as paraquat may contribute to an increased incidence of PD.76 Oxidative stress and chronic inflammation are also essential elements that lead to the demise of dopaminergic neurons.76 Currently, no effective cure has been developed for PD.
After 1-methyl-4-phenylpyridinium (MPP+) treatment, expression levels of malondialdehyde (MDA, an indicator of lipid peroxidation), NO, and ROS were significantly lower in primary mesencephalic glia/neuron cultures derived from GMFB-KO mice compared with WT controls (24 hours after 5, 10, and 20 μM MPP+ treatment, MDA level was 1.74±0.12, 2.39±0.09, and 2.98±0.03 μmol/mg protein in the GMFB-KO group, while it was 4.09±0.37, 5.95±0.23, and 8.29±0.94 μmol/mg protein in the WT control; level of total NO was 6.54±0.17, 8.17±0.85, and 11.31±0.19 μmol/L in the GMFB-KO group, while it was 15.82±0.53, 30.92±1.06, and 46.21±5.88 μmol/L in the WT control), while glutathione (an endogenous antioxidant) level was higher in GMFB-KO cells.43 These evidence revealed an alleviated oxidative stress in GMFB-KO cells after MPP+ stimulation. Also, diminished pro-inflammatory cytokines/chemokines (TNF-α and IL-1β) secretion subsequent to the decreased activation of NF-κB in GMFB-KO cells was observed, indicating the alleviated inflammatory response.43 Taken together, it seems that GMFB-KO neurons/glia are more tolerant of MPP+ toxicity. A proteome analysis of human SNc has reported that GMFB expression was significantly higher in PD samples compared to healthy controls.28 However, expression of GMFB was downregulated in the striatum of maneb–paraquat-induced PD model compared with control groups.78 This may result from the difference between animal models and real PD patients. Further research studies are warranted to explain such discrepancy.
In general, GMFB may contribute to the pathogenesis of PD by enhancing oxidative stress and inflammatory response just like its role in AD and MS. Aside from astrocytes and microglia, GMFB can also mediate pathologic responses of mast cells, another type of immunocyte involved in PD.79 Since limited research studies have been carried out to study the role of GMFB in PD, and further investigations are warranted to elucidate the participation of GMFB in PD.
CNS injury
As a growth and differentiation factor, GMFB is also actively implicated in brain injury. It has been reported that GMFB immunoreactivity increased in mouse astrocytes after cryogenic brain injury while an increase in the serum concentration of GMFB was also observed (serum GMFB peaked early at 1 day after cryogenic injury [0.98±0.12 μg/mL], while the sham-operated group remained at 0.30±0.10 μg/mL).80 Additionally, GMFB has been found to be actively secreted in the wound cavity after aspiration lesion, instead of simply being released passively from fractured cells.20 Furthermore, the application of GMFB to the wound cavity significantly promoted dendritic outgrowth as well as the hypertrophy of selective neurons.81 These evidence have cast light on the possible role of GMFB in the regeneration of neurons.
It has been reported that the overexpression of GMFB using an adenovirus vector in primary astrocytes derived from embryonic rat brains led to increased BDNF and NGF secretion.33 Nevertheless, GMFB is responsible for the upregulation of GM-CSF and some pro-inflammatory cytokines/chemokines.80 These evidence have underlined a multidimensional role of GMFB after brain injury. It may promote the regeneration of neurons and at the meantime exacerbate inflammation responses that are detrimental to wound recovery.
Conclusion and perspectives
In summary, current data have shown that GMFB may play both protective and detrimental roles in the pathogenesis and progression of various neuroinflammatory and neurodegenerative diseases such as AD, PD, and MS. GMFB could function through multiple pathways, and the relationship between GMFB and Cu-Zn SOD, GM-CSF, NTs as well as apoptosis has been discussed in this review. Considering the successful case in which intravenous injection of anti-GMFB antibody significantly mitigated the onset of EAE in mice,74 it is inspiring to propose that GMFB may be a promising therapeutic target for neuroinflammatory and neurodegenerative diseases.
Notably, some concerns on GMFB need to be voiced. As discussed previously, GMFB is expressed on the cell surface of astrocytes and can be secreted under certain conditions.21 Also, many in vitro studies have treated culture cells by adding GMFB into the media.23,79,82 Functioning as an extracellular signaling ligand, GMFB must have its receptors, but little is known about its candidate receptor. Besides, the exact signaling pathway of GMFB and how GMFB expression is triggered during neuroinflammation and neurodegeneration remain to be elucidated. Further investigation will help us understand the nature of GMFB and its relationship with neuroinflammation and neurodegeneration, thus casting some light on the novel therapeutic strategy of neuroinflammatory and neurodegenerative diseases.
Disclosure
The authors report no conflicts of interest in this work.
References
Lim R. Neural Development Part II. Moscona AA, Monroy AB, editors. Vol 16. New York: Academic Press; 1980:305–322. | ||
Lim R, Miller JF, Zaheer A. Purification and characterization of glia maturation factor beta: a growth regulator for neurons and glia. Proc Natl Acad Sci U S A. 1989;86(10):3901–3905. | ||
Zaheer A, Fink BD, Lim R. Expression of glia maturation factor beta mRNA and protein in rat organs and cells. J Neurochem. 1993;60(3):914–920. | ||
Kaimori J, Takenaka M, Nakajima H, et al. Induction of glia maturation factor-beta in proximal tubular cells leads to vulnerability to oxidative injury through the p38 pathway and changes in antioxidant enzyme activities. J Biol Chem. 2003;278(35):33519–33527. | ||
Inagaki M, Aoyama M, Sobue K, et al. Sensitive immunoassays for human and rat GMFB and GMFG, tissue distribution and age-related changes. Biochim Biophys Acta. 2004;1670(3):208–216. | ||
Lim R, Hicklin DJ, Miller JF, Williams TH, Crabtree JB. Distribution of immunoreactive glia maturation factor-like molecule in organs and tissues. Brain Res. 1987;430(1):93–100. | ||
Zaheer A, Haas JT, Reyes C, Mathur SN, Yang B, Lim R. GMF-knockout mice are unable to induce brain-derived neurotrophic factor after exercise. Neurochem Res. 2006;31(4):579–584. | ||
Lim R, Zaheer A, Khosravi H, et al. Impaired motor performance and learning in glia maturation factor-knockout mice. Brain Res. 2004;1024(1–2):225–232. | ||
Zaheer A, Yang B, Cao X, Lim R. Decreased copper-zinc superoxide dismutase activity and increased resistance to oxidative stress in glia maturation factor-null astrocytes. Neurochem Res. 2004;29(8):1473–1480. | ||
Kempuraj D, Khan MM, Thangavel R, Xiong Z, Yang E, Zaheer A. Glia maturation factor induces interleukin-33 release from astrocytes: implications for neurodegenerative diseases. J Neuroimmune Pharmacol. 2013;8(3):643–650. | ||
Barreto GE, Gonzalez J, Capani F, Morales L. Neurodegenerative Diseases – Processes, Prevention, Protection and Monitoring. Chang RCC, editor. Croatia: Intech; 2011:257–272. | ||
Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11(5):400–407. | ||
Buschini E, Piras A, Nuzzi R, Vercelli A. Age related macular degeneration and drusen: neuroinflammation in the retina. Prog Neurobiol. 2011;95(1):14–25. | ||
Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–372. | ||
Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. | ||
Thangavel R, Stolmeier D, Yang X, Anantharam P, Zaheer A. Expression of glia maturation factor in neuropathological lesions of Alzheimer’s disease. Neuropathol Appl Neurobiol. 2012;38(6):572–581. | ||
Lim R, Zaheer A. Phorbol ester stimulates rapid intracellular phosphorylation of glia maturation factor. Biochem Biophys Res Commun. 1995;211(3):928–934. | ||
Rubartelli A, Cozzolino F, Talio M, Sitia R. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J. 1990;9(5):1503–1510. | ||
Stolmeier D, Thangavel R, Anantharam P, Khan MM, Kempuraj D, Zaheer A. Glia maturation factor expression in hippocampus of human Alzheimer’s disease. Neurochem Res. 2013;38(8):1580–1589. | ||
Nieto-Sampedro M, Lim R, Hicklin DJ, Cotman CW. Early release of glia maturation factor and acidic fibroblast growth factor after rat brain injury. Neurosci Lett. 1988;86(3):361–365. | ||
Lim R, Liu YX, Zaheer A. Cell-surface expression of glia maturation factor beta in astrocytes. FASEB J. 1990;4(15):3360–3363. | ||
Utsuyama M, Shiraishi J, Takahashi H, Kasai M, Hirokawa K. Glia maturation factor produced by thymic epithelial cells plays a role in T cell differentiation in the thymic microenvironment. Int Immunol. 2003;15(5):557–564. | ||
Turriff DE, Lim R. Glia maturation factor increases cyclic GMP in glioblasts. Brain Res. 1979;166(2):436–440. | ||
Wang BR, Zaheer A, Lim R. Polyclonal antibody localizes glia maturation factor beta-like immunoreactivity in neurons and glia. Brain Res. 1992;591(1):1–7. | ||
Nishiwaki A, Asai K, Tada T, et al. Expression of glia maturation factor during retinal development in the rat. Brain Res Mol Brain Res. 2001;95(1–2):103–109. | ||
Lim R, Hicklin DJ, Ryken TC, Miller JF, Bosch EP. Endogenous immunoreactive glia maturation factor-like molecule in cultured rat Schwann cells. Brain Res. 1988;468(2):277–284. | ||
Li YL, Ye F, Cheng XD, et al. Identification of glia maturation factor beta as an independent prognostic predictor for serous ovarian cancer. Eur J Cancer. 2010;46(11):2104–2118. | ||
Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci. 2008;6:8. | ||
Haynes EM, Asokan SB, King SJ, Johnson HE, Haugh JM, Bear JE. GMF beta controls branched actin content and lamellipodial retraction in fibroblasts. J Cell Biol. 2015;209(6):803–812. | ||
Boczkowska M, Rebowski G, Dominguez R. Glia maturation factor (GMF) interacts with Arp2/3 complex in a nucleotide state-dependent manner. J Biol Chem. 2013;288(36):25683–25688. | ||
Zaheer A, Lim R. In vitro inhibition of MAP kinase (ERK1/ERK2) activity by phosphorylated glia maturation factor (GMF). Biochemistry. 1996;35(20):6283–6288. | ||
Lim R, Zaheer A. In vitro enhancement of p38 mitogen-activated protein kinase activity by phosphorylated glia maturation factor. J Biol Chem. 1996;271(38):22953–22956. | ||
Zaheer A, Yorek MA, Lim R. Effects of glia maturation factor overexpression in primary astrocytes on MAP kinase activation, transcription factor activation, and neurotrophin secretion. Neurochem Res. 2001;26(12):1293–1299. | ||
Zaheer A, Zaheer S, Sahu SK, et al. A novel role of glia maturation factor: induction of granulocyte-macrophage colony-stimulating factor and pro-inflammatory cytokines. J Neurochem. 2007;101(2):364–376. | ||
Lee SC, Liu W, Brosnan CF, Dickson DW. GM-CSF promotes proliferation of human fetal and adult microglia in primary cultures. Glia. 1994;12(4):309–318. | ||
Tarkowski E, Wallin A, Regland B, Blennow K, Tarkowski A. Local and systemic GM-CSF increase in Alzheimer’s disease and vascular dementia. Acta Neurol Scand. 2001;103(3):166–174. | ||
Lim R, Zaheer A, Yorek MA, Darby CJ, Oberley LW. Activation of nuclear factor-kappaB in C6 rat glioma cells after transfection with glia maturation factor. J Neurochem. 2000;74(2):596–602. | ||
Tainer JA, Getzoff ED, Richardson JS, Richardson DC. Structure and mechanism of copper, zinc superoxide dismutase. Nature. 1983;306(5940):284–287. | ||
Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA. Reactive oxygen species, cell signaling, and cell injury. Free Radic Biol Med. 2000;28(10):1456–1462. | ||
Haider L. Inflammation, iron, energy failure, and oxidative stress in the pathogenesis of multiple sclerosis. Oxid Med Cell Longev. 2015;2015:725370. | ||
Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-α gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18(5):1633–1641. | ||
Zaheer A, Sahu SK, Wu Y, et al. Diminished cytokine and chemokine expression in the central nervous system of GMF-deficient mice with experimental autoimmune encephalomyelitis. Brain Res. 2007;1144:239–247. | ||
Khan MM, Zaheer S, Nehman J, Zaheer A. Suppression of glia maturation factor expression prevents 1-methyl-4-phenylpyridinium (MPP+)-induced loss of mesencephalic dopaminergic neurons. Neuroscience. 2014;277:196–205. | ||
Bhat NR, Feinstein DL, Shen Q, Bhat AN. p38 MAPK-mediated transcriptional activation of inducible nitric-oxide synthase in glial cells: roles of nuclear factors, nuclear factor κB, cAMP response element-binding protein, CCAAT/enhancer-binding protein-β, and activating transcription factor-2. J Biol Chem. 2002;277:29584–29592. | ||
Zaheer S, Wu Y, Bassett J, Yang B, Zaheer A. Glia maturation factor regulation of STAT expression: a novel mechanism in experimental autoimmune encephalomyelitis. Neurochem Res. 2007;32(12):2123–2131. | ||
Dell’Albani P, Santangelo R, Torrisi L, Nicoletti VG, de Vellis J, Giuffrida Stella AM. JAK/STAT signaling pathway mediates cytokine-induced iNOS expression in primary astroglial cell cultures. J Neurosci Res. 2001;65(5):417–424. | ||
Pantazis NJ, Zaheer A, Dai D, Zaheer S, Green SH, Lim R. Transfection of C6 glioma cells with glia maturation factor upregulates brain-derived neurotrophic factor and nerve growth factor: trophic effects and protection against ethanol toxicity in cerebellar granule cells. Brain Res. 2000;865(1):59–76. | ||
Zaheer A, Weiss JL, Goyal P, Lim R. Enhanced expression of neurotrophic factors by C6 rat glioma cells after transfection with glia maturation factor. Neurosci Lett. 1999;265(3):203–206. | ||
Jackson GR, Apffel L, Werrbach-Perez K, Perez-Polo JR. Role of nerve growth factor in oxidant–antioxidant balance and neuronal injury. I. Stimulation of hydrogen peroxide resistance. J Neurosci Res. 1990;25(3):360–368. | ||
Zaheer A, Knight S, Zaheer A, Ahrens M, Sahu SK, Yang B. Glia maturation factor overexpression in neuroblastoma cells activates glycogen synthase kinase-3beta and caspase-3. Brain Res. 2008;1190:206–214. | ||
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270(5240):1326–1331. | ||
Kolko M, Vosborg F, Henriksen UL, et al. Lactate transport and receptor actions in retina: potential roles in retinal function and disease. Neurochem Res. 2016;41(6):1229–1236. | ||
de Hoz R, Rojas B, Ramirez AI, et al. Retinal macroglial responses in health and disease. Biomed Res Int. 2016;2016:2954721. | ||
Bringmann A, Pannicke T, Grosche J, et al. Muller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25(4):397–424. | ||
Cascella R, Ragazzo M, Strafella C, et al. Age-related macular degeneration: insights into inflammatory genes. J Ophthalmol. 2014;2014:582842. | ||
Jin N, Gao L, Fan X, Xu H. Friend or foe? Resident microglia vs bone marrow-derived microglia and their roles in the retinal degeneration. Mol Neurobiol. 2017;54(6):4094–4112. | ||
Xi H, Katschke KJ Jr, Li Y, et al. IL-33 amplifies an innate immune response in the degenerating retina. J Exp Med. 2016;213(2):189–207. | ||
Bosch EP, Zhong W, Lim R. Axonal signals regulate expression of glia maturation factor-beta in Schwann cells: an immunohistochemical study of injured sciatic nerves and cultured Schwann cells. J Neurosci. 1989;9(10):3690–3698. | ||
Conforti L, Gilley J, Coleman MP. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci. 2014;15(6):394–409. | ||
Geden MJ, Deshmukh M. Axon degeneration: context defines distinct pathways. Curr Opin Neurobiol. 2016;39:108–115. | ||
Tramutola A, Lanzillotta C, Perluigi M, Butterfield DA. Oxidative stress, protein modification and Alzheimer disease. Brain Res Bull. 2017;133:88–96. | ||
Thangavel R, Kempuraj D, Stolmeier D, Anantharam P, Khan M, Zaheer A. Glia maturation factor expression in entorhinal cortex of Alzheimer’s disease brain. Neurochem Res. 2013;38(9):1777–1784. | ||
Zaheer S, Thangavel R, Sahu SK, Zaheer A. Augmented expression of glia maturation factor in Alzheimer’s disease. Neuroscience. 2011;194:227–233. | ||
Zaheer S, Thangavel R, Wu Y, Khan MM, Kempuraj D, Zaheer A. Enhanced expression of glia maturation factor correlates with glial activation in the brain of triple transgenic Alzheimer’s disease mice. Neurochem Res. 2013;38(1):218–225. | ||
Zaheer A, Zaheer S, Thangavel R, Wu Y, Sahu SK, Yang B. Glia maturation factor modulates β-amyloid-induced glial activation, inflammatory cytokine/chemokine production and neuronal damage. Brain Res. 2008;1208:192–203. | ||
Zaheer A, Mathur SN, Lim R. Overexpression of glia maturation factor in astrocytes leads to immune activation of microglia through secretion of granulocyte–macrophage-colony stimulating factor. Biochem Biophys Res Commun. 2002;294(2):238–244. | ||
Ntranos A, Lublin F. Diagnostic criteria, classification and treatment goals in multiple sclerosis: the chronicles of time and space. Curr Neurol Neurosci Rep. 2016;16(10):90. | ||
Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14(2):183–193. | ||
Yadav SK, Mindur JE, Ito K, Dhib-Jalbut S. Advances in the immunopathogenesis of multiple sclerosis. Curr Opin Neurol. 2015;28(3):206–219. | ||
Procaccini C, De Rosa V, Pucino V, Formisano L, Matarese G. Animal models of multiple sclerosis. Eur J Pharmacol. 2015;759:182–191. | ||
Zaheer A, Zaheer S, Sahu SK, Yang B, Lim R. Reduced severity of experimental autoimmune encephalomyelitis in GMF-deficient mice. Neurochem Res. 2007;32(1):39–47. | ||
Zaheer S, Wu Y, Yang X, Ahrens M, Sahu SK, Zaheer A. Clinical course of myelin oligodendrocyte glycoprotein 35–55 induced experimental autoimmune encephalomyelitis is aggravated by glia maturation factor. Neurochem Int. 2012;60(3):215–219. | ||
Zaheer S, Wu Y, Sahu SK, Zaheer A. Overexpression of glia maturation factor reinstates susceptibility to myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis in glia maturation factor deficient mice. Neurobiol Dis. 2010;40(3):593–598. | ||
Zaheer S, Wu Y, Sahu SK, Zaheer A. Suppression of neuro inflammation in experimental autoimmune encephalomyelitis by glia maturation factor antibody. Brain Res. 2011;1373:230–239. | ||
Fontana A, Weber E, Grob PJ, Lim R, Miller JF. Dual effect of glia maturation factor on astrocytes: differentiation and release of interleukin-1 like factors. J Neuroimmunol. 1983;5(3):261–269. | ||
Dexter DT, Jenner P. Parkinson disease: from pathology to molecular disease mechanisms. Free Radic Biol Med. 2013;62:132–144. | ||
Betarbet R, Sherer TB, Greenamyre JT. Animal models of Parkinson’s disease. Bioessays. 2002;24(4):308–318. | ||
Patel S, Sinha A, Singh MP. Identification of differentially expressed proteins in striatum of maneb- and paraquat-induced Parkinson’s disease phenotype in mouse. Neurotoxicol Teratol. 2007;29(5):578–585. | ||
Kempuraj D, Thangavel R, Yang E, et al. Dopaminergic toxin 1-methyl-4-phenylpyridinium, proteins α-synuclein and glia maturation factor activate mast cells and release inflammatory mediators. PLoS One. 2015;10(8):e0135776. | ||
Hotta N, Aoyama M, Inagaki M, et al. Expression of glia maturation factor beta after cryogenic brain injury. Brain Res Mol Brain Res. 2005;133(1):71–77. | ||
Lim R, Huang L. Glia maturation factor-beta promotes the appearance of large neurofilament-rich neurons in injured rat brains. Brain Res. 1989;504(1):154–158. | ||
Lim R, Nakagawa S, Arnason BG, Turriff DE. Glia maturation factor promotes contact inhibition in cancer cells. Proc Natl Acad Sci U S A. 1981;78(7):4373–4377. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.