Back to Journals » Journal of Inflammation Research » Volume 19
Ginsenoside Rg3 Mitigates LPS-Induced Injury in Human Bronchial Epithelial Cells by Restoring Autophagic Flux and Inhibiting the TLR4/NF-κB-Mediated Inflammatory Response
Authors Tao X, Liu L, Gu X, Jia B, Yu Q, Wei Y, Zhang W, Zhou J
Received 20 July 2025
Accepted for publication 25 December 2025
Published 20 January 2026 Volume 2026:19 555053
DOI https://doi.org/10.2147/JIR.S555053
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Xingyu Tao,1– 4,* Lingjiao Liu,1– 3,* Xiaoke Gu,5 Baohui Jia,6 Qi Yu,1– 3 Yingying Wei,1– 3 Wei Zhang,1– 3 Jing Zhou1– 3
1Department of Respiratory and Critical Care Medicine, Jiangxi Medical College, Nanchang University, The First Affiliated Hospital, Jiangxi Institute of Respiratory Disease, Nanchang, Jiangxi, 330006, People’s Republic of China; 2Jiangxi Clinical Research Center for Respiratory Diseases, Nanchang, Jiangxi, 330006, People’s Republic of China; 3China-Japan Friendship Jiangxi Hospital, National Regional Center for Respiratory Medicine, Nanchang, Jiangxi, 330200, People’s Republic of China; 4Department of Respiratory and Critical Care Medicine, The Third Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, People’s Republic of China; 5Department of Anesthesia, Sun Yat-Sen University Sixth Affiliated Hospital, Guangzhou, Guangdong, 510000, People’s Republic of China; 6Department of Respiratory and Critical Care Medicine, The Fourth Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, 330006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jing Zhou, Department of Respiratory and Critical Care Medicine, Jiangxi Medical College, Nanchang University, The First Affiliated Hospital, Jiangxi Institute of Respiratory Disease, Nanchang, Jiangxi, 330006, People’s Republic of China, Email [email protected] Xingyu Tao, Department of Respiratory and Critical Care Medicine, Jiangxi Medical College, Nanchang University, The First Affiliated Hospital, Jiangxi Institute of Respiratory Disease, Nanchang, Jiangxi, 330006, People’s Republic of China, Email [email protected]; [email protected]
Purpose: To elucidate the molecular mechanism by which ginsenoside Rg3 (G-Rg3) protects human bronchial epithelial (HBE) cells against lipopolysaccharide (LPS)-induced injury, focusing on its regulation of autophagic flux and the TLR4/NF-κB-mediated inflammatory pathway.
Methods: HBE cells were treated with LPS (1– 100 ng/mL) to induce autophagy dysregulation and inflammation. G-Rg3 (2– 16 μM) was administered to evaluate its protective effects. Western blotting was used to detect autophagy-related proteins (ATG4B, ATG7, PIK3C3, LC3B, p62) and TLR4/NF-κB signaling molecules; ELISA quantified proinflammatory cytokines (TNF-α, IL-1β, IL-2, IL-6, IL-8); PI staining and flow cytometry analyzed cell death and apoptosis.
Results: LPS dose-dependently upregulated the expression of autophagy-related proteins (ATG4B, ATG7, PIK3C3, p62, LC3B-II), with accumulated p62 and LC3B-II indicating impaired clearance of autophagic substrates. Additionally, G-Rg3 inhibited LPS-induced TLR4/NF-κB activation, suppressed proinflammatory cytokine secretion, and attenuated HBE cell apoptosis/necrosis.
Conclusion: G-Rg3 mitigates LPS-induced HBE cell injury by dual mechanisms: restoring impaired autophagic flux and inhibiting the TLR4/NF-κB inflammatory cascade. These findings identify G-Rg3 as a promising therapeutic agent targeting the crosstalk between autophagy and inflammation in respiratory diseases such as COPD and acute lung injury.
Keywords: Ginsenoside Rg3, autophagy, LPS, HBE, cell death
Introduction
A cornerstone of traditional Chinese medicine, ginseng (genus Panax), has long been studied and characterized for its wide range of therapeutic effects, including immunomodulatory, antioxidant, and anti-inflammatory effects. Ginsenosides, the primary active constituents of ginseng, which include more than 40 structurally different types, are divided into protopanaxadiol (PPD) and protopanaxatriol (PPT) saponins,1,2 and the pharmacological benefits of ginseng are attributed mainly to these active compounds. The ginsenoside Rg3 (G-Rg3), a PPD-type minor ginsenoside, is abundant in red ginseng and produced by steaming and drying fresh ginseng roots. Due to its potent biological activities, including anticancer, anti-inflammatory, and antiapoptotic effects,3,4 G Rg3 has garnered significant attention.5 However, most studies on G-Rg3’s protective effects against respiratory injury focus on systemic inflammation or animal models, with limited exploration of its direct regulatory role in human bronchial epithelial (HBE) cells—the frontline barrier of the respiratory tract.
Chronic obstructive pulmonary disease (COPD), acute lung injury (ALI), and acute respiratory distress syndrome (ARDS) are often accompanied by excessive, uncontrolled inflammation and epithelial cell death.1,6 In research, lipopolysaccharide (LPS), a structural component of the cell walls of gram-negative bacteria, is widely used to model bacterial infections and induce inflammatory responses, especially in airway epithelial cells.7 LPS can activate Toll-like receptor 4 (TLR4) when inhaled or administered systemically, leading to excessive production of inflammatory cytokines, including tumor necrosis factor-alpha (TNF), IL-1β, IL-6, and IL-8.6 Excessive autophagy or impaired autophagic flux (blocked autophagic flux; impaired autophagosome-lysosome fusion) exacerbates inflammation and cell death.8 Epithelial injury, increased vascular permeability, and subsequent respiratory complications are all key contributors to the proinflammatory mediators listed above.9,10
A highly conserved cellular degradation process, autophagy, has been shown to regulate inflammation and cell survival under stress conditions.11 This process results in the synthesis of double-membrane autophagosomes that fuse with lysosomes, degrading damaged organelles and proteins. Under pathological conditions, dysregulated autophagy can exacerbate cellular injury or promote apoptosis; under normal physiological conditions, autophagy maintains homeostasis.12 Accumulating evidence suggests that LPS modulates autophagic activity and, in bronchial epithelial cells, may exert either protective or detrimental effects, depending on the context and dose.13,14 For example, some studies have reported that moderate levels of autophagy can help block bacterial clearance and cell survival. Moreover, excessive or blocked autophagic flux may contribute to cytotoxicity and inflammation.8
According to recent studies, natural compounds, such as ginsenosides, reportedly modulate autophagy and inflammatory signaling pathways. In particular, G-Rg3 can inhibit nuclear factor kappa B (NF-kβ) signaling and cytokine release in macrophages and epithelial cells stimulated with LPS.15,16 Furthermore, in animal models, G-Rg3 has been shown to protect against LPS-mediated acute lung injury by inhibiting oxidative stress and decreasing neutrophil infiltration.17,18 In addition, its antiapoptotic effects have been demonstrated in cardiomyocytes, hepatocytes, and renal tubular cells, primarily through the modulation of the PI3K/Akt/mTOR and AMPK pathways.19,20
LPS induces autophagy and apoptosis through a pro-inflammatory, epithelial barrier-disrupting process in human bronchial epithelial (HBE) cells; however, the regulatory role of G-Rg3 in this process is poorly understood. While studies have demonstrated the anti-inflammatory effects of G-Rg3 in macrophages and cancer cells,21,22 the impact of G-Rg3 and its modulation of the autophagy-related proteins ATG4B, ATG7, LC3B, and p62, as well as PIK3C3, on the airway epithelium is unknown. This relationship is well understood and particularly relevant because epithelial cell death and inflammation are key targets for managing respiratory diseases.
Furthermore, G Rg3 suppresses LPS-induced COX-2 and nitric oxide production in RAW 264.7 cells.23 G-Rg3 also exhibits anti-inflammatory activity and has been reported to protect against reactive oxygen species (ROS)-induced damage in human dermal fibroblasts and keratinocytes,24 suggesting a broad range of cytoprotective roles. These findings warrant further studies to assess potential therapeutics for bronchial epithelial cells under inflammatory conditions.
Therefore, this study aimed to examine the effects of G-Rg3 on LPS-induced inflammation, autophagy dysregulation, and HBE cell death. We will utilize protein expression assays, cytokine quantification, and apoptosis detection techniques to elucidate the molecular mechanisms underlying the protective effects of G-Rg3. These findings may yield new approaches for the therapeutic use of G-Rg3 for treating airway epithelial damage/inflammation triggered by bacterial endotoxins.
Methods
Cell Culture
Human bronchial epithelial (HBE) cells (ATCC, Cat# CCL-202) were used as the in vitro model, with strict control of cell handling and culture conditions to ensure experimental consistency: remove cryopreserved cells from −80°C storage, immediately immerse the cryovial in a 37°C water bath (Thermo Fisher, Model WB2000) and gently agitate for 1–2 minutes until only a small ice crystal remains, disinfect the vial surface with 75% ethanol (Solarbio, Cat# 10009218), transfer the cell suspension to a 15 mL centrifuge tube (Corning, Cat# 430791) containing 5 mL pre-warmed (37°C) complete Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Cat# c11965500BT) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, Cat# 10099141) and 1% penicillin-streptomycin (100 U/mL penicillin, 100 μg/mL streptomycin; Solarbio, Cat# P1400), centrifuge at 1000×g for 5 minutes at room temperature (RT), discard the supernatant, resuspend the cell pellet in 5 mL complete DMEM, count cells using a hemocytometer (Thermo Fisher, Cat# 02–671-5), seed at a density of 5×105 cells per T75 flask (Corning, Cat# 430641), and culture in a humidified incubator (Thermo Fisher, Model 3111) set to 37°C, 5% CO2, and 95% relative humidity; when cells reach 80–90% confluence (typically 48–72 hours post-seeding), perform passaging by aspirating the culture medium, rinsing twice with 5 mL pre-warmed PBS (Solarbio, Cat# P1020), adding 2 mL 0.25% trypsin-EDTA (Gibco, Cat# 25200056) and incubating at 37°C for 2–3 minutes (monitoring cell detachment under an inverted microscope), neutralizing trypsin with 4 mL complete DMEM, centrifuging at 1000×g for 5 minutes, discarding the supernatant, resuspending the cell pellet in 5 mL complete DMEM, counting again, and seeding at the required density, with only cells at passages 3–8 used for experiments (to avoid cellular senescence) and cell viability confirmed to be >95% via trypan blue staining (Solarbio, Cat# T8154) before each experiment.
Reagents and Antibodies
Ginsenoside Rg3 (G-Rg3) with ≥98% purity was purchased from TargetMol (Cat# T3402), dissolved in dimethyl sulfoxide (DMSO; Sigma, Cat# C6628) to prepare a 10 mM stock solution, and stored at −80°C (avoiding repeated freeze-thaw cycles, with final DMSO concentration in cell culture ≤0.1% to eliminate cytotoxicity); lipopolysaccharide (LPS) from Escherichia coli 055:B5 was obtained from Solarbio (Beijing, China; Cat# L8880), reconstituted in sterile phosphate-buffered saline (PBS; Solarbio, Cat# P1020) to a 1 mg/mL stock solution, filtered through a 0.22 μm sterile filter (Millipore, Cat# SLGP033RB), and stored at −20°C; chloroquine (autophagy inhibition positive control) was purchased from Sigma (Cat# RNBH9960), prepared as a 50 mM stock solution in PBS, and stored at 4°C; primary antibodies against autophagy-related proteins and internal reference were sourced as follows: β-actin (Cat# 66009-1-Ig) and ATG4B (Cat# 11306-1-AP) from Proteintech, ATG7 (Cat# PA5-114334) from Thermo Fisher, PIK3C3 (also known as VPS34; Cat# 21656-1-AP), LC3B (Cat# 18725-1-AP), and p62 (also known as SQSTM1; Cat# 18420-1-AP) from Proteintech, all stored at −20°C after reconstitution according to the manufacturer’s instructions; horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-rabbit: Cat# ab6721, anti-mouse: Cat# ab6789) were obtained from Abcam, stored at 4°C, and used at the recommended dilution ratio; other reagents including RIPA lysis buffer (Millipore, Cat# 20–188), protease inhibitor cocktail (Roche, Cat# 04693159001), PMSF (MedChemExpress, Cat# HY-B0496), BCA protein assay kit (Thermo Fisher, Cat# 23225), 5× SDS loading buffer (Solarbio, Cat# P1040), acrylamide (Sigma, Cat# A9099), Tris-HCl (Solarbio, Cat# T8154), SDS (Solarbio, Cat# S8010), ammonium persulfate (APS; Solarbio, Cat# A1060), TEMED (Solarbio, Cat# T8132), PVDF membranes (Millipore, Cat# IPVH00010), 5% non-fat skim milk (Solarbio, Cat# D8340), and enhanced chemiluminescence (ECL) reagent (Yeasen, Cat# 36208ES) were of analytical grade, stored at the recommended temperatures (4°C or −20°C), and used before their expiration dates.
Western Blot Analysis
Cell apoptosis was quantified using an Annexin V-FITC/Propidium Iodide (PI) Apoptosis Detection Kit (Elabscience, Cat# E-CK-A211) and flow cytometry, with precise control of each step to ensure accurate results: seed HBE cells in 6-well plates at a density of 6×105 cells per well, culture them for 24 hours (until reaching 70–80% confluence), then replace the medium with fresh complete DMEM containing the corresponding treatments (control: no treatment; LPS: 100 ng/mL; G-Rg3: 16 μM; LPS + G-Rg3: 100 ng/mL LPS + 16 μM G-Rg3) and incubate for another 24 hours at 37°C, 5% CO2; after treatment, aspirate the culture medium, rinse the cells twice with cold PBS, add 1 mL of 0.25% trypsin-EDTA (Gibco, Cat# 25200056) to each well, incubate at 37°C for 2–3 minutes until cells detach, neutralize the trypsin with 2 mL of complete DMEM, collect the cell suspension into a 15 mL centrifuge tube, centrifuge at 1000×g for 5 minutes at 4°C, discard the supernatant, wash the cell pellet twice with cold PBS (centrifuging at 1000×g for 5 minutes each time to remove residual medium and trypsin), resuspend the pellet in 100 μL of ice-cold binding buffer (provided in the detection kit), add 5 μL of Annexin V-FITC and 10 μL of PI solution (both provided in the kit) to the cell suspension, mix gently by pipetting, and incubate at room temperature in the dark for 15 minutes (avoiding prolonged incubation to prevent non-specific staining of viable cells); after incubation, add 400 μL of binding buffer to each sample to dilute the staining solution, and immediately analyze the samples using a BD FACSCanto II flow cytometer (BD Biosciences, San Jose, CA) with BD FACSDiva software (Version 8.0.1); set the detection parameters as follows: excitation wavelength of 488 nm, emission wavelength of 525 nm for Annexin V-FITC (FITC channel) and 620 nm for PI (PI channel), collect 10,000 events per sample, and use forward scatter (FSC) and side scatter (SSC) gating to exclude cell debris and non-single cells; classify the cells into four quadrants based on fluorescence signals: Q1 (necrotic cells: Annexin V-negative/PI-positive), Q2 (late apoptotic cells: Annexin V-positive/PI-positive), Q3 (viable cells: Annexin V-negative/PI-negative), and Q4 (early apoptotic cells: Annexin V-positive/PI-negative), then calculate the total apoptotic rate as the sum of the percentages of Q2 and Q4 cells.
Cell Apoptosis Assay
Cell apoptosis was quantified using an Annexin V-FITC/Propidium Iodide (PI) Apoptosis Detection Kit (Elabscience, Cat# E-CK-A211) and flow cytometry, with precise control of each step to ensure accurate results: seed HBE cells in 6-well plates at a density of 6×105 cells per well, culture them for 24 hours (until reaching 70–80% confluence), then replace the medium with fresh complete DMEM containing the corresponding treatments (control: no treatment; LPS: 100 ng/mL; G-Rg3: 16 μM; LPS + G-Rg3: 100 ng/mL LPS + 16 μM G-Rg3) and incubate for another 24 hours at 37°C, 5% CO2; after treatment, aspirate the culture medium, rinse the cells twice with cold PBS, add 1 mL of 0.25% trypsin-EDTA (Gibco, Cat# 25200056) to each well, incubate at 37°C for 2–3 minutes until cells detach, neutralize the trypsin with 2 mL of complete DMEM, collect the cell suspension into a 15 mL centrifuge tube, centrifuge at 1000×g for 5 minutes at 4°C, discard the supernatant, wash the cell pellet twice with cold PBS (centrifuging at 1000×g for 5 minutes each time to remove residual medium and trypsin), resuspend the pellet in 100 μL of ice-cold binding buffer (provided in the detection kit), add 5 μL of Annexin V-FITC and 10 μL of PI solution (both provided in the kit) to the cell suspension, mix gently by pipetting, and incubate at room temperature in the dark for 15 minutes (avoiding prolonged incubation to prevent non-specific staining of viable cells); after incubation, add 400 μL of binding buffer to each sample to dilute the staining solution, and immediately analyze the samples using a BD FACSCanto II flow cytometer (BD Biosciences, San Jose, CA) with BD FACSDiva software (Version 8.0.1); set the detection parameters as follows: excitation wavelength of 488 nm, emission wavelength of 525 nm for Annexin V-FITC (FITC channel) and 620 nm for PI (PI channel), collect 10,000 events per sample, and use forward scatter (FSC) and side scatter (SSC) gating to exclude cell debris and non-single cells; classify the cells into four quadrants based on fluorescence signals: Q1 (necrotic cells: Annexin V-negative/PI-positive), Q2 (late apoptotic cells: Annexin V-positive/PI-positive), Q3 (viable cells: Annexin V-negative/PI-negative), and Q4 (early apoptotic cells: Annexin V-positive/PI-negative), then calculate the total apoptotic rate as the sum of the percentages of Q2 and Q4 cells.
PI Staining for Cell Death
Propidium iodide (PI) staining was used to assess necrotic or late-stage cell death, with standardized operation to ensure consistency and reproducibility: first, sterile circular coverslips (diameter 12 mm, thickness 0.13–0.17 mm; Solarbio, Cat# C0301) were placed at the bottom of each well in 6-well plates and UV-sterilized under a biosafety cabinet for 30 minutes to eliminate contamination, after which HBE cells were seeded directly onto the pre-sterilized coverslips at a density of 5×105 cells per well and cultured in a humidified 37°C, 5% CO2 incubator for 24 hours until reaching 70–80% confluence; next, cells on coverslips were treated with the same experimental groups as the apoptosis assay (control, LPS, G-Rg3, LPS + G-Rg3) for 24 hours, and after treatment, the culture medium was aspirated, cells (and coverslips) were rinsed twice with pre-warmed PBS to remove residual medium and reagents, then 1 mL of co-staining solution (containing 10 μg/mL PI [Yeasen, Cat# 40711ES] and 1 μg/mL Hoechst 33342 [Solarbio, Cat# H1399] in PBS) was added to each well—Hoechst 33342 is a membrane-permeable dye that penetrates both viable and dead cells to embed in double-stranded DNA, emitting bright blue fluorescence to label all cell nuclei, while PI only penetrates cells with compromised membranes (necrotic/late apoptotic cells) to emit red fluorescence; the plate was then incubated at 37°C in the dark for 30 minutes to ensure uniform staining, after which the co-staining solution was aspirated, cells were rinsed twice with PBS to remove unbound dyes, coverslips were carefully lifted with forceps (cell-side down) and mounted on glass slides (Solarbio, Cat# S8010) with a drop of anti-fluorescence quenching mounting medium (Solarbio, Cat# S2100), and observed under an inverted fluorescence microscope (Invitrogen, Model EVOS FL Auto 2) using a Texas Red filter (for PI, excitation 535 nm/emission 617 nm) and a DAPI filter (for Hoechst 33342, excitation 350 nm/emission 460 nm); images were captured at 4×, 10×, and 20× magnifications (three random fields per coverslip), with red fluorescent cells defined as dead/dying cells and blue fluorescent cells representing all cells (viable + dead), and ImageJ software was used to count red and blue fluorescent cells in each field to calculate the cell death rate as (number of red fluorescent cells / number of blue fluorescent cells) × 100%.
ELISA for Cytokine Quantification
Enzyme-linked immunosorbent assay (ELISA) was used to quantify the levels of proinflammatory cytokines (TNF-α, IL-1β, IL-2, IL-6, IL-8) in HBE cell culture supernatants, with strict adherence to kit instructions and standardized operation to ensure accuracy: seed HBE cells in 96-well plates at a density of 1×105 cells per well (200 μL of cell suspension per well) to ensure uniform cell distribution, culture for 24 hours, then replace the medium with 200 μL of fresh complete DMEM containing the experimental treatments (control, LPS, G-Rg3, LPS + G-Rg3) and incubate for 24 hours; after treatment, collect the culture supernatants from each well into 1.5 mL centrifuge tubes (Eppendorf, Cat# 0030122226), centrifuge at 3000×g for 10 minutes at 4°C to remove cell debris and floating cells, and transfer the supernatant to new centrifuge tubes (stored at −80°C if not tested immediately); use human-specific ELISA kits (Neobioscience, China) for each cytokine (TNF-α: Cat# EH0101, IL-1β: Cat# EH0102, IL-2: Cat# EH0103, IL-6: Cat# EH0106, IL-8: Cat# EH0108) and follow the manufacturer’s protocol for detection: add 100 μL of standard solutions (with concentrations ranging from 0 to 1000 pg/mL, prepared by serial dilution according to the kit) or diluted samples (supernatants diluted 1:2 with sample dilution buffer provided in the kit) to the corresponding wells of the pre-coated ELISA plates, incubate the plate at 37°C for 90 minutes, aspirate the liquid from each well, add 100 μL of biotin-labeled detection antibody (diluted according to the kit’s instructions) to each well, incubate at 37°C for 60 minutes, wash the plate three times with washing buffer (each time adding 300 μL of buffer and incubating for 30 seconds before aspirating), add 100 μL of streptavidin-HRP conjugate to each well, incubate at 37°C for 30 minutes, wash the plate five times with washing buffer (same as above), add 100 μL of TMB substrate solution to each well, incubate at 37°C in the dark for 15–20 minutes (until the standard wells show a gradient of blue color), add 50 μL of stop solution (1 M H2SO4) to each well to terminate the reaction (the color changes from blue to yellow), immediately measure the absorbance at 450 nm using a microplate reader (Bio-Rad, Model 680); draw a standard curve with the concentration of the standard solution as the x-axis and the corresponding absorbance value as the y-axis, calculate the concentration of each cytokine in the sample supernatant based on the standard curve and the dilution factor, and express the results in pg/mL; each experimental group was set up with three biological replicates and three technical replicates per biological replicate to ensure the reliability of the data.
Statistical Analysis
All experiments were independently repeated at least three times to ensure reproducibility, with the resulting data collated and expressed as the mean ± standard deviation (SD) to reflect the central tendency and variability of the data; statistical analysis was performed using GraphPad Prism 6.0 software (GraphPad Software, Inc., La Jolla, CA, USA), with the choice of statistical method based on the number of comparison groups: for comparisons between two independent groups (control group vs single treatment group), Student’s t-test (two-tailed) was used to determine the significance of differences; for comparisons among three or more groups (control, LPS, G-Rg3, LPS + G-Rg3 groups), one-way analysis of variance (ANOVA) was first used to test whether there was an overall significant difference among the groups, and if a significant overall difference was found (p < 0.05), Tukey’s post hoc test was further used to perform pairwise comparisons between groups to identify specific differences; the level of statistical significance was defined as follows: *p < 0.05, **p < 0.01, and ***p < 0.001, with all p-values less than 0.05 considered statistically significant; before performing parametric tests (t-test and ANOVA), the normality of the data distribution was verified using the Shapiro–Wilk test, and the homogeneity of variances was verified using Levene’s test, with no violations of test assumptions observed in the dataset, ensuring the validity of the statistical results.
Results
LPS Can Activate Autophagy While Inhibiting Autophagic Flux
To investigate how different concentrations of LPS regulate autophagy in HBE cells, we assessed the expression of autophagy-related proteins. As shown in Figure 1, representative Western blot bands (Figure 1A) revealed that, compared with the control (0 ng/mL LPS), LPS treatment gradually increased the gray intensity of ATG4B, ATG7, PIK3C3, p62, and LC3B bands as its concentration rose (1–100 ng/mL). In contrast, the internal control β-actin remained stable Quantitative analysis (Figure 1B–F) further confirmed that LPS significantly upregulated the relative expression (normalized to β-actin) of these proteins in a concentration-dependent manner: for instance, the 100 ng/mL LPS group exhibited ~1.75-fold higher expression of each protein than the control group, with statistically significant differences (P < 0.05). These results suggest that LPS enhances the expression of autophagy-related proteins to promote autophagic vesicle formation and maturation; meanwhile, the concurrent elevation of p62 and LC3B (a marker of autophagosome accumulation) indicates that LPS also inhibits the fusion of autophagosomes with lysosomes, thereby blocking autolysosome formation. Collectively, LPS upregulates autophagy-related protein expression in a concentration-dependent manner while suppressing autolysosome formation in HBE cells.
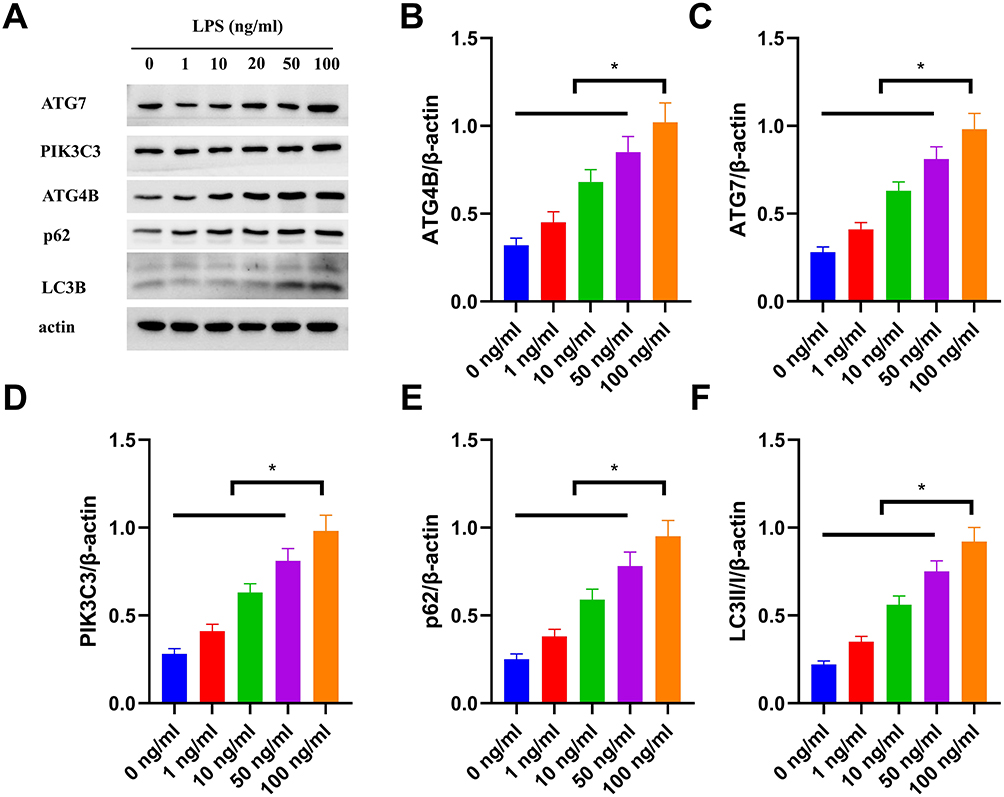 |
Figure 1 LPS activates autophagy-related protein expression in HBE cells in a concentration-dependent manner. (A) Representative Western blot bands showing the expression of ATG7, PIK3C3, ATG4B, p62, LC3B, and β-actin (loading control) in HBE cells treated with 0, 1, 10, 50, or 100 ng/mL LPS. (B–F) Quantitative analysis of relative protein expression (normalized to β-actin) for ATG4B (B), ATG7 (C), PIK3C3 (D), p62 (E), and LC3B-II (F). Data are presented as mean ± SD (n=3 independent experiments). *P<0.05 vs 0 ng/mL group. |
In the LPS-induced inflammatory model, we first investigated the regulatory effect of G-Rg3 on the AMPK/mTOR signaling pathway (Figure S1): In the control (Ctrl) group, the expression of total AMPK (A) and phosphorylated AMPK (p-AMPK, B) maintained basal levels; treatment with G-Rg3 alone (Rg3 group) had no significant effect on AMPK or p-AMPK expression. After LPS stimulation (LPS+Rg3 group), p-AMPK expression was significantly upregulated, while the phosphorylation level of mTOR (p-mTOR, D) was markedly decreased (total mTOR (C) showed no noticeable change). When combined with an AMPK inhibitor (LPS+Rg3+CC group), both the upregulation of p-AMPK and downregulation of p-mTOR were reversed, indicating that G-Rg3 can inhibit mTOR phosphorylation by activating AMPK.
Next, we analyzed the impact of G-Rg3 on the NF-κB pathway (Figure S2): In the Ctrl group, p-p65 (A) expression was low, and Rg3 treatment alone did not cause a significant change. After LPS stimulation (LPS+Rg3 group), p-p65 expression was significantly increased, accompanied by enhanced degradation of IκBα (C). However, when combined with the AMPK inhibitor (LPS+Rg3+CC group), both the upregulation of p-p65 and degradation of IκBα were suppressed; total p65 (B) showed no significant difference across groups. These results demonstrate that G-Rg3 inhibits LPS-induced NF-κB pathway activation in an AMPK-dependent manner.
Finally, we verified the mechanism by which G-Rg3 exerts anti-inflammatory effects by autophagy (Figure S3): First, ATG5 knockdown (si-ATG5) effectively reduced ATG5 expression (A). In the Ctrl group, the ratio of the autophagy marker LC3-II/I (B) was low, while SQSTM1/p62 (D) expression was high. After LPS stimulation (LPS+Rg3 group), the LC3-II/I ratio increased, and SQSTM1/p62 expression decreased, indicating autophagy activation. However, after ATG5 knockdown (LPS+Rg3+si-ATG5 group), both the upregulation of LC3-II/I and downregulation of SQSTM1/p62 were reversed. Meanwhile, when combined with the AMPK inhibitor (LPS+Rg3+CC group, C, E), G-Rg3-induced increases in LC3-II/I and decreases in SQSTM1/p62 were also suppressed. These data suggest that G-Rg3 mediates anti-inflammatory effects by regulating ATG5-dependent autophagy via the AMPK pathway.
Rg3 Effectively Antagonizes the Activation of Autophagy Induced by LPS
G-Rg3, an active ginseng-derived component, exerts prominent regulatory effects on LPS-stimulated HBE cells. To clarify its function, we first treated HBE cells with 100 ng/mL LPS combined with gradient concentrations of G-Rg3 (0, 2, 4, 8, 16 µM) and detected autophagy-related protein expression as shown in Figure 2A–D. As presented in Figure 2A, compared with the LPS-only group (0 µM G-Rg3), G-Rg3 dose-dependently reduced the expression of ATG4B, ATG7, PIK3C3, and p62, with the most significant downregulation observed at 16 µM G-Rg3 (quantified in Figure 2C, P < 0.05). These results confirm that G-Rg3 effectively attenuates LPS-induced upregulation of autophagy-related proteins, thereby alleviating the pathological changes caused by excessive autophagy activation.
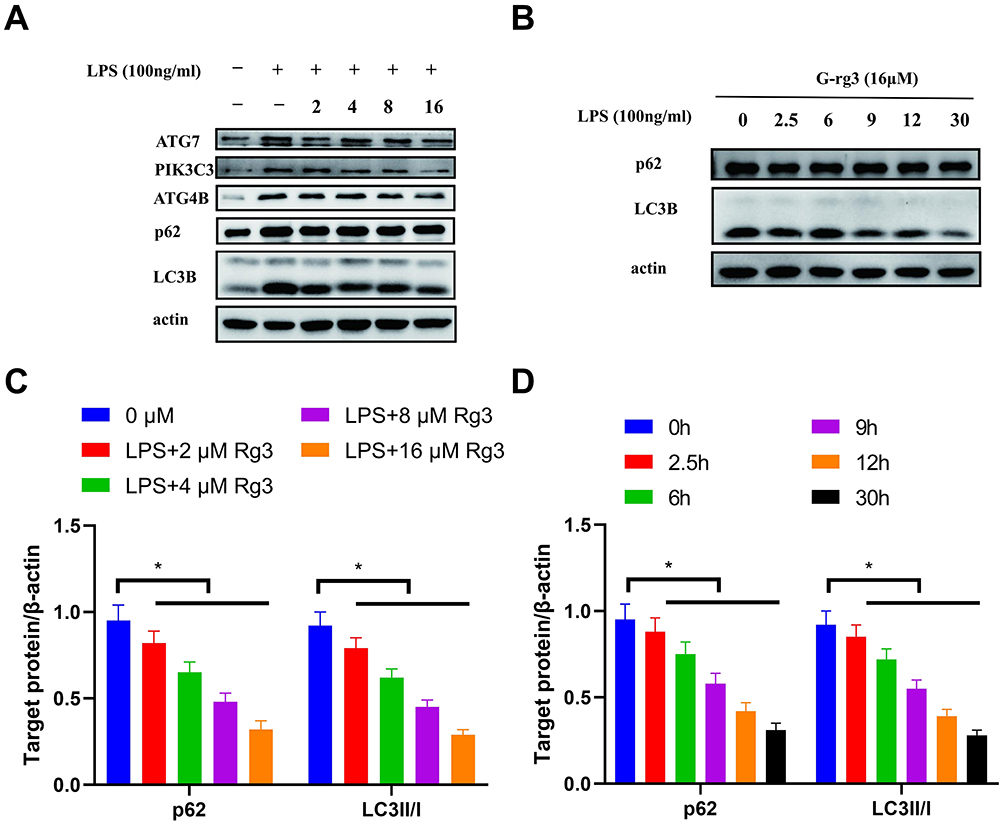 |
Figure 2 G-Rg3 Antagonizes LPS-Induced Autophagy in HBE Cells in Concentration- and Time-Dependent Manners. (A) Representative Western blot bands of ATG7, PIK3C3, ATG4B, p62, LC3B, and β-actin (loading control) in HBE cells treated with LPS (100 ng/mL) alone or co-treated with 2–16 μM G-Rg3. (B) Representative Western blot bands of p62, LC3B, and β-actin in HBE cells treated with LPS (100 ng/mL) + 16 μM G-Rg3 for 0–30 h. (C) Quantitative analysis of relative p62 and LC3B-II/I expression (normalized to β-actin) in LPS-treated cells with different G-Rg3 concentrations. (D) Quantitative analysis of relative p62 and LC3B-II/I expression (normalized to β-actin) in LPS + 16 μM G-Rg3-treated cells at different time points. Data are presented as mean ± SD (n = 3 independent experiments). *P < 0.05 vs the corresponding control group. |
To further explore the time-dependent effect of G-Rg3, we treated cells with 100 ng/mL LPS plus 16 µM G-Rg3 for varying durations (0, 2.5, 6, 9, 12, 30 h): Figure 2B (and its quantification in Figure 2D) shows that as treatment time extended, the expression levels of LC3B and p62 gradually decreased (with significant reductions at later time points, P < 0.05). This indicates that G-Rg3’s inhibitory effect on LPS-induced autophagy strengthens progressively with prolonged exposure.
Ginsenoside Rg3 Effectively Antagonizes Cell Death Induced by LPS-Mediated Autophagy Activation
To further investigate the effects of LPS on cellular viability and the subsequent impact of cotreatment with Rg3, we treated the cells with LPS, G-Rg3, or a combination of both for 24 hours. Following treatment, the cells were stained with propidium iodide (PI) dye, and the intensity of red fluorescence was observed via a fluorescence-inverted microscope. The results showed no significant increase in red fluorescence in G-Rg3-treated cells compared with the control group. In contrast, cells treated with LPS presented a marked increase in red fluorescence. In contrast, cotreatment with LPS and G-Rg3 significantly reduced red fluorescence intensity (Figure 3A). Flow cytometry further confirmed that the LPS group showed greater cell death than the control group. In contrast, the LPS+G-Rg3 group showed reduced cell death (Figure 3B). These findings underscore that LPS promotes cell death, whereas G-Rg3 effectively mitigates it.
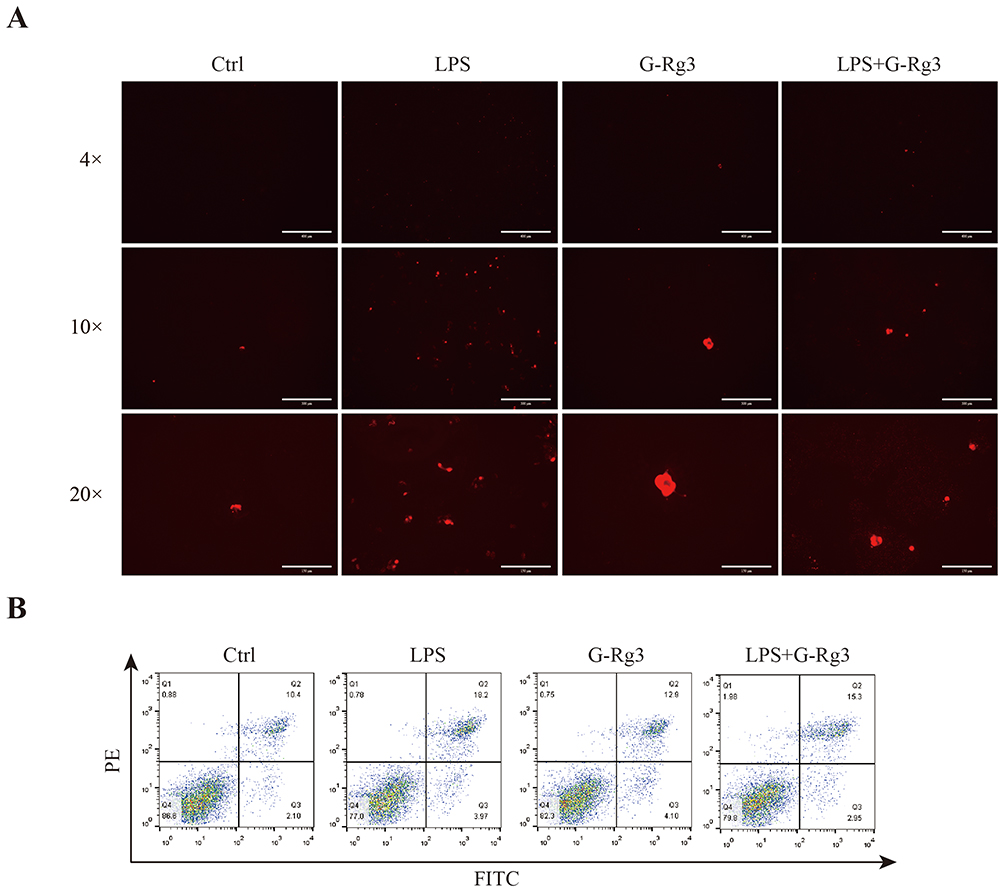 |
Figure 3 Ginsenoside Rg3 mitigates LPS-induced HBE cell death mediated by autophagy activation. (A) HBE cells were treated with LPS (100 ng/mL) alone, G-Rg3 (16 μM) alone, or their combination for 24 hours, followed by propidium iodide (PI) staining. Red fluorescence indicates cells with compromised membrane integrity (necrotic or late apoptotic cells); images were captured with an inverted fluorescence microscope at 4×, 10×, and 20× magnification. (B) After 24 hours of the same treatment as (A), HBE cells were stained with Annexin V-FITC/PI and analyzed by flow cytometry. Quadrant definitions: Q1 = necrotic cells (PI-positive/Annexin V-negative), Q2 = late apoptotic cells (PI-positive/Annexin V-positive), Q3 = viable cells (PI-negative/Annexin V-negative), Q4 = early apoptotic cells (PI-negative/Annexin V-positive). |
Rg3 Ginsenosides Inhibit the LPS-Induced Increase in the Levels of Inflammatory Factors in Cells
To elucidate the effects of G-Rg3 on the levels of inflammatory factors induced by LPS, we employed ELISA to compare the concentrations of these factors before and after G-Rg3 treatment in LPS-stimulated HBE cells. The results indicated that, compared with the control group, levels of proinflammatory factors TNF-α, IL-1β, IL-2, IL-6, and IL-8 were significantly elevated in the LPS group. Conversely, the G-Rg3+LPS group demonstrated significant downregulation of these factors (Figure 4A–E). Therefore, G-Rg3 effectively reduces the release of proinflammatory factors induced by LPS.
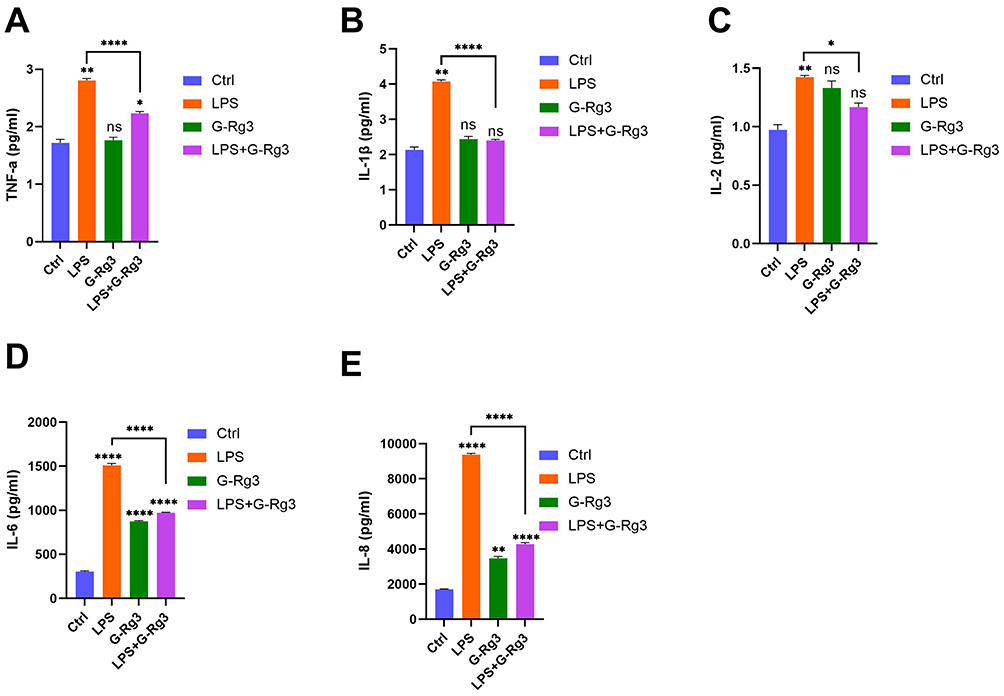 |
Figure 4 Effect of Ginsenoside Rg3 Rg3 (G-Rg3) on lipopolysaccharide (LPS)-induced inflammatory cytokine secretion. Cytokine concentrations (pg/mL) in four treatment groups (Ctrl: control; LPS: LPS stimulation; G-Rg3: G-Rg3 treatment; LPS+G-Rg3: LPS combined with G-Rg3) are shown for (A) tumor necrosis factor-α (TNF-α), (B) interleukin-1β (IL-1β), (C) interleukin-2 (IL-2), (D) interleukin-6 (IL-6), and (E) interleukin-8 (IL-8). Data represent mean ± standard error; statistical significance is indicated as *P < 0.05, **P < 0.01, ****P < 0.0001, while “ns” denotes no significant difference (P > 0.05). |
Discussion
Our study indicates that G-Rg3’s regulation of autophagy, inhibition of inflammation, and alleviation of cell death are not isolated parallel events but involve a coordinated interplay with both causal links and complementary independent mechanisms: First, LPS-induced impairment of autophagic flux acts as an upstream pathological driver—dysregulated autophagy not only disrupts cellular homeostasis but also promotes the overproduction of proinflammatory mediators and exacerbates cellular stress leading to apoptosis/necrosis, and G-Rg3’s restoration of autophagic balance (by downregulating ATG4B/ATG7/PIK3C3 and reducing p62/LC3B-II accumulation) directly mitigates this upstream trigger, thereby contributing to its anti-inflammatory and anti-cell death effects. Second, G-Rg3 also exerts an independent anti-inflammatory effect by inhibiting the LPS-activated TLR4/NF-κB signaling pathway to suppress the secretion of proinflammatory cytokines (TNF-α, IL-1β, IL-2, IL-6, IL-8)—a mechanism parallel to autophagy regulation and synergizing with it to enhance protection against HBE cell injury. Our experimental data support this: G-Rg3 simultaneously modulates autophagy-related protein expression, reduces cytokine levels, and improves cell survival, with the reduction in inflammation and cell death closely correlating with restored autophagic flux. At the same time, the inhibition of TLR4/NF-κB provides an additional anti-inflammatory layer independent of autophagy.
LPS is well known to induce the production of inflammatory cytokines, including TNF-α, IL-6, IL-1β, IL-2, and IL-8,21 which are involved in epithelial dysfunction and acute lung injury. Park et al also demonstrated that airway epithelial cells exposed to LPS upregulate TLR4 expression and activate the NF-kB and MAPK pathways, leading to a cytokine storm,22 findings similar to ours. Interestingly, treatment with G-Rg3 significantly inhibited these cytokines, presumably by crippling TLR4 downstream signaling. This finding is consistent with earlier observations that G-Rg3 prevents LPS-induced inflammatory responses in microglia by blocking NF-κB translocation.23 Together, these findings highlight the immunomodulatory potential of G‒Rg3 broadly across different cell types.
In addition, this study revealed that LPS-induced autophagy in HBE cells is not purely protective but rather pathogenic, especially when the autophagic flux is incomplete. In addition to disrupted autophagosome clearance, which has been reported in alveolar epithelial cells subjected to endotoxin stress,24 autophagy impairs p62 and LC3B-II accumulation. Incomplete autophagy may serve as a double-edged sword: it initially aims to degrade damaged components but, if hindered, eventually leads to inflammation and cell death.25,26 G Rg 3 treatment reduced the expression of autophagy-related markers, suggesting that G Rg 3 may not completely shut off autophagy but may restore its balance. Other models also show this fine-tuning effect; in renal tubular cells under diabetic conditions, G-Rg3 stimulates autophagosome-lysosome fusion.27
These experiments revealed that G-Rg3 significantly attenuated apoptosis, as determined by flow cytometry and PI staining. LPS-induced lung injury is associated with apoptosis in the airway epithelium, which is also tightly connected to increased oxidative stress and mitochondrial dysfunction.28 Although our study did not measure ROS levels or mitochondrial membrane potential, previous studies have shown that G-Rg3 reduces oxidative damage by enhancing antioxidant enzyme activity and mitochondrial integrity.29,30 G-Rg3 has been reported to reduce cytochrome c release and inhibit caspase activation in cardiac and hepatic tissues,31 which has been correlated with the preservation of cell viability. This is plausible; therefore, the oxidative and mitochondrial pathways involved in HBE cells should be closely investigated.
Moreover, the G-Rg3-mediated inhibition of the release of proinflammatory cytokines. TNF-α, IL-1β, and IL-6 are not only markers of inflammation but also act as components of tissue remodeling, fibrosis, and epithelial-mesenchymal transition (EMT).32 These cytokines are persistently elevated in chronic airway diseases (asthma and COPD).33 Interestingly, G-Rg3 treatment has been reported to decrease IL-6 and TGF-β in fibrotic lung models, suggesting that G-Rg3 may also help inhibit EMT and airway remodeling.34 Thus, G-Rg3 provides acute protection and has long-term therapeutic implications in chronic pulmonary pathology.
Additionally, G-Rg3 may affect signaling pathways beyond autophagy and inflammation. Recent reports suggest that G-Rg3 activates the Nrf2/ARE pathway, leading to the upregulation of antioxidant genes, including HO-1 and NQO1.35 The lungs rely on this pathway for cellular defense against oxidative stress. For example, Nrf2 activation has been linked to autophagy regulation, suggesting that G-Rg3 may act on two fronts by connecting molecular nodes.36 In addition, G-Rg3 modulates SIRT1 and AMPK, the opposing two energy sensors of autophagy, inflammation, and aging.37,38
Combined with the known multi-target pharmacological effects of G-Rg3, we speculate that its protective effects against LPS-induced HBE cell injury may be mediated by synergistic regulation of a complex signaling network involving potential molecular targets such as glucocorticoid receptors (GR), histone deacetylase 3 (HDAC3), and PI3K/AKT/mTOR pathways—known to be regulated by G-Rg3 in anti-tumor and anti-inflammatory contexts. Previous studies have confirmed that G-Rg3 can inhibit NF-κB activation, downregulate HDAC3 expression to enhance p53 acetylation, and modulate PI3K/AKT/mTOR signaling; notably, GR is closely intertwined with these pathways, as GR activation can suppress NF-κB-mediated inflammation and regulate autophagic flux through PI3K/AKT/mTOR. Given that G-Rg3 has been shown to exert anti-inflammatory effects by targeting NF-κB and modulating epigenetic modifications via HDAC3, we hypothesize that G-Rg3 may interact with GR to form a GR-NF-κB/HDAC3 regulatory axis, which not only explains its ability to restore impaired autophagic flux but also clarifies its inhibition of proinflammatory cytokine secretion. This speculation is consistent with G-Rg3’s known multi-target pharmacological characteristics. It establishes a deeper connection between our in vitro findings and the broader research context of G-Rg3, moving beyond mere repetition of experimental results to offer a more comprehensive molecular-mechanism hypothesis.
These findings are promising, yet we must concede to limitations. This was an in vitro study, eschewing the complexity of in vivo lung physiology (immune cell interactions, vascular permeability, and mechanical stress). Furthermore, the pharmacokinetics and lung bioavailability of G-Rg3 are poorly understood. Accordingly, G-Rg3 has been reported to have poor oral bioavailability due to poor solubility and fast metabolism.39 Yet, nanoparticle-based delivery systems currently hold promise for enhancing systemic absorption and targeting specific tissues.39 In vivo models of lung inflammation, future pharmacokinetic studies of G-Rg3, and investigations into whether G-Rg3 has synergistic effects with existing corticosteroids or bronchodilators should be the focus of future studies. A final limitation of this study is the insufficient depth of mechanistic research into the direct molecular targets of G-Rg3. Although existing evidence confirms that G-Rg3 regulates autophagy and inflammation to exert protective effects, the initiating mechanism of this regulatory role remains unclear—specifically, the direct binding partners of G-Rg3 and its upstream signaling pathways have not been experimentally verified.
Conclusion
This study finds that ginsenoside Rg3 (G-Rg3) alleviates lipopolysaccharide (LPS)-induced autophagic dysregulation, proinflammatory cytokine (TNF-α, IL-1β, IL-2, IL-6, IL-8) release, and cell death in human bronchial epithelial (HBE) cells. These results provide a preliminary basis for further exploring the role of G-Rg3 in respiratory inflammatory diseases.
Data Sharing Statement
All datasets used and analyzed in the current study are available on reasonable request from the corresponding authors Jing Zhou and Xingyu Tao.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Jiangxi Provincial Natural Science Foundation, China (Project No. 20202BABL216002) and the Administration of Traditional Chinese Medicine of Jiangxi Province, China (Project No. 2020A0382).
Disclosure
Xingyu Tao and Lingjiao Liu are co-first authors for this study. The authors declare no conflicts of interest in this work.
References
1. Ratan ZA, Haidere MF, Hong YH, et al. Pharmacological potential of ginseng and its major component ginsenosides. J Ginseng Res. 2021;45(2):199–13. doi:10.1016/j.jgr.2020.02.004
2. Yang W, Qiao X, Li K, Fan J, Bo T, Guo D. Identification and differentiation of Panax ginseng, Panax quinquefolium, and Panax notoginseng by monitoring multiple diagnostic chemical markers. Acta Pharm Sin B. 2016;6(6):568–575. doi:10.1016/j.apsb.2016.05.005
3. Kim J-H. Pharmacological and medical applications of Panax ginseng and ginsenosides: a review for use in cardiovascular diseases. J Ginseng Res. 2018;42(3):264–269. doi:10.1016/j.jgr.2017.10.004
4. Tian L, Shen D, Li X, Shan X, Wang X, Zhao J. Ginsenoside Rg3 inhibits epithelial-mesenchymal transition (EMT) and invasion of lung cancer by downregulating FUT4. Oncotarget. 2016;7(2):1619–1632. doi:10.18632/oncotarget.6451
5. Barnes PJ, Ware LB, Zimmerman GA. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Clin Invest. 2012;122(8):2731–2740. doi:10.1172/JCI60331
6. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1(2):135–145. doi:10.1038/35100529
7. Guo RF, Ward PA. Role of cytokines in septic shock. Crit Care Med. 2005;33(12 Suppl):S478–S481. doi:10.1097/01.ccm.0000191725.59611.14
8. Lam GY, Cemma M, Muise AM, Higgins DE, Brumell JH. Host autophagy proteins target Salmonella to the lysosome and clear infection in a murine typhoid fever model. Cell Host Microbe. 2013;13(6):759–768. doi:10.1016/j.chom.2013.05.003
9. Ichikawa A, Kuba K, Morita M, et al. CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am J Respir Crit Care Med. 2013;187(1):65–77. doi:10.1164/rccm.201203-0508OC
10. Lu L, Tian W, Ying X, Hongqing, C, Shan, Y, Xiaofei, S. Sea buckthorn extract mitigates chronic obstructive pulmonary disease by suppression of ferroptosis via scavenging ROS and blocking p53/MAPK pathways. J Ethnopharmacol. 2025;336(118726). doi:10.1016/j.jep.2024.118726
11. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. doi:10.1038/nature06639
12. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi:10.1016/j.cell.2007.12.018
13. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–737. doi:10.1038/nri3532
14. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi:10.1038/nature09663
15. Shin Y-M, Jung H-J, Choi W-Y, Lim C-J. Antioxidative, anti-inflammatory, and matrix metalloproteinase inhibitory activities of 20(S)-ginsenoside Rg3 in cultured mammalian cell lines. Mol Biol Rep. 2013;40(1):269–279. doi:10.1007/s11033-012-2058-1
16. Kang L-J, Choi Y-J, Lee S-G. Stimulation of TRAF6/TAK1 degradation and inhibition of JNK/AP-1 signaling by ginsenoside Rg3 attenuates hepatitis B virus replication. Int J Biochem Cell Biol. 2013;45(12):2612–2621. doi:10.1016/j.biocel.2013.08.016
17. Yang J, Li S, Wang L, Du F, Zhou X, Li C. Ginsenoside Rg3 attenuates lipopolysaccharide-induced acute lung injury via MerTK-dependent activation of the PI3K/AKT/mTOR pathway. Front Pharmacol. 2018;9:850. doi:10.3389/fphar.2018.00850
18. Xiaoying W, Lili C, Ting W, et al. Ginsenoside Rg3 antagonizes adriamycin-induced cardiotoxicity by improving endothelial dysfunction from oxidative stress via upregulating the Nrf2-ARE pathway through the activation of akt. Phytomedicine. 2015;22(10):875–84. doi:10.1016/j.phymed.2015.06.010
19. Zhang C, Yu H, Ye J, Tong H, Wang M, Zhao M. Ginsenoside Rg3 protects against diabetic cardiomyopathy and promotes adiponectin signaling via activation of PPAR-γ. Int J Mol Sci. 2023;24(13):16736. doi:10.3390/ijms242316736
20. Kim D, Yang KE, Kim DW, Hwang H, Kim J, Choi H. Activation of Ca2+-AMPK-mediated autophagy by ginsenoside Rg3 attenuates cellular senescence in human dermal fibroblasts. Clin Transl Med. 2021;11(1):e521. doi:10.1002/ctm2.521
21. Kee J-Y, Hong S-H. Ginsenoside Rg3 suppresses mast cell–mediated allergic inflammation via mitogen-activated protein kinase signaling pathway. J Ginseng Res. 2019;43(2):282–290. doi:10.1016/j.jgr.2018.02.008
22. Kim HS, Kim DH, Kim BK, et al. Effects of topically applied Korean red ginseng and its genuine constituents on atopic dermatitis-like skin lesions in NC/Nga mice. Int Immunopharmacol. 2011;11(2):280–285. doi:10.1016/j.intimp.2010.11.022
23. Bae E-A, Han MJ, Shin Y-W, Kim D-H. Inhibitory effects of Korean red ginseng and its genuine constituents ginsenosides Rg3, Rf, and Rh2 in mouse passive cutaneous anaphylaxis reaction and contact dermatitis models. Biol Pharm Bull. 2006;29(9):1862–1867. doi:10.1248/bpb.29.1862
24. Xiong J, Yuan H, Fei S, et al. The preventive role of the red ginseng ginsenoside Rg3 in the treatment of lung tumorigenesis induced by benzo(a)pyrene. Sci Rep. 2023;13:4528. doi:10.1038/s41598-023-31710-9
25. Donna ED. The role of the epithelium in airway remodeling in asthma. Proc Am Thorac Soc. 2009;6(8):678–82. doi:10.1513/pats.200907-067DP
26. Yingfang W, Zhouyang L, Lingling D, et al. Inactivation of MTOR promotes autophagy-mediated epithelial injury in particulate matter-induced airway inflammation. Autophagy. 2020;16(3):435–450. doi:10.1080/15548627.2019.1628536
27. Yan G, Jiaqing Y, Juntong L, et al. Ginsenoside Rg3 ameliorates acetaminophen-induced hepatotoxicity by suppressing inflammation and oxidative stress. J Pharm Pharmacol. 2021;73(3):322–331. doi:10.1093/jpp/rgaa069
28. Chunzhou T, Keran L, Qing Y, et al. Activation of Nrf2 by Ginsenoside Rh3 protects retinal pigment epithelium cells and retinal ganglion cells from UV. Free Radic Biol Med. 2018;117:238–246. doi:10.1016/j.freeradbiomed.2018.02.001
29. Kang J, Lee J, Lee J, et al. Autophagic dysfunction contributes to LPS-induced acute lung injury. J Inflam. 2022;19(1):2. doi:10.1186/s12950-022-00299-7
30. Janice K, Kianfan C, Charalambos M, et al. The role of mitochondria in eosinophil function: implications for severe asthma pathogenesis. Front Cell Develop Biol. 2024;12:
31. Tomasz MS, Marcin KK, et al. Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radic Biol Med. 2011;50(9):1186–95. doi:10.1016/j.freeradbiomed.2011.01.033
32. Li Y, Zhao Y, Feng X, Hu Y, Li X. Airway epithelial apoptosis in LPS-induced acute lung injury. Respir Res. 2020;21(1):175. doi:10.1186/s12931-020-01423-y
33. Yuan S, Bo L, Wei M, et al. PGE2 downregulates LPS-induced inflammatory responses via the TLR4-NF-κB signaling pathway in bovine endometrial epithelial cells. Prostaglandins Leukot Essent Fatty Acids. 2018;129(25):25–31. doi:10.1016/j.plefa.2018.01.004
34. Yuan S, Bo L, Wei M, et al. PGE2 downregulates LPS-induced inflammatory responses via the TLR4-NF-κB signaling pathway in bovine endometrial epithelial cells. Prostaglandins Leukot Essent Fatty Acids. 2018;129(25):25–31. doi:10.1016/j.plefa.2018.01.004
35. Peng J, Dong S, Xie Q, et al. Ginsenoside Rg3 attenuates pulmonary fibrosis via inhibition of TGF-β/Smad signaling. J Ethnopharmacol. 2022;284:114781. doi:10.1016/j.jep.2021.114781
36. Erjin W, Xingxuan H, Ming, Z. The Role of S1P and the Related Signaling Pathway in the Development of Tissue Fibrosis. Front Pharmacol. 2019;9:1504. doi:10.3389/fphar.2018.01504
37. Sun Z, Zhang X, Wang J, et al. Ginsenoside Rg3 restores autophagic balance in diabetic nephropathy. Phytomedicine. 2023;112:154712. doi:10.1016/j.phymed.2023.154712
38. Chen G, Qingxi H, Yisa W, et al. Therapeutic application of natural products: NAD+ metabolism as potential target. Phytomedicine. 2023;114:154768. doi:10.1016/j.phymed.2023.154768
39. Zhigang R, Xinmei C, Liangjie H, et al. Nanoparticle Conjugation of Ginsenoside Rg3 Inhibits Hepatocellular Carcinoma Development and Metastasis. Small. 2020;16(2):e1905233. doi:10.1002/smll.201905233
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
STK11 Alleviates Pulmonary Inflammation During Acute Lung Injury by Phosphorylating AMPK to Activate Autophagy in A549 Cell Model
Xiao R, Qi Z, Chen T, Wei F
Journal of Inflammation Research 2025, 18:17789-17802
Published Date: 19 December 2025