Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
Ginseng Saponin Rb1 Attenuates Cigarette Smoke Exposure-Induced Inflammation, Apoptosis and Oxidative Stress via Activating Nrf2 and Inhibiting NF-κB Signaling Pathways
Authors Li Z, Li L, Lv X, Hu Y, Cui K
Received 10 May 2023
Accepted for publication 17 August 2023
Published 28 August 2023 Volume 2023:18 Pages 1883—1897
DOI https://doi.org/10.2147/COPD.S418421
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Zhizheng Li,1 Li Li,1 Xiaohui Lv,1 Yingqian Hu,1 Kun Cui2
1Department of Respiratory and Critical Care Medicine, Tangshan Gongren Hospital, Tangshan, People’s Republic of China; 2Respiratory Medicine, Tangshan Gongren Hospital, Tangshan, People’s Republic of China
Correspondence: Kun Cui, Email [email protected]
Objective: Cigarette smoke exposure is one of the major risk factors for the development of chronic obstructive pulmonary disease (COPD). Ginseng saponin Rb1 (Rb1) is a natural extract from ginseng root with anti-inflammatory and anti-oxidant effects. However, the underlying mechanism of the Rb1 in COPD remains unknown. Therefore, we sought to explore the role of Rb1 in cigarette smoke-induced damage and to reveal the potential mechanism.
Methods: The cell viability and lactose dehydrogenase (LDH) activity were analyzed using cell counting kit-8 (CCK-8) and LDH release assays. We further investigated the inflammation, apoptosis and oxidative stress markers and analyzed the nuclear factor-kappa B (NF-κB) and nuclear factor erythroid-2-related factor 2 (Nrf2) pathways in BEAS-2B cells and COPD rat model following cigarette smoke extract (CSE) exposure.
Results: Our results showed that CSE promoted inflammation, apoptosis and oxidative stress in BEAS-2B cells. Rb1 suppressed the inflammatory response by inhibiting expression of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and IL-1β and inhibiting the NF-κB signaling pathway. Rb1 possessed the ability to hinder cell apoptosis induced by CSE. In addition, Rb1 concurrently reduced CSE-induced oxidative reactions and promoted Nrf2 translocation to nucleus. For in vivo study, Rb1 treatment alleviated CSE-induced lung injury, apoptosis, reactive oxygen species (ROS) release and inflammatory reactions. Also, Rb1 treatment activated Nrf2 signaling and inactivated NF-κB signaling in COPD rats.
Conclusion: Rb1 attenuates CSE-induced inflammation, apoptosis and oxidative stress by suppressing NF-κB and activating Nrf2 signaling pathways, which provides novel insights into the mechanism underlying CSE-induced COPD.
Keywords: ginseng saponin Rb1, cigarette smoking exposure, inflammation, oxidative stress, NF-κB, Nrf2
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive chronic lung disease that is characterized by progressive and airflow obstruction.1 COPD is a common disease and a major cause of morbidity and mortality worldwide.2 As the population increases, the environment deteriorates, and the problem of aging becomes more pronounced, the number of people with COPD is also increasing.3 Oxidative stress and inflammation are the crucial hallmarks of COPD development. Lung inflammatory and structural cells represent sources of endogenous oxidants that enhance inflammatory gene expression, which involve in the pathogenesis of COPD.4 Although recent studies have provided insights into the pathogenesis of COPD, there are currently no effective treatments that can fully reverse the damage to the lungs caused by the disease. Therefore, it is urgent to investigate novel therapeutic targets and develop novel anti-COPD drugs.
Cigarette smoke exposure is widely accepted to be a major reason for the pathogenesis and progression of COPD, which contributes to the excessive inflammatory response in airways, alveoli, and microvasculature.5 Patients with COPD showed increased levels of pro-inflammatory cytokines such as interleukin-23 (IL-23), interleukin-1β (IL-1β) and interleukin-6 (IL-6).6 Increasing evidence suggested that NF-κB (nuclear factor-kappa B) is closely relevant to COPD occurrence.7,8 NF-κB is an important transcription factor, which plays a key role in the regulation of the stress-associated immune response and inflammation.9 When a cell receives a stimulus from the outside environment, inhibitor of NF-κB (IκBs) would be phosphorylated and degraded, and subsequent the NF-κB translocates to the nucleus, where it regulates the expression of inflammatory cytokines.10 Emerging evidence has manifested that tobacco smoke can lead to oxidative stress, thereby affecting airway remodeling under certain pathological conditions in COPD.11 Nuclear factor erythroid-2-related factor 2 (Nrf2) is one of the critical regulators of the cellular response to defend against oxidative stress.12 Under oxidative stress conditions, Nrf2 is isolated from Kelch-like ECH-associated protein 1 (Keap1) and translocate into the nucleus from the cytoplasm.13 Upon entering the nucleus, Nrf2 regulates the expressions of downstream antioxidant enzymes by binding to the antioxidant response element (ARE).14 Sussan et al15 reported that Nrf2-ARE pathway related antioxidant system was impaired in tobacco smoke-induced COPD mice model.
Ginsenoside Rb1 is the major bioactive component in ginseng, which exhibited potential pharmacologic and immunologic effects.16,17 Increasing evidence suggested that Rb1 has the potential anti-inflammatory and antioxidative effects.18 Zhou et al reported that Rb1 pretreatment alleviated TNF-α-induced inflammatory injury in endothelial cells by inhibiting NF-κB, JNK and p38 Signaling Pathways.19 Apoptosis and mitochondrial dysfunction of muscle stem cells can be attenuated by Rb1 through the activation of NF-κB signaling pathway.20 In addition, Rb1 effectively alleviates triptolide-induced cytotoxicity in HL-7702 cells through activation of the Keap1/Nrf2/ARE antioxidant pathway.21 Based on this, it is speculated that Rb1 might possess anti-inflammatory and antioxidative effect which might be mediated by NF-κB and Nrf2 pathways. To further confirm this hypothesis, we investigated the effect of Rb1 against cigarette smoke extract (CSE)-induced cytotoxicity in BEAS-2B cells. We also examined the inflammation, oxidative stress, and apoptosis markers in BEAS-2B cells. Additionally, we assessed the Nrf2 and NF-κB pathways to better understand the mechanism behind the anti-inflammatory and antioxidative effect of Rb1 on CSE-induced BEAS-2B cells. Furthermore, in vivo experiments were conducted to demonstrate the effects of Rb1 on CSE-induced inflammation, oxidative stress, and apoptosis as well as Nrf2/and NF-κB signaling pathways in COPD rats.
Materials and Methods
Preparation of Cigarette Smoke Extracts (CSE)
Two unfiltered commercial cigarettes (Beidaihe filter-tipped cigarettes; Zhangjiakou Cigarette Factory, Zhangjiakou, China), each containing 0.8 mg of nicotine and 10 mg of tar per cigarette, were burned and used to collect smoke into 10 mL Dulbecco’s modified Eagle’s medium (DMEM; Gibco, USA) at a speed of 2 min per cigarette. The medium was sterilized using a 0.22 μm filter. This solution was considered as a 100% CSE solution and stored at −80°C for subsequent experiments. This CSE was diluted with medium to different concentration for study.
Cell Culture
Human bronchial epithelial cells (BEAS-2B) were purchased from ATCC and cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (Gibco; USA) and supplement with 1% penicillin/streptomycin. Cells were maintained at 37°C in a humidified atmosphere with 5% CO2 at 37°C. The cells were grown to approximately 90% confluence before experiments.
Drug Administration
The Rb1 was purchased from Shanghai Ye Yuan Biotechnology Co. Ltd. (Shanghai, China) and the chemical structures of the compounds are shown in Figure 1A. To assess the toxicity effects of Rb1 on cells, cells were exposed to different concentrations (1μM, 5μM, 10μM, 20μM) of Rb1 for 6h. According to the results of the cytotoxicity assay, BEAS-2B cells were pretreated with the non-cytotoxic Rb1 doses for 6h, and then performed CSE treatment. Once the optimal treatment concentration had been determined of Rb1, cells were divided into the following four groups for the experiment: control group, Rb1 group, CSE group and CSE+Rb1 group.
 |
Figure 1 Rb1 ameliorated CSE-induced cell viability and LDH leakage. (A) Structures of Rb1. (B) Cell viability of BEAS-2B cells treated with different concentration of CSE. (C) Cell viability of BEAS-2B cells treated with different concentration Rb1. (D) Rb1 attenuated the CSE-induced decrease in cellular viability in BEAS-2B cells. (E) Rb1 attenuated the release of LDH induced by CSE. *P<0.05, **P<0.01. Abbreviation: ns, not significant. |
Animals
Twenty-eight male Sprague-Dawley rats (weighted 160 ± 10 g) were purchased from Beijing Weitong Lihua Experimental Animals Co. LTD (Beijing, China) and used in the controlled condition at 22±2°C with 50–60% humidity under 12-h light/dark cycle. Animals were acclimated for 7 days with free access to food and water. The rats were randomly divided into control, Rb1, COPD, and COPD+Rb1 groups (n = 7 in each group).
COPD Model Establishment
The COPD rat model was established by cigarette smoke exposure combined with endotracheal injection of lipopolysaccharide (LPS) as described previously.22 Briefly, on day 1 and day 14, rat was instilled 200μg LPS (0.4 μg/μL, 50 μL) through the throat of rats after anesthesia with 5% isoflurane. To ensure uniform distribution of LPS in the lungs, the rats were gently swayed on a board for 20 seconds after each instillation. Subsequently, on days 2 to 13 and 29 to 30, the rats were confined in a covered box (70 cm × 60 cm × 60 cm) and exposed to the smoke of eight continuously burning commercial cigarettes (Beidaihe filter-tipped cigarettes; Zhangjiakou Cigarette Factory, Zhangjiakou, China) for a duration of 30 minutes each time. The rats in the control group and Rb1 group were intratracheally injected with an equal amount of sterile water at the same time point, and they were also exposed to air using a similar procedure. In the Rb1 group and COPD+Rb1 group, Rb1 (20 mg/kg) was intraperitoneally injected from the 15th day and up to day 28.23 Rats in the Control and COPD group were intraperitoneally given an equal volume of saline. At the end of experiments, rats were sacrificed by dislocation and lung tissues were immediately collected for further experiments.
Pathological Examination
Pathological examination was performed by haematoxylin eosin (HE) staining in lung tissues. Lung tissues were immerged in 4% paraformaldehyde (Beyotime, China) for fixation and embedded in paraffin. Slides with 5-μm thickness were cut, then deparaffinated with xylene and rehydrated with descending concentration of ethanol (100%, 95%, 80%, and 70% for 2 min, respectively). Next, slides were stained with haematoxylin solution for 5 min and with 0.5% eosin (Beyotime, China) for 1 min. Pathological changes of lung tissues were evaluated under a microscope (Nikon, Japan).
Cell Viability Assay
Cell viability assay was conducted using the Cell Counting Kit-8 (CCK-8). Briefly, Beas-2B cells were seeded into 96-well plates and placed in an incubator at temperature of 37°C with 5% CO2 and later incubated with CSE and at different concentrations. Then, 10 μL CCK-8 solutions was added to the cells and incubated for 1 h in the dark. Finally, the absorbance values were measured at 450 nm by a microplate reader (Thermo Fisher).
Lactate Dehydrogenase (LDH) Assay
BEAS-2B cells were plated onto a 96-well plate and then exposed to varying concentrations of Rb1. The LDH cytotoxicity assay kit (Dojindo Laboratories, Japan) was employed to examine the levels of LDH in accordance with the manufacturer’s instructions. Immediately afterward, the optical densities at 490 nm were measured by means of a microplate reader in order to gauge absorbance.
Quantitative Real-Time PCR (qRT-PCR)
TRIzol (Tiangen, China) was used to extract total RNA from the cells or lung tissues. Next, the total RNA was reverse transcribed using a reverse transcription kit (Tiangen, China) following the manufacturer’s instructions. qRT-PCR was carried out in a CFX96 instrument (Biorad, USA) using the SuperReal PreMix (SYBR Green) qRT-PCR kit (Tiangen, China). The expression levels were analyzed using the 2-ΔΔCt method after normalization to β-actin. The qRT-PCR primers can be found in Table 1.
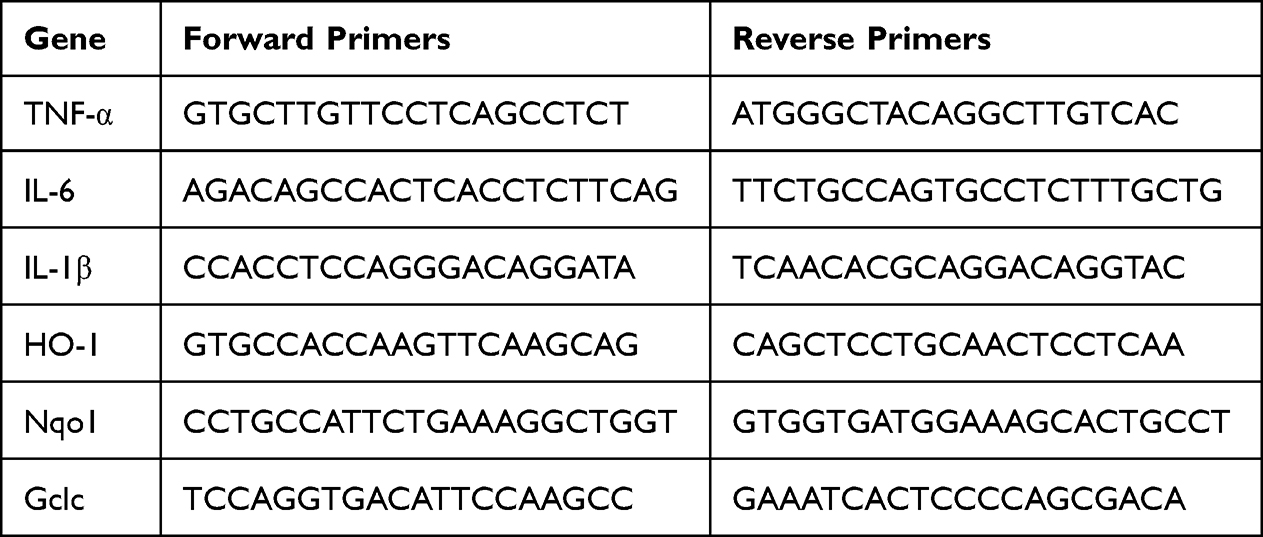 |
Table 1 The Primer Sequences Used for qPCR Analysis |
Western Blot
Total protein was extracted from cells or lung tissues using RIPA lysis, and concentration was determined via BCA assay kit. Equal amounts of protein were then separated via SDS-PAGE and transferred to a PVDF membrane. Blots were incubated with primary antibodies overnight. The primary antibodies are as follows: anti-Cleaved Caspase-3 (1:500; ab2302; Abcam), anti-Bax (1:500; ab32503; Abcam), anti-Bcl-2 (1:500; ab182858; Abcam), anti-β actin (1:500; ab8226; Abcam), anti-Nrf2 (1:500; ab137550; Abcam), anti-Histone H3 (1:500; ab1791; Abcam), p-IκBα (1:500; sc-8404 Santa Cruze), IκBα (1:500; ab32518; Abcam), and NF-κB (1:500; 10,745-1-AP; Proteintech). HRP-conjugated goat anti-rabbit IgG (1:1000; 7074; Cell signaling) was used as secondary antibody. β-actin and Histone H3 was used as the internal reference and quantification of the band densities was obtained using the ImageJ software.
Measurement of Intracellular Reactive Oxygen Species (ROS) Levels
The level of intracellular ROS was measured using the fluorescent probe DCFH-DA and Dihydroethidium (DHE) staining. For DCFH-DA testing, cells were seeded onto a 6-well plate for 24 h. Following drug treatment, cells were incubated with 2 μM DCFH-DA in the absence of light at a temperature of 37°C for 20 min. Further, cells were washed twice using cold PBS. For DHE staining, cells were rinsed twice using PBS, incubated with 2µM DHE at 37°C for 30 min, and then washed with PBS and mounted with DAPI. In lung tissues, oxidative stress was assessed by measuring the intracellular ROS via staining with DHE as previously reported.24 Lastly, fluorescence images of cells were observed using fluorescence microscopy (Olympus, Japan).
Biochemical Analysis
The level of malondialdehyde (MDA) content was determined through employment of the thiobarbituric acid reactive substances (TBARS) assay. Additionally, the enzyme activities of superoxide dismutase (SOD) and catalase (CAT) were appraised using colorimetric kits, respectively (Nanjing Jiancheng Bioengineering Institute, China). The specific processes entailed in each assay were performed in conformance with the instructions in the corresponding assay kits.
Terminal Deoxynucleotidyl Transferase Mediated dUTP Nick-End Labeling (TUNEL) Staining
TUNEL staining was used to detect the cell apoptosis. In brief, cells were fixed via utilization of a 4% formaldehyde solution and permitted to remain so for duration of 30 min following culture. For lung tissue, slides were digested using proteinase K (20 µg/mL, Sigma) at 37°C for 1 h after dewaxing with xylene and rehydration with descending concentrations of ethanol. Subsequently, cells and slides were added with TUNEL reagent (Roche, Shanghai, China) and incubated at a temperature of 37°C for 1 h. After that, the nuclei were counterstained employing DAPI (Beyotime, Shanghai, China) and cells were washed utilizing PBS solution. Lastly, apoptotic cells were observed with a fluorescent microscope (Olympus, Japan).
Statistical Analysis
The data were presented as mean ± SEM, and the statistical analyses were conducted using SPSS 23.0 software (IBM Corporation, Armonk, NY, USA). To determine the intergroup differences, one-way ANOVA followed by Duncan’s multiple range tests was performed. Significance was set at P<0.05.
Results
Effects of CSE and Ginseng Saponin Rb1 on Cell Viability
To verify the effect of different CSE or Rb1 doses on the viability of BEAS-2B cells, and CCK-8 assay was performed. CCK-8 assay revealed that BEAS-2B cell viability was decreased markedly by CSE treatment in a concentration-dependent manner and reduced to ∼58% at 6% concentration (Figure 1B). We next sought to validate the cytotoxic of Rb1 on BEAS-2B cells. Administration of Rb1 at concentrations of 1, 5, and 10 µM exhibited no cytotoxicity, whereas a higher concentration at 20 µM significantly increased cell viability (Figure 1C). In addition, we found that Rb1 (1, 5 and 10 μM) significantly enhanced CSE-induced BEAS-2B cell viabilities, partly in a dose-specific manner (Figure 1D). Moreover, in the LDH release assays, there was a markedly suppressed of Rb1 treatment (Figure 1E). Based on the results, Rb1 treatment at 10 μM was used for further assays.
Rb1 Abolished CSE-Induced Inflammation Responses in BEAS-2B Cells
Inflammation plays an important role in the progression of COPD. In the present study, we evaluated the effect of Rb1 on CSE-induced inflammation. As shown in Figure 2A–C, CSE significantly increased the gene expression, including TNF-α, IL-6, and IL-1β compared with control group, while its increasing effect attenuated significantly by Rb1 treatment. Then, the secretion levels of inflammatory factors were measured via ELISA. The content of TNF-α, IL-6, and IL-1β were rapidly increased in response to the CSE treatment compared with the levels found in the control group. Interestingly, Rb1 treatment significantly attenuated the CSE-induced elevation of inflammation factors level in BEAS-2B cells (Figure 2D–F).
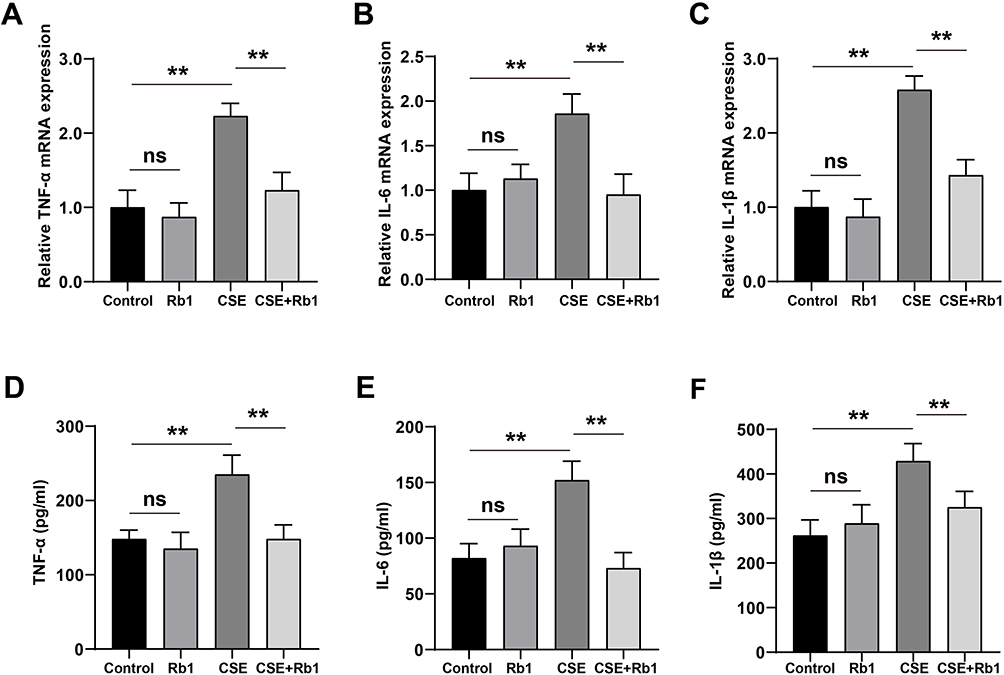 |
Figure 2 Rb1 ameliorated CSE-induced inflammation in BEAS-2B cells. (A) Relative mRNA expression level of TNF-α. (B) Relative mRNA expression level of IL-6. (C) Relative mRNA expression level of IL-1β. (D) The concentration of TNF‐α in BEAS-2B cells detected via ELISA. (E) The concentration of IL-6 in BEAS-2B cells detected via ELISA. (F) The concentration of IL-1β in BEAS-2B cells detected via ELISA. **P<0.01. Abbreviation: ns, not significant. |
Rb1 Reduced CSE-Induced Cell Apoptosis in BEAS-2B Cells
We further focused on the apoptotic to understand the possible mechanisms of BEAS-2B cells apoptosis after Rb1 treatment. The protein expressions of cleaved-Caspase-3, Bcl2 and Bax were first performed by Western blot. As shown in Figure 3A–C, CSE significantly increased Cleaved-Caspase-3 protein expression and decreased the ratio of Bcl2/Bax compared with control group, whereas Rb1 pre-treatment decreased the expression of Cleaved-Caspase-3 and increased the ratio expression of Bcl2/Bax compared with CSE-treated cells. To further investigate the impact of Rb1 on the apoptosis of CSE-induced BEAS-2B cells, we performed flow cytometry analysis using Annexin V-FITC. In comparison with the control group, the rate of apoptosis was significantly increased in the CSE group, however, treatment with Rb1, the rate of apoptosis induced by CSE was reduced from 12% to 7% (Figure 3D and E). Additionally, our results indicated that the apoptotic BEAS-2B cells were markedly increased after CSE, whereas this increase in CSE-induced TUNEL-positive was attenuated by Rb1 administration (Figure 3F and G).
 |
Figure 3 Rb1 ameliorated CSE-induced apoptosis in BEAS-2B cells. (A) Western blot was used to detect the effects of Rb1 on cleaved caspase-3, Bax and Bcl-2 protein expression. (B) Histogram of cleaved caspase-3 protein expression. (C) Histogram of Bcl-2 / Bax protein ratio. (D) Flow cytometry analysis of apoptosis. (E) Apoptosis rate of BEAS-2B cells. (F) TUNEL staining was used to assess the effects of Rb1 on the apoptosis of BEAS-2B cells. (G) Percentage of TUNEL positive cells. **P<0.01. Abbreviation: ns, not significant. |
Rb1 Inhibited CSE-Induced Oxidative Stress in BEAS-2B Cells
To investigate the effects of Rb1 against oxidative stress induced by CSE in BEAS-2B cells, the ROS levels were first measured by DCFH-DA staining. We found that the cells treated with CSE exhibited brighter green fluorescence than control cells, implying that CSE treatment led to an increase in intracellular ROS levels. However, Rb1 significantly reduced the fluorescence intensity of ROS induced by CSE (Figure 4A and B). In addition, DHE staining was further used to determine the levels of ROS in BEAS-2B cells. As shown in the Figure 4C and D, Rb1 also reduced the CSE-induced ROS production as evidenced by the DHE immunofluorescence. Next, we determined MDA content as well as SOD and CAT activities in BEAS-2B cells. We found that CSE stimulation significantly enhanced MDA levels in BEAS-2B cells compared with control group (P<0.01), and this effect was reversed by Rb1 treatment (Figure 4E). Meanwhile, CSE stimulation reduced SOD and CAT expression in BEAS-2B cells compared with control group and these treads were similar reversed by Rb1 exposure (Figure 4F and G).
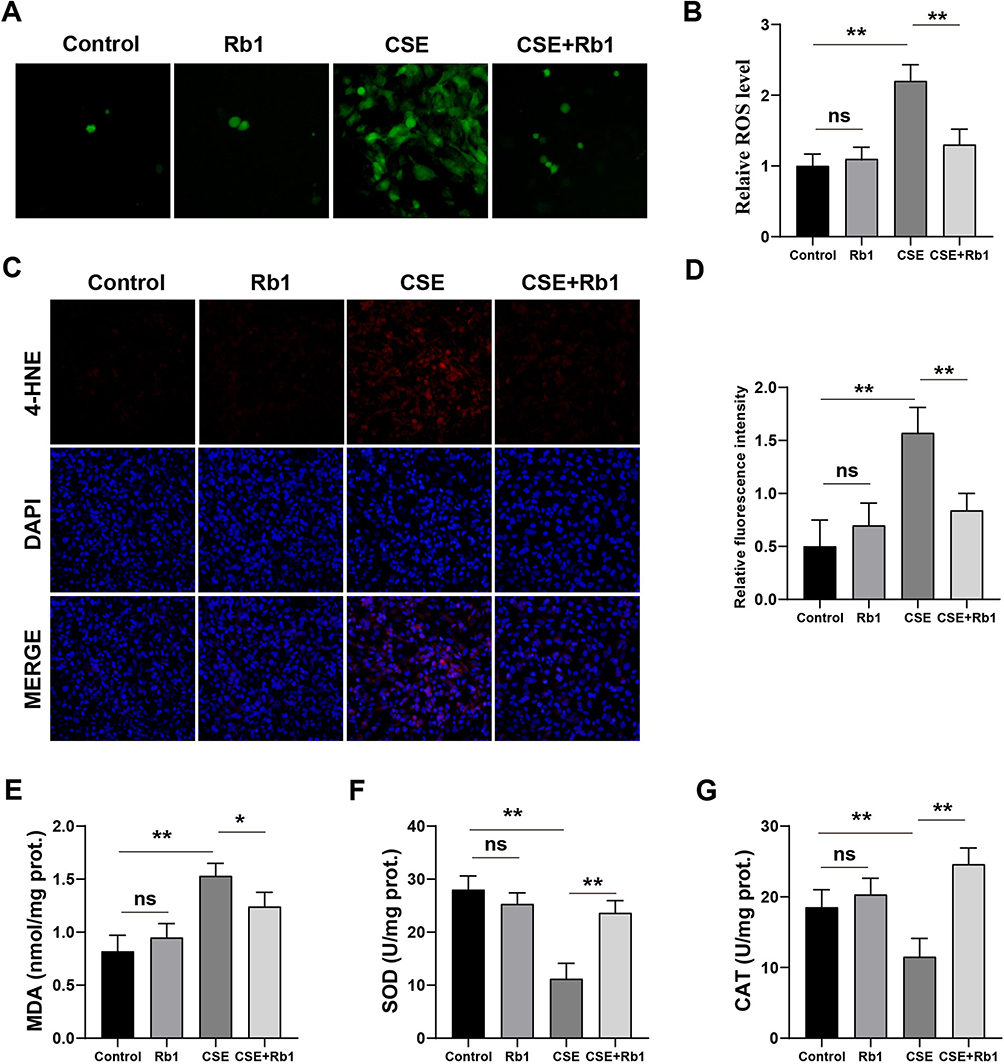 |
Figure 4 Rb1 ameliorated CSE-induced oxidative stress in BEAS-2B cells. (A) ROS was measured using DCFH-DA fluorescence probe in BEAS-2B cells after Rb1 treatment. (B) Quantification of intensity of the DCFH-DA fluorescence. (C) Representative images of DHE staining. (D) Quantitative analysis of DHE-positive cells. (E–G) The levels of MDA, SOD and CAT. *P<0.05, **P<0.01. Abbreviation: ns, not significant. |
Rb1 Activated Nrf2 and Inhibited NF-κb Signaling Pathways in BEAS-2B Cells
To explore the potential anti-inflammatory mechanisms of Rb1, we used Western blot analysis to determinate the levels of IκB phosphorylation and nuclear translocation of NF-κB in BEAS-2B cells. CSE significantly induced the phosphorylation of IκB compared with control group, resulting in the degradation of total IκB and the increased phosphorylation of NF-κB, while treatment with Rb1 inhibited the phosphorylation of IκB (Figure 5A) and NF-κB nuclear translocation induced by CSE (Figure 5B and C). In order to investigate the antioxidant mechanism of Rb1, we measured the levels of Nrf2 nucleus transport in the BEAS-2B cells. As shown in Figure 5D–F, compared with the control group, the levels of Nrf2 in cytoplasm and nucleus from the Rb1 group were no significantly difference. However, CSE led to a significant reduction in the expression of Nrf2 in the nucleus than that of control group, while treatment with Rb1 increased the nuclear translocation of Nrf2 compared with CSE-treated cells. Then, the mRNA levels of Nrf2 target genes NADPH quinone dehydrogenase 1 (Nqo1), heme oxygenase 1 (HO1) and glutamate-cysteine ligase catalytic subunit (Gclc) were evaluated by qPCR. Consist with the expression levels of Nrf2 in the nucleus, a significantly decreased trend of HO-1, NQO-1 and Gclc expression was observed in the CSE group compared with control group (P<0.01), whereas the expression of these genes was significantly increased in the Rb1 treatment group compared with CSE group (P<0.01) (Figure 5G–I).
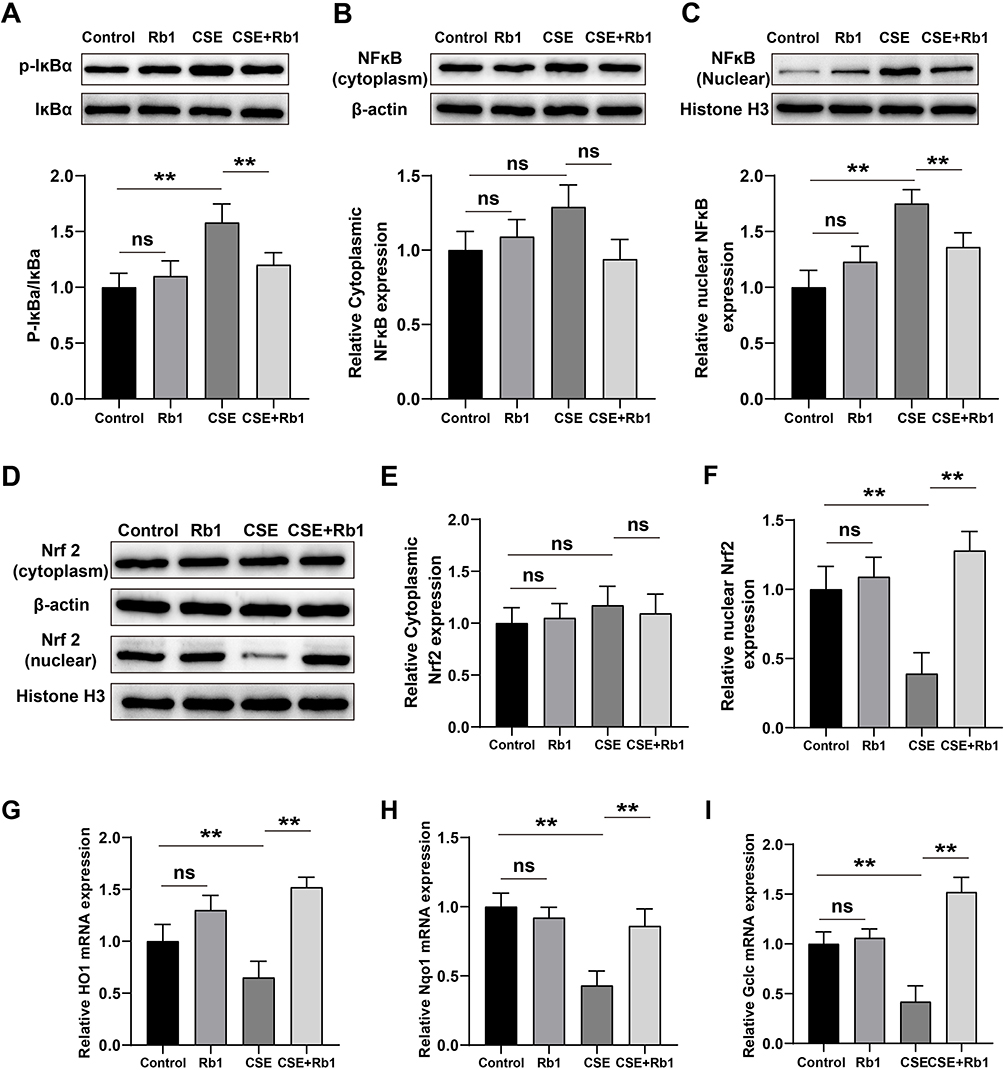 |
Figure 5 Rb1 abolished the effects of CSE on NF-κB/Nrf2 signaling pathways in BEAS-2B cells. (A) Representative blots and statistical graphs of relative protein expression of p-IκBα and IκBα. (B and C) Representative blots and statistical graphs of relative protein expression of NF-κB in cytoplasm and nucleus. (D), Representative blots of Western blot for cytoplasmic and nuclear Nrf2. (E and F) Statistical graphs of cytoplasmic and nuclear Nrf2 protein. (G–I), Relative mRNA expression levels of HO-1. Nqo1 and Gclc. **P<0.01. Abbreviation: ns, not significant. |
Rb1 Alleviated CSE-Induced Lung Injury, Apoptosis, Oxidative Stress and Inflammation in Rats
To investigate the effect of Rb1 on CSE-induced COPD, the rat COPD model was established and pathological changes of lung tissues were evaluated by HE staining. As shown in Figure 6A, rats in COPD group exhibited significant enlarged alveolar air spaces and lung parenchyma destruction compared with rats in control group and Rb1 group; while Rb1 treatment remarkably alleviated lung injury in COPD rats. The results from TUNEL deciphered that CSE promoted cell apoptosis, whereas treatment of Rb1 reduced apoptotic cells (Figure 6B). Also, the treatment of Rb1 significantly abolished CSE-induced ROS release (Figure 6C). Additionally, injection of CSE extremely enhanced the accumulation of TNF-α, IL-6, and IL-1β compared with control group; application of Rb1 reversed the pro-inflammatory effects of CSE by inhibiting the production of TNF-α, IL-6, and IL-1β (Figure 6D–F).
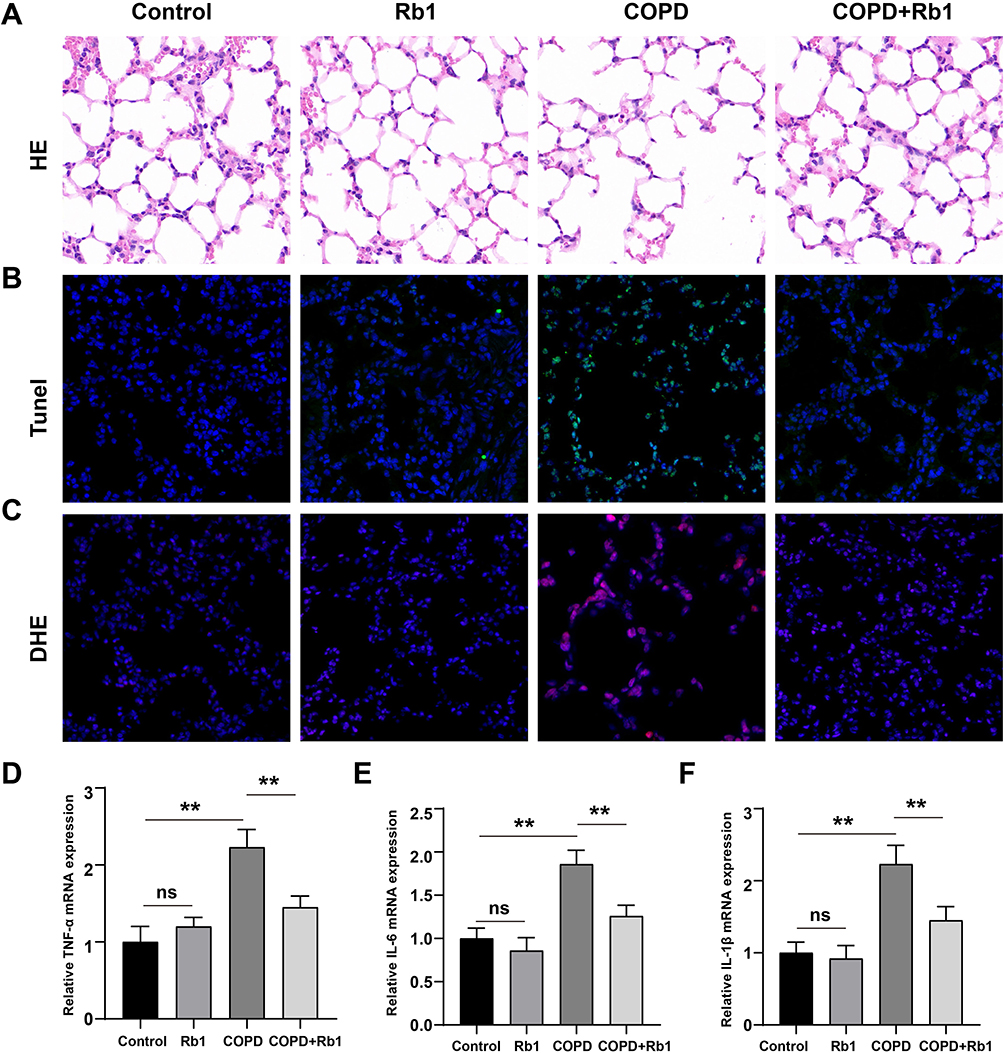 |
Figure 6 Rb1 alleviated CSE-induced lung injury, apoptosis, oxidative stress and inflammation in rats. (A) Pathological changes of lung tissues after injection of CSE and treatment of Rb1 using HE staining. (B) TUNEL staining detecting cell apoptosis after injection of CSE and Rb1 treatment using HE staining. (C) DHE staining measuring intracellular ROS in COPD and Rb1 treatment rats. (D–F) Relative mRNA expression levels pro-inflammatory cytokines including TNF-α, IL-6, and IL-1β. **P<0.01. Abbreviation: ns, not significant. |
Rb1 Activated Nrf2 and Inactivated NF-κb Signaling Pathways in COPD Rats
Subsequently, the effect of Rb1 on NF-κb signaling pathway in COPD rats was evaluated using Western blot analysis. Application of CSE remarkably promoted the phosphorylation of IκB and nuclear translocation of NF-κB compared with control group, whereas Rb1 treatment significantly decreased the phosphorylation of IκB and NF-κB nuclear translocation (Figure 7A–C). Additionally, we investigated the anti-oxidative effect of Rb1 by measuring the levels of Nrf2 nucleus transport in COPD rats. As displayed in Figure 7E and F, the expression of nuclear Nrf2 was inhibited by CSE compared with control group, whereas Rb1 treatment enhanced the nuclear translocation of Nrf2 in CSE-induced COPD rats. Meanwhile, CSE significantly decreased the production of HO-1, NQO-1 and Gclc compared with control group (P<0.01), while Rb1 treatment reversed the effects of CSE on HO-1, NQO-1 and Gclc in COPD rats (Figure 7G–I).
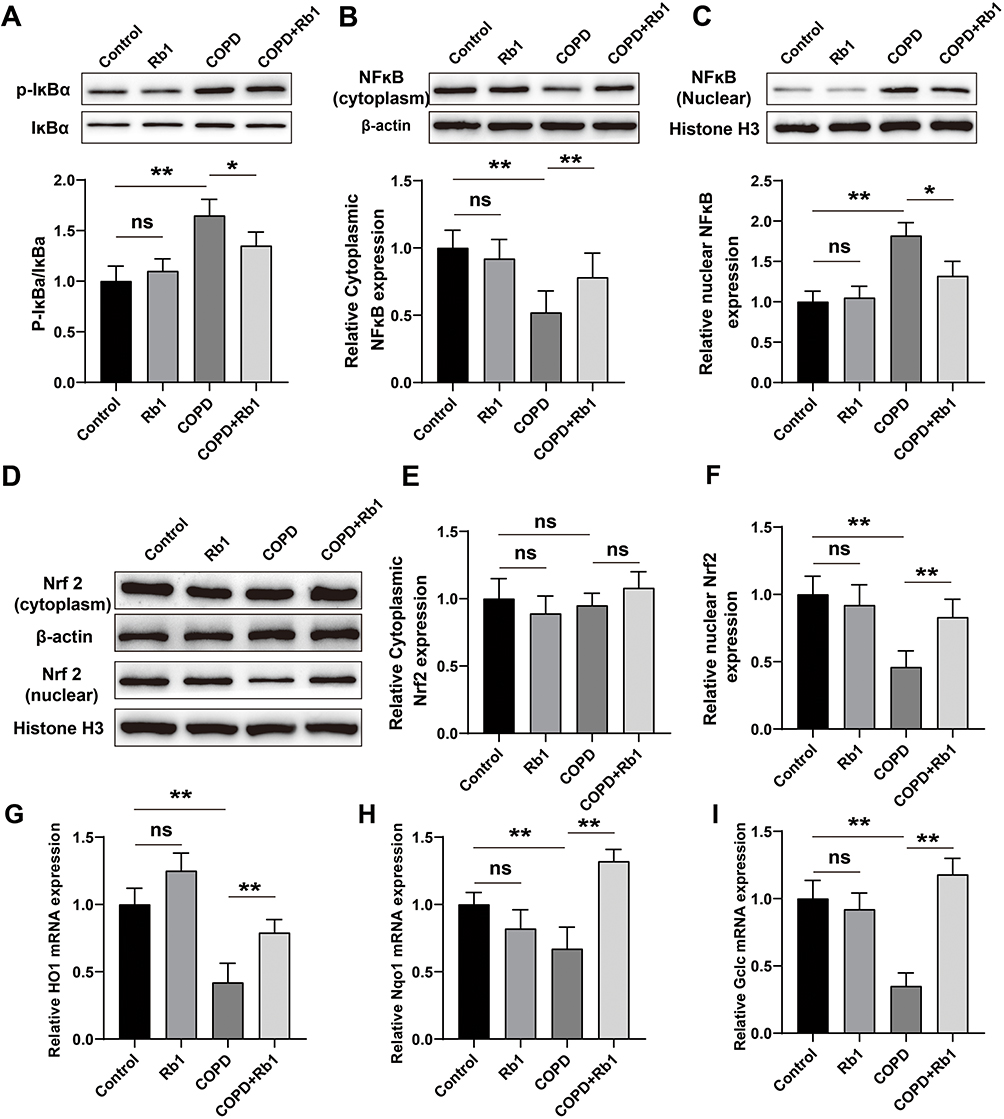 |
Figure 7 Rb1 activated Nrf2 and inactivated NF-κb signaling pathways in COPD rats. (A) Representative blots and statistical graphs of relative protein expression of p-IκBα and IκBα. (B and C) Representative blots and statistical graphs of relative protein expression of cytoplasmic and nuclear NF-κB. (D) Representative blots of Western blot analysis for cytoplasmic and nuclear Nrf2. (E and F) Statistical graphs of cytoplasmic and nuclear Nrf2 protein. (G–I) Relative mRNA expression levels of HO-1, Nqo1 and Gclc determined by qRT-PCR. **P<0.01, *P<0.05. Abbreviation: ns, not significant. |
Discussion
Reactive oxygen species (ROS) plays a pivotal role in promoting inflammatory response via activating and phosphorylating mitogen-activated protein kinases (MAPKs) and redox-sensitive transcription factors including NF-κB and activator protein-1, which lead to harmful events such as increase in apoptotic alveolar epithelial and endothelial cells, hypersecretion of mucus and enhanced epithelial permeability during the development of COPD.25,26 Over the last few years, many studies have displayed that Rb1 has various pharmacological effects including antioxidant, anti-inflammation and anti-apoptosis.27,28 Herein, our study exhibited that Rb1 could improve CSE-induced oxidative stress, inflammation and apoptosis in the BEAS-2B cells and rats. Additionally, our study showed that the potential anti-inflammation and antioxidant effects of Rb1 might be related to its ability to regulate the NF-κB and Nrf2 signaling pathways.
The adverse effects of CSE on human health and the environment are gotten increasing public attention.29 CSE have increased risk of developing diseases such as lung and other cancers, COPD, cardiovascular disease, stroke as well as periodontitis.30 In addition, CSE is a major contributor to chronic diseases, largely due to the inflammation response.31 It was mentioned that. CSE also induced upregulation of TNF-α and IL-1β in the lungs and macrophages.32 Jin et al reported that lung tissues are continuously exposed to CSE could result in serum concentrations of TNF-α.33 Compelling evidence has reported that CSE could induce death or apoptosis. Exposure to CSE (in vitro and in vivo) triggers a strong inflammatory and pro-apoptotic response in the epithelium,34 In additional, CSE-induced apoptosis and inflammation in BEAS-2B cells.35 Rb1 has anti-inflammatory effects in Staphylococcus aureus (S. aureus)-induced acute lung injury both in vivo and in RAW 264.7 macrophage cells.36 Moreover, Piao et al reported that Rb1 has a significant protective effect against PM2.5-induced skin cell apoptosis.37 Given the repeatedly documented anti-inflammatory and anti-apoptosis effects of Rb1, we presume that Rb1 might inhibit CSE-induced inflammation and apoptosis. Our results were in agreement with the previous findings, which demonstrated that CSE induced inflammatory cytokine release and cell apoptosis in BEAS-2B cells after exposure to CSE, including TNF-α, IL-6, and IL-1β, whereas Rb1 treatment suppressed CSE-induced inflammatory factors and inhibited cell apoptosis.
NF-κB is composed of P50/P65 heterodimer and plays a critical role in inflammation.38 Under normal physiological condition, NF-κB is located in the cytoplasm where it is present inactive state by bounding to the protein IκB.39 In response to stimulation, IκB kinase complex (IKK) is activated and phosphorylates IκBα which causes ubiquitination and degradation of IκBα. Subsequently, p65-p50 subunit is released from the p65-p50/IκBα complex and translocased into the nucleus, thereby promoting the production of pro-inflammatory cytokines. Therefore, we examined the level of IκB and p65 in BEAS-2B cells in order to evaluate the impact of Rb1 on the NF-κB signaling pathway after CSE. The result indicated that Rb1 inhibit NF-κB activation by decreasing the levels of p-IκB and preventing nuclear translocation of NF-κB p65, which suggests that Rb1 exhibits anti-inflammatory effects in COPD by modulating the NF-κB signaling pathway.
Oxidative stress is a result of an imbalance between ROS production and consumption, and maintaining cell redox balance is crucial for cell survival and tissue homeostasis.40 The study found that CSE induces activation of oxidative stress, which can be assessed by increased production of ROS.41 In the present study, we also found that ROS production was increased. 4-HNE is a product of lipid peroxidation, which is considered as indicator of oxidative stress.42 Our data revealed that 4-HNE levels were significantly reduced after Rb1 treatment. In addition, our results showed an increase in MDA a decrease in SOD and CAT enzymes, and a decrease in SOD and CAT enzymes, implying oxidative stress activation. However, the administration of Rb1 was found to have an antioxidant stress effect, suppressing the increase in MDA and the reduction of both CAT and SOD in BEAS-2B cells induced by CSE. These observations suggested that oxidative stress is involved in COPD pathogenesis and Rb1 has an excellent antioxidant stress effect.
It has been demonstrated that Nrf2 is a crucial transcription factor to protect the cells from oxidative stress.43 Upon being triggered by oxidative stress, Nrf2 will journey to the nucleus where it will attach to the antioxidant element known as ARE to enhance the expression of antioxidant genes.44 Hence, we evaluated the activation of Nrf2 by measuring its levels in both the nucleus and cytoplasm following CSE exposure. Our results demonstrated that Rb1 activates the Nrf2 signaling pathway, which protects against oxidative stress in BEAS-2B cells. Furthermore, we assessed the levels of Nrf2-mediated downstream antioxidant products, like HO-1 and NQO-1, to verify the activation state of Nrf2 signaling. Our data suggest that the mRNA levels of HO-1, NQO-1 and Gclc also increased with Nrf2 activation after Rb1 treatment. These results illustrate well the fact that Rb1 could decrease CSE-induced oxidative damage through activation of the Nrf2 signaling pathway.
In conclusion, our present study demonstrated that administration of Rb1 is effective in preventing CSE-induced COPD. Rb1 can reduce CSE-induced inflammatory response by inhibiting NF-κB signaling pathway and abolish oxidative stress response by stimulating Nrf2 signaling pathway. Our results provide more insights into the pathogenesis of COPD and the protective effects of Rb1.
Limitations
There are some limitations to our study that need to be addressed in future studies. First, although we have demonstrated that treatment with Rb1 could alleviate CSE-induced inflammation, apoptosis, and oxidative stress via activating Nrf2 and inhibiting NF-κB signaling pathway, the effect of Rb1 on the upstream and downstream key regulatory genes or proteins of Nrf2/NF-κB signaling pathway remains unclarified; hence, it is essential to investigate the upstream and downstream regulatory targets of Nrf2/NF-κB signaling pathways. Second, we only demonstrated the effect of Rb1 using in vivo and in vitro experiments; further studies should be conducted to validate our results in clinical trials.
Data Sharing Statement
All data can be accessed upon reasonable inquiry to the corresponding author.
Disclosure
There is no conflict of interest.
References
1. Liao YH, Chen LY, Liao KM, Chen CY. Drug safety of benzodiazepines in asian patients with chronic obstructive pulmonary disease. Front Pharmacol. 2020;11:592910. doi:10.3389/fphar.2020.592910
2. García-Sanz MT, Cánive-Gómez JC, Senín-Rial L, et al. One-year and long-term mortality in patients hospitalized for chronic obstructive pulmonary disease. J Thorac Dis. 2017;9(3):636–645. doi:10.21037/jtd.2017.03.34
3. Le Thi Bich P, Nguyen Thi H, Dang Ngo Chau H, et al. Allogeneic umbilical cord-derived mesenchymal stem cell transplantation for treating chronic obstructive pulmonary disease: a pilot clinical study. Stem Cell Res Ther. 2020;11(1):60. doi:10.1186/s13287-020-1583-4
4. Nucera F, Mumby S, Paudel Keshav R, et al. Role of oxidative stress in the pathogenesis of COPD. Minerva Med. 2022;113. doi:10.23736/S0026-4806.22.07972-1
5. Yao RQ, Ren C, Xia ZF, Yao YM. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. 2021;17(2):385–401. doi:10.1080/15548627.2020.1725377
6. Wu JH, Zhou M, Jin Y, et al. Generation and Immune Regulation of CD4 + CD25 - Foxp3 + T cells in chronic obstructive pulmonary disease. Front Immunol. 2019;10:220. doi:10.3389/fimmu.2019.00220
7. Lee H, Park JR, Kim WJ, et al. Blockade of RAGE ameliorates elastase-induced emphysema development and progression via RAGE-DAMP signaling. FASEB J. 2017;31(5):2076–2089. doi:10.1096/fj.201601155R
8. Zhang D, Hu S, Li W, et al. Schisandra A ameliorates cigarette smoke extract and lipopolysaccharide-induced oxidative stress in lung epithelial cells. J Thorac Dis. 2020;12(3):394–402. doi:10.21037/jtd.2020.01.24
9. Orlowski SK, Cauble R, Tabler T, et al. Processing evaluation of random bred broiler populations and a common ancestor at 55 days under chronic heat stress conditions. Poult Sci. 2020;99(7):3491–3500. doi:10.1016/j.psj.2020.03.036
10. Zhang L, Xu C, Hu W, Wu P, Qin C, Zhang J. Anti-inflammatory effects of Lefty-1 in renal tubulointerstitial inflammation via regulation of the NF-κB pathway. Int J Mol Med. 2018;41(3):1293–1304. doi:10.3892/ijmm.2017.3327
11. Leberl M, Kratzer A, Taraseviciene-Stewart L. Tobacco smoke induced COPD/emphysema in the animal model-are we all on the same page? Front Physiol. 2013;4:91. doi:10.3389/fphys.2013.00091
12. Lombard DB, Kohler WJ, Guo AH, et al. High-throughput small molecule screening reveals Nrf2-dependent and -independent pathways of cellular stress resistance. Sci Adv. 2020;6(40). doi:10.1126/sciadv.aaz7628
13. Cui C, Wang C, Jin F, et al. Calcitriol confers neuroprotective effects in traumatic brain injury by activating Nrf2 signaling through an autophagy-mediated mechanism. Molecul Med. 2021;27(1):118. doi:10.1186/s10020-021-00377-1
14. Wang J, Zhang J, Chen L, et al. Combination of broccoli sprout extract and zinc provides better protection against intermittent hypoxia-induced cardiomyopathy than monotherapy in mice. Oxid Med Cell Longev. 2019;2985901. doi:10.1155/2019/2985901
15. Sussan TE, Rangasamy T, Blake DJ, et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc Natl Acad Sci USA. 2009;106(1):250–255. doi:10.1073/pnas.0804333106
16. Lim S, Park J, Um JY. Ginsenoside Rb1 induces beta 3 adrenergic receptor-dependent lipolysis and thermogenesis in 3T3-L1 adipocytes and db/db mice. Front Pharmacol. 2019;10:1154. doi:10.3389/fphar.2019.01154
17. Wu Y, Huang XF, Bell C, Yu Y. Ginsenoside Rb1 improves leptin sensitivity in the prefrontal cortex in obese mice. CNS Neurosci Ther. 2018;24(2):98–107. doi:10.1111/cns.12776
18. Li M, Lan J, Li X, et al. Novel ultra-small micelles based on ginsenoside Rb1: a potential nanoplatform for ocular drug delivery. Drug Deliv. 2019;26(1):481–489. doi:10.1080/10717544.2019.1600077
19. Zhou P, Lu S, Luo Y, et al. Attenuation of TNF-α-induced inflammatory injury in endothelial cells by ginsenoside Rb1 via inhibiting NF-κB, JNK and p38 signaling pathways. Front Pharmacol. 2017;8:464. doi:10.3389/fphar.2017.00464
20. Dong W, Chen W, Zou H, et al. Ginsenoside Rb1 prevents oxidative stress-induced apoptosis and mitochondrial dysfunction in muscle stem cells via NF- κ B pathway. Oxid Med Cell Longev. 2022;2022:9159101. doi:10.1155/2022/9159101
21. Peng H, You L, Yang C, et al. Ginsenoside Rb1 attenuates triptolide-induced cytotoxicity in HL-7702 cells via the activation of Keap1/Nrf2/ARE pathway. Front Pharmacol. 2021;12:723784. doi:10.3389/fphar.2021.723784
22. Feng F, Du J, Meng Y, Guo F, Feng C. Louqin zhisou decoction inhibits mucus hypersecretion for acute exacerbation of chronic obstructive pulmonary disease rats by suppressing EGFR-PI3K-AKT signaling pathway and restoring Th17/treg balance. Evid Based Complement Alternat Med. 2019;2019:6471815. doi:10.1155/2019/6471815
23. Dong C, Liu P, Wang H, Dong M, Li G, Li Y. Ginsenoside Rb1 attenuates diabetic retinopathy in streptozotocin-induced diabetic rats1. Acta Cir Bras. 2019;34(2):e201900201. doi:10.1590/s0102-8650201900201
24. Shimouchi A, Yokota H, Ono S, et al. Neuroprotective effect of water-dispersible hesperetin in retinal ischemia reperfusion injury. Jpn J Ophthalmol. 2016;60:51–61. doi:10.1007/s10384-015-0415-z
25. Rahman I. Oxidative stress in pathogenesis of chronic obstructive pulmonary disease: cellular and molecular mechanisms. Cell Biochem Biophys. 2005;43:167–188. doi:10.1385/CBB:43:1:167
26. Song Q, Chen P, Liu X-M. The role of cigarette smoke-induced pulmonary vascular endothelial cell apoptosis in COPD. Respir Res. 2021;22:1–15. doi:10.1186/s12931-021-01630-1
27. Ke L, Guo W, Xu J, Zhang G, Wang W, Huang W. Ginsenoside Rb1 attenuates activated microglia-induced neuronal damage. Neural Regen Res. 2014;9(3):252–259. doi:10.4103/1673-5374.128217
28. Wang Y, Li Y, Yang W, et al. Ginsenoside Rb1 inhibit apoptosis in rat model of Alzheimer’s disease induced by Aβ 1-40. Am J Transl Res. 2018;10(3):796–805.
29. Yu G, Phillips S, Gail MH, et al. The effect of cigarette smoking on the oral and nasal microbiota. Microbiome. 2017;5(1):3. doi:10.1186/s40168-016-0226-6
30. Murphy JD, Liu B, Parascandola M. Smoking and HIV in Sub-Saharan Africa: a 25-country analysis of the demographic health surveys. Nicotine Tob Res. 2019;21(8):1093–1102. doi:10.1093/ntr/nty176
31. Ye D, Gajendra S, Lawyer G, et al. Inflammatory biomarkers and growth factors in saliva and gingival crevicular fluid of e-cigarette users, cigarette smokers, and dual smokers: a pilot study. J Periodontol. 2020;91(10):1274–1283. doi:10.1002/jper.19-0457
32. Guan R, Wang J, Li Z, et al. Sodium tanshinone IIA sulfonate decreases cigarette smoke-induced inflammation and oxidative stress via blocking the activation of MAPK/HIF-1α signaling pathway. Front Pharmacol. 2018;9:263. doi:10.3389/fphar.2018.00263
33. Jin Y, Wan Y, Chen G, et al. Treg/IL-17 ratio and Treg differentiation in patients with COPD. PLoS One. 2014;9(10):e111044. doi:10.1371/journal.pone.0111044
34. Murray LA, Dunmore R, Camelo A, et al. Acute cigarette smoke exposure activates apoptotic and inflammatory programs but a second stimulus is required to induce epithelial to mesenchymal transition in COPD epithelium. Respir Res. 2017;18(1):82. doi:10.1186/s12931-017-0565-2
35. Liu X, Wang J, Luo H, Xu C, Chen X, Zhang R. MiR-218 Inhibits CSE-induced apoptosis and inflammation in BEAS-2B by Targeting BRD4. Int J Chron Obstruct Pulmon Dis. 2020;15:3407–3416. doi:10.2147/copd.S278553
36. Shaukat A, Guo YF, Jiang K, et al. Ginsenoside Rb1 ameliorates staphylococcus aureus-induced acute lung injury through attenuating NF-κB and MAPK activation. Microb Pathog. 2019;132:302–312. doi:10.1016/j.micpath.2019.05.003
37. Piao MJ, Kang KA, Zhen AX, et al. Particulate Matter 2.5 mediates cutaneous cellular injury by inducing mitochondria-associated endoplasmic reticulum stress: protective effects of ginsenoside Rb1. Antioxidants. 2019;8(9):383. doi:10.3390/antiox8090383
38. Lee SH, Choi BY, Kho AR, et al. Combined treatment of dichloroacetic acid and pyruvate increased neuronal survival after seizure. Nutrients. 2022;14(22):4804. doi:10.3390/nu14224804
39. Park M, Lim JW, Kim H. Docoxahexaenoic acid induces apoptosis of pancreatic cancer cells by suppressing activation of STAT3 and NF-κB. Nutrients. 2018;10(11):1621. doi:10.3390/nu10111621
40. Canli Ö, Alankuş YB, Grootjans S, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127(1):139–148. doi:10.1182/blood-2015-06-654194
41. Reddy AT, Lakshmi SP, Banno A, Reddy RC. Role of GPx3 in PPARγ-induced protection against COPD-associated oxidative stress. Free Radic Biol Med. 2018;126:350–357. doi:10.1016/j.freeradbiomed.2018.08.014
42. Suthammarak W, Numpraphrut P, Charoensakdi R, et al. Antioxidant-enhancing property of the polar fraction of mangosteen pericarp extract and evaluation of its safety in humans. Oxid Med Cell Longev. 2016;2016:1293036. doi:10.1155/2016/1293036
43. Zhu G, Xin X, Liu Y, Huang Y, Li K, Wu C. Geraniin attenuates LPS-induced acute lung injury via inhibiting NF-κB and activating Nrf2 signaling pathways. Oncotarget. 2017;8(14):22835–22841. doi:10.18632/oncotarget.15227
44. Tan XL, Marquardt G, Massimi AB, Shi M, Han W, Spivack SD. High-throughput library screening identifies two novel NQO1 inducers in human lung cells. Am J Respir Cell Mol Biol. 2012;46(3):365–371. doi:10.1165/rcmb.2011-0301OC
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.