Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Ginaton improves neurological function in ischemic stroke rats via inducing autophagy and maintaining mitochondrial homeostasis
Authors Li X, Zhang D, Bai Y, Xiao J, Jiao H, He R
Received 16 February 2019
Accepted for publication 20 May 2019
Published 3 July 2019 Volume 2019:15 Pages 1813—1822
DOI https://doi.org/10.2147/NDT.S205612
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yu-Ping Ning
Xiaoqiang Li,1 Deli Zhang,1 Yinliang Bai,1 Jiyuan Xiao,1 Haisheng Jiao,1 Rongxia He2
1Department of Pharmacy, Lanzhou University Second Hospital, Lanzhou 730030, People’s Republic of China; 2Department of Gynecology, Lanzhou University Second Hospital, Lanzhou 730030, People’s Republic of China
Purpose: The present study was carried out to confirm the protective effect of extract of Ginkgo biloba (Ginaton) against ischemic neuronal damage post-treatment at 24 h after reperfusion in rats with middle cerebral artery occlusion (MCAO) and further reveal its possible mechanisms.
Methods: Adult male Sprague-Dawley rats were modeled by MCAO for 2 h. The rats were divided into three groups: sham, model, and Ginaton (50 mg/kg). All animals received treatment once a day for 14 days from 24 h after reperfusion. Modified neurological severity score test was performed in 1, 7 and 14 days after MCAO, and beam walking test was performed only 14 days after MCAO. Hematoxylin-eosin straining was implemented to measure infarct volume and immunohistochemical analysis was performed to calculate the number of neurons in ischemic cortex penumbra. Western blot was used to evaluate the expression of autophagy (Beclin1, LC3, AMPK, mTOR, ULK), mitochondrial dynamic protein (Parkin, DRP1, OPA1) and apoptosis (Bcl-2, Bax).
Results: Post-treatment with Ginaton for 14 days decreased neurological deficit score, promoted the recovery of motor function, and noticeably reduced infarct size. Besides, Ginaton also alleviated the loss of NeuN-positive cells in ischemic cortex penumbra. In ischemic cortex, Ginaton increased the expression of Beclin1 and LC3-Ⅱ, elevated the AMPK, mTOR and ULK1, and induced autophagy. Moreover, Ginaton treatment upregulated Parkin, DRP1, and OPA1, and elevated the ratio of Bcl-2/Bax in 14 days after MCAO reperfusion injury.
Conclusion: Ginaton exhibited obvious neuroprotective effects in MCAO rats with initial administered 24 h after MCAO. The mechanism of Ginaton included induction of autophagy via activation of the AMPK pathway, maintenance of mitochondrial homeostasis and inhibition of apoptosis.
Keywords: Ginaton, ischemic stroke, middle cerebral artery occlusion, autophagy, apoptosis, mitochondrial function
Introduction
Stroke – one of the leading causes of death globally – is characterized by high morbidity, high disability and a high mortality rate.1 Ischemic stroke accounts for ~85% of all strokes and exerts a profoundly negative impact on both patients and society.2,3 However, tissue plasminogen activator is so far the only approved therapeutic drug in clinical use for ischemic stroke.4 Despite encouraging results emerging from preclinical tests, none of the alternative candidates evaluated has resulted in consistent clinical improvements.5–7 Multiple mechanisms are involved in the ischemic process that ultimately leads to neuron death, including mitochondrial dysfunction, apoptosis, inflammation, oxidative stress, and excitotoxity.8–10 Drugs that work on multiple deleterious pathways are more likely to be efficacious clinically.
Ginaton, also known as “extract of Ginkgo biloba” (EGB), is derived from the leaves of Ginkgo biloba, which is widely used for the treatment of acute ischemic stroke in China and shows excellent clinical effects in many cases.11–13 It is also approved in Germany for the treatment of cerebral insufficiency.14 EGB contains terpenoids, flavonoids, alkylphenols, polyprenols, and organic acids.15 The main active constituents in EGB are flavonoid glycosides (flavonoid fraction, 22–24%), primarily composed of quercetin, kaempferol, and isorhamnetin, which have demonstrated efficacy against hydroxyl radicals.16,17 Rat studies showed that terpenoids rapidly reached maximum plasma concentrations. Ginkgolide A and B and biobalide are detectable in the plasma of humans and animals shortly after oral application of EGB.18 Further studies showed that EGB exhibits pleiotropic biological activities – such as anti-apoptotic, anti-oxidative, anti-Ca2+-overload, anti-inflammatory, and anti-glutamate toxicity – and improves damaged neuronal energy metabolism activities.19–21 Further, there is substantial evidence to indicate that EGB exerts neuroprotective effects, including reducing ischemic brain damage, delaying hippocampal neuronal death and apoptosis, ameliorating cognitive deficits and emotional disorders, and promoting motor function after ischemic stroke.22–24 But all these effects were observed in studies involving pre-treatment or post-treatment (up to 4 h after ischemia) with EGB. Whether EGB could exert a protective effect over longer therapeutic time periods remains unknown
In the present study, we evaluated the neuroprotective effects of Ginaton in rats modeled by middle cerebral artery occlusion (MCAO) 24 h after ischemia and further revealed the underlying molecular mechanism of Ginaton treatment on ischemia reperfusion.
Materials and methods
Animals
Adult male Sprague-Dawley rats weighing 260–280 g were purchased from the Academy of Military Medical Sciences (Beijing, China). The animal study was conducted in accordance with the Guide for Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health. All experimental protocols were approved by the Animal Care and Use Committee of Lanzhou University. All rats were housed in a temperature-controlled environment with a 12 h light/dark cycle and ad libitum access to feed and water, in which they were acclimated for 1 week before surgery.
Middle cerebral artery occlusion model
After a 12-h fast, the middle cerebral artery occlusion (MCAO) surgery was conducted in rats as previously described, with some modifications.25 All rats were anesthetized with 1% pentobarbital sodium (50 mg/kg) by intraperitoneal injection and the right common carotid artery was carefully isolated. Then a silicone-coated nylon monofilament was inserted from the external carotid artery stump into the internal carotid artery. Two hours later, the suture was withdrawn to recover blood circulation. The rats in the sham group underwent the same surgery without ligating the arteries. After surgery, rats were randomly divided into three groups (n=10 each): the sham group, the model group and the Ginaton (50 mg/kg) group. Ginaton injection solution was purchased from Chi Sheng Chemical Corporation (Taiwan, China). The rats in the sham and model groups received equivalent volumes of saline. Saline and Ginaton were injected intraperitoneally from the start of reperfusion once a day for 14 days until the rats were sacrificed.
Neurological function assessment
For all animals, the neurological function assessment was performed 1, 7 and 14 days after MCAO by an investigator who was blinded to the experimental design. Neurological deficits were evaluated as described previously,26 which included a set of modified Neurological Severity Scores (NSS) involving a series of motor, sensory, reflex, and balance tests.27 In the test, neurological function was graded from 0 to 18 (normal score, 0; maximal deficit score, 18).
Beam walking test
The beam walking test was carried out 14 days after MCAO, as reported previously, with some modifications.28 The task involved training rats to walk across a narrow wooden beam (4 cm wide and 105 cm long), which was placed 80 cm above the ground. The first 20 cm on the beam was taken as the starting area, and a horizontal line was drawn at a distance of 20 cm from the staring zone. Each rat was placed at the starting zone and a stopwatch started immediately upon release of the animal. Total times taken to move across the beam were recorded. All rats were pretrained three times a day for 2 days before the test. The maximal time allowed for the task was 2 min.
Tissue preparations
After behavioral tests, three rats in each group were sacrificed and the brain of each rat was rapidly removed. Each section of brain was quenched in liquid nitrogen and stored at −80 °C in preparation for Western blot analysis. The other rats in each group were anesthetized with 1% pentobarbital sodium (50 mg/kg) intraperitoneally and perfused firstly with 0.1 mol/L PBS, then 4% paraformaldehyde (pH 7.4) through the left ventricle. The whole brains were removed and postfixed in 4% paraformaldehyde solution at 4 °C for at least 48 h, then gradually equilibrated in 0.1 mol/L PBS containing 15%, 20% and 30% sucrose at 4 °C, respectively. Coronal sections were cut using a cryostat. Sections for hematoxylin-eosin (HE) staining were cut into 10 μm-thick sections and chosen from five segments;, each section was 2 mm apart from frontal to occipital pole. Additionally, 35-μm sections were cut for immunohistochemical analysis.
Infarct volume assessment
One section from each segment mentioned above was stained with HE to measure the infarct volume.29 The sections were photographed using a digital camera and analyzed using Image-Pro Plus 6.0. The infarct volumes of the lesion structures were expressed as a percentage of the volume of the structures in the control hemispheres by using the formula [(VC-VL)/VC] ×100%, where VC is the volume of control hemisphere and VL is the volume of non-infarcted tissue in the lesion hemisphere.30 The total infarct volume of each brain was calculated as the sum of the infarct volumes of the five brain slides.
Immunohistochemical analysis
The slices were pretreated with 3% H2O2 solution to reduce endogenous peroxide activity for 10 min at room temperature away from light. After washing three times in PBST, all slices were blocked in 10% bovine serum for 1 h and then incubated with mouse anti-NeuN antibody (Merk Millipore, Germany) at 4 °C overnight. The primary antibody was then removed, washing the slices in PBS another three times, then incubated with HRP-goat anti-mouse secondary antibody according to the manufacturer’s instructions (Beijing Zhong Shan Biotechnology Co., Beijing, China). DAB substrate kits (Beijing Zhong Shan Biotechnology Co., Beijing, China) were used to develop color. Images were captured at objective magnification 20× using an Olympus microscope (Olympus Corporation, Japan). The number of NeuN positive cells were counted using Image-Pro Plus software by a blinded investigator. In each slice, three fields evenly distributed throughout the areas of interest were analyzed.
Western blotting
The total proteins were extracted from the ischemic cortex in rats. The protein concentration was measured using the Enhanced BCA Protein Assay Kit according to manufacturer’s instructions (Beyotime Institute of Biotechnology, Shanghai, China). Proteins of each group were separated by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride (PVDF) membrane. The membranes were blocked in 5% non-fat milk for 1 h at room temperature and then incubated with primary antibodies overnight at 4 °C. The primary antibodies used in the study were listed as follows: rabbit anti-Beclin1, rabbit anti-LC3, rabbit anti-AMPK, rabbit anti-mTOR, rabbit anti-ULK1, mouse anti-Parkin, rabbit anti-DRP1, rabbit anti-Bcl-2, rabbit anti-Bax antibody (Cell Signaling Technology, MA, USA), rabbit anti-OPA1 antibody (Abcam, Cambridge, UK), and mouse anti-β-actin antibody (Applygen, Beijing, China). On the second day, the membranes were washed three times in TBST and then incubated with secondary antibody which conjugated to horseradish peroxidase for 1 h. Immunoblot was visualized with enhanced chemiluminescence and analyzed with GelPro software. β-actin served as a loading control.
Statistical analysis
All data were analyzed by one-way analysis of variance (ANOVA) using SPSS 17.0 software. Multiple-comparison post hoc tests between groups were performed with the least-significant difference test. Data were presented as mean ± standard error of mean (SEM), and differences between groups were considered significant at P<0.05.
Results
Ginaton improved neurological functional recovery, promoted motor function, and reduced the infarct volume after cerebral ischemia reperfusion in MCAO rats
Firstly, we evaluated the neuroprotective effect of Ginaton in MCAO rats with Ginaton administered intraperitoneally at 24 h after cerebral ischemia reperfusion. Results of mNSS test showed that the neurological deficit scores of MCAO rats were significantly higher than sham group consistently. Treatment with Ginaton did not change the deficit at 1 day and 7 days after reperfusion, whereas the scores were significantly reduced by Ginaton treatment at 14 days after reperfusion (Figure 1A). Additionally, in the beam walking test, rats treated with Ginaton for 14 days expended shorter time (3.94 ± 0.83 s) to walk across the whole beam compared with the model group (12.04 ± 2.45 s) (Figure 1B), implying better locomotion and motor coordination capacity. Along with the functional improvement, post-treatment with Ginaton significantly decreased infarct volumes from 46.85 ± 6.50% to 33.13 ± 2.14%, as shown in Figure 1C and D. The results demonstrated that post-treatment with Ginaton at 24 h after reperfusion exerted a neuroprotective effect in MCAO rats, suggesting a high potential value for clinical applications.
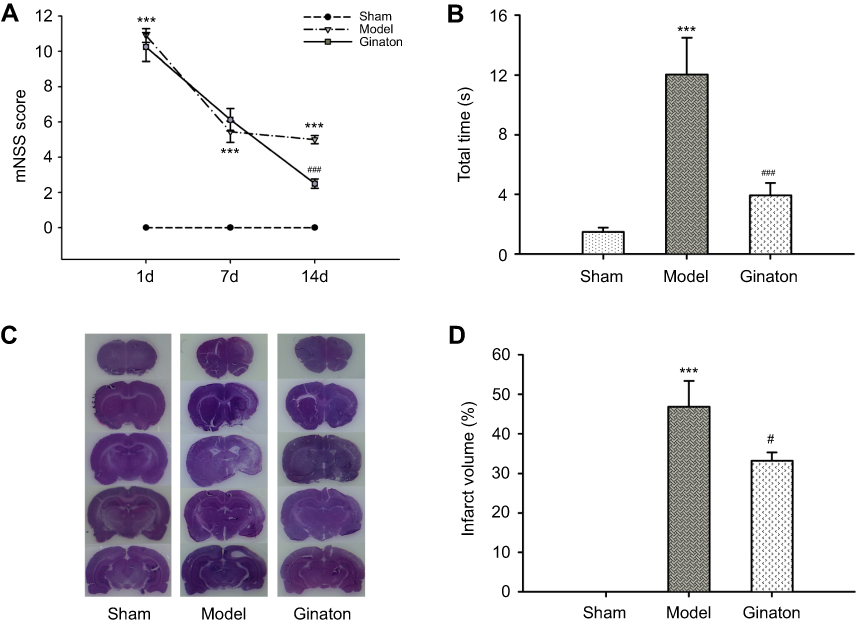 |
Figure 1 Ginaton promoted the recovery of neurological function and reduced infarct volume after cerebral ischemia reperfusion in MCAO rats. Notes: (A) mNSS scores of each group at 1 d, 7 d and 14 d after cerebral ischemia reperfusion; (B) time to walk across the whole beam in the beam-walking test; (C) representative images of infarct volume were shown by HE staining at 14 days after reperfusion; (D) quantitative evaluation of infarct volumes ratio in each group. Data were expressed as mean ± SEM. ***P<0.001 vs Sham group; #P<0.05, ###P<0.001 vs Model group; n=8–10. Abbreviations: MCAO, middle cerebral artery occlusion; mNSS, modified neurological severity scores; HE, hematoxylin-eosin; SEM, standard error of mean. |
Ginaton reduced the loss of neurons in ischemic cortex penumbra after MCAO in rats
We further observed obvious loss of neurons labeled by NeuN in ischemic cortex penumbra using immunohistochemical analysis. As shown in Figure 2A, the number of NeuN was obviously decreased in ischemic cortex of MCAO rats, while Ginaton treatment noticably prevented this change and increased the number of neurons at 14 days after reperfusion (Figure 2A and C), hence protecting the brain against ischemic damage. In the contralateral ischemic brain, there were no differences in number of neurons between all groups (Figure 2 A and B).
 |
Figure 2 Ginaton reduced loss of NeuN-positive neurons in ischemic cortex penumbra after MCAO in rats. Notes: (A) Representative immunohistochemical images of NeuN labeling in ischemic cortex penumbra and contralateral cortex in MCAO rats (magnification 200×, scale bar 50 μm); (B) quantification of NeuN-positive cells in contralateral cortex in different groups; (C) quantification of NeuN-positive cells in ischemic cortex in different groups. Data were expressed as mean ± SEM. *P<0.05 vs Sham group; #P<0.05 vs Model group; n=3. Abbreviations: MCAO, middle cerebral artery occlusion; SEM, standard error of mean. |
Ginaton induced autophagy via activating AMPK pathway in ischemic cortex of MCAO rats
There is increasing evidence to suggest that activation of autophagy in ischemic brain may contribute to neuroprotection.31 To investigate whether autophagic processes are involved in Ginaton-mediated protection, we examined the expression of Beclin1 and LC3-II – both recognized as important molecular markers of autophagy that are responsible for formation of autophagosome.32–34 Western blot results showed that a significant decrement in Beclin1 and LC3-II formation took place in the ischemic cortex at 14 days after reperfusion, indicating inhibitory autophagy induced by ischemic reperfusion; while Ginaton increased the expression of Beclin1 and LC3-II, activating the autophagy process (Figure 3A). In addition, we found that Ginaton elevated the expression of proteins including phosphorylation of AMP-activated protein kinase (p-AMPK), mammalian target of rapamycin (mTOR), and unc-51-like kinase 1 (ULK1), which play vital roles in autophagy signaling pathway,35,36 as shown in Figure 3B. The results suggested that Ginaton induced autophagy by activating the AMPK pathway in ischemic cortex of MCAO rats.
 |
Figure 3 Ginaton induced autophagy via AMPK pathway in ischemic cortex of MCAO rats. Notes: (A) Representative images and relative band intensity ratio of Beclin1 and LC3; (B) representative images and relative band intensity ratio of p-AMPKα, AMPKα, mTOR, and ULK1. The intensity of each band was normalized to that of β-actin. Data were expressed as mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 vs Sham group; #P<0.05, ##P<0.01 ###P<0.001 vs Model group; n=3. Abbreviations: AMPK, adenosine 5’-monophosphate (AMP)-activated protein kinase; MCAO, middle cerebral artery occlusion; LC3, light chain 3; mTOR, mammalian target of rapamycin; ULK1, unc-51-like kinase 1; SEM, standard error of mean. |
Ginaton maintained mitophagy and mitochondrial dynamics in ischemic cortex of MCAO rats
A strong association between autophagy and mitochondrial function has been hypothesized.37 Autophagy regulates mitochondrial turnover, and mitochondrial dysfunction can also influence autophagic activities.38 Since Ginaton induced autophagy in MCAO rats, we further investigated the effect of Ginaton on mitochondrial function, including mitophagy and mitochondrial dynamics. At 14 days after reperfusion, ischemia resulted in decreased expression of Parkin (Figure 4A and B), dynamin-related protein 1 (DRP1) (Figure 4A and C), and optic atrophy 1 (OPA1) (Figure 4A and D) in ischemic cortex, which play key roles in mitophagy, mitochondrial fission and fusion, respectively.39–41 But the decrement effects were significantly attenuated by Ginaton treatment (Figure 4).
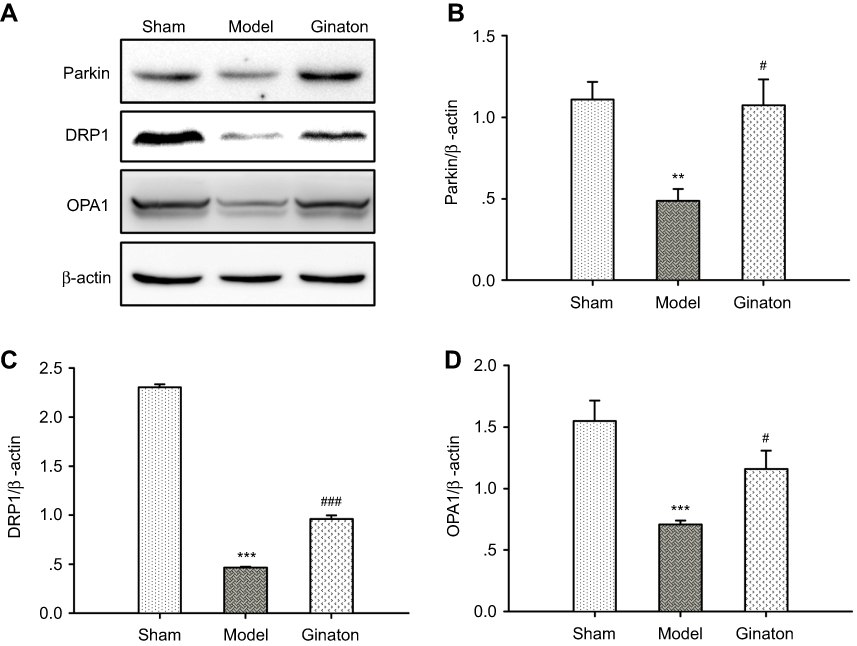 |
Figure 4 Ginaton maintained mitophagy and mitochondrial dynamics in ischemic cortex of MCAO rats. Notes: (A) Representative images of immunoblots of Parkin, DRP1, OPA1 and β-actin; (B) relative band intensity ratio of Parkin/β-actin; (C) relative band intensity ratio of DRP1/β-actin; (D) relative band intensity ratio of OPA1/β-actin. The intensity of each band was normalized to that of β-actin. Data were expressed as mean ± SEM. **P<0.01, ***P<0.001 vs Sham group; #P<0.05, ###P<0.001 vs Model group; n=3. Abbreviations: MCAO, middle cerebral artery occlusion; DRP1, dynamin-related protein 1; OPA1, optic atrophy 1; SEM, standard error of mean. |
Ginaton inhibited apoptosis in ischemic cortex induced by MCAO in rats
Disturbance in autophagy and mitochondrial dysfunction both result in apoptosis of neurons, which is one of the major pathways driving neuronal cell death after ischemia.42,43 To clarify the anti-apoptotic effect of Ginaton, we detected the expression of apoptotic proteins-Bcl-2 and Bax in ischemic cortex. Our data showed that the Bcl-2 was downregulated and Bax was upregulated, hence the ratio of Bcl-2/Bax dropped significantly after MCAO reperfusion. Though Ginaton did not change the decline of Bcl-2 (Figure 5A and B), it noticeably inhibited the level of Bax (Figure 5A and C) and elevated the ratio of Bcl-2/Bax (Figure 5A and D), indicating the anti-apoptotic effects of Ginaton in MCAO reperfusion injury.
 |
Figure 5 Ginaton inhibited apoptosis in ischemic cortex induced by MCAO in rats. Notes: (A) Representative images of immunoblots of Bcl-2, Bax, and β-actin; (B) relative band intensity ratio of Bcl-2/β-actin; (C) relative band intensity ratio of Bax/β-actin; (D) relative band intensity ratio of Bcl-2/Bax. The intensity of each band was normalized to that of β-actin. Data were expressed as mean ± SEM. *P<0.05, **P<0.01 vs Sham group #P<0.05, ###P<0.001 vs Model group; n=3. Abbreviations: MCAO, middle cerebral artery occlusion; SEM, standard error of mean. |
Discussion
Many studies have reported that EGB has a significant neuroprotective effect on the experimental models of stroke in animals.19,22 Yin and colleagues reviewed the studies of EGB systematically. Though analysis of the route of EGB administration affected the variability of the effect size, they suggested that immediate treatment with EGB after ischemia yielded a lower effect size than did treatment at other time points.19 Combination with the actual situation in stroke treatment that many patients arrive in hospital several hours after their stroke onset, we evaluated the potential protective effect of Ginaton administered 24 h after MCAO. Results showed that, even when administered 24 h after MCAO reperfusion, Ginaton exhibited strong neuroprotective effects: promoting an improvement in neurological function, accelerating the recovery of motor function, and reducing infarct size at day 14 after MCAO reperfusion. Moreover, Ginaton also alleviated the loss of neurons in ischemic cortex penumbra, while having no influence on the contralateral of ischemic brain, indicating its restorative effects on neurons. This wide therapeutic time window makes Ginaton an attractive drug for use in patients who cannot receive immediate treatment.
Previous studies have found that EGB could induce autophagy in Lewis lung cancer,44 since the disturbances in autophagy may play an important role in pathogenesis of ischemic brain injury; and many studies have shown that autophagy can protect cells from death by apoptosis.45,46 We evaluated whether autophagy was involved in the effect of Ginaton. Data illustrated that Ginaton significantly induced autophagy in ischemic cortex via reactivating the AMPK/mTOR/ULK1 signaling pathway, the key molecular pathways that regulate autophagy, in which AMPK negatively regulates mTOR, leading to ULK1–AMPK interaction to stimulate autophagy.47
Autophagy occurs in mitochondria to selectively remove the damaged mitochondria called mitophagy. As mentioned above, changes in autophagy also influence the mitochondria function. Once disrupted, damaged mitochondria would accumulate in cellular and lead to releasing kinds of mitochondrial protein from mitochondrial matrix to cytoplasm, which turning the neurons to apoptosis process. Neurons in the central nervous system are more sensitive to mitochondrial dysfunction. Dynamic mitochondrial changes in their shape and populations are crucial for normal cellular energy homeostasis.
Studies have suggested that Parkin, a ubiquitin ligase that mediates mitophagy, DRP1 and OPA1, the modulators promoting mitochondrial fission and fusion, are all critical molecules to maintain the integrity of the mitochondrial network.48 In our study, cerebral ischemia reperfusion injury caused decline of Parkin, DRP1, and OPA1 in ischemic cortex at 14 days after MCAO, while these effects were all reversed in the Ginaton treatment group. Further, we also found that Ginaton significantly increased the ratio of Bcl-2/Bax, which was important to apoptosis,49 indicating the anti-apoptotic ability of Ginaton. Taken together, these findings support the modulating role of Ginaton on autophagy, mitochondrial function and apoptosis in the ischemic brain.
Conclusion
In conclusion, the present study firstly demonstrated the neuroprotective effect of Ginaton on ischemic stroke with Ginaton initial administered 24 h after MCAO, which showed a wide therapeutic time window. The effect of Ginaton was partially due to the induction of autophagy via activating the AMPK pathway, maintaining themitochondrial homeostasis, and inhibiting apoptosis. The wide therapeutic time window and multiple effects of Ginaton make it potentially useful in clinical practice.
Acknowledgments
This study was supported by Cuiying Scientific and Technological Innovation Program of Lanzhou University Second Hospital (Grant No.CY2018-QN15) and Administration of Traditional Chinese Medicine Scientific Research Funds of Gansu Province (Grant No. GZK-2017-53).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Guzik A, Bushnell C. Stroke epidemiology and risk factor management. Continuum (Minneap Minn). 2017;23(1, Cerebrovascular Disease):15–39. doi:10.1212/CON.0000000000000416
2. Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371(9624):1612–1623. doi:10.1016/S0140-6736(08)60694-7
3. Feigin VL, Lawes CM, Bennett DA, Anderson CS. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003;2(1):43–53.
4. Broussalis E, Killer M, McCoy M, Harrer A, Trinka E, Kraus J. Current therapies in ischemic stroke. Part A. Recent developments in acute stroke treatment and in stroke prevention. Drug Discov Today. 2012;17(7–8):296–309. doi:10.1016/j.drudis.2011.11.005
5. Fisher M. New approaches to neuroprotective drug development. Stroke. 2011;42(1 Suppl):S24–S27. doi:10.1161/STROKEAHA.110.592394
6. Xing C, Arai K, Lo EH, Hommel M. Pathophysiologic cascades in ischemic stroke. Int J Stroke. 2012;7(5):378–385. doi:10.1111/j.1747-4949.2012.00839.x
7. Moretti A, Ferrari F, Villa RF. Neuroprotection for ischaemic stroke: current status and challenges. Pharmacol Ther. 2015;146:23–34. doi:10.1016/j.pharmthera.2014.09.003
8. Pandya RS, Mao L, Zhou H, et al. Central nervous system agents for ischemic stroke: neuroprotection mechanisms. Cent Nerv Syst Agents Med Chem. 2011;11(2):81–97.
9. Sims NR, Anderson MF. Mitochondrial contributions to tissue damage in stroke. Neurochem Int. 2002;40(6):511–526. doi:10.1016/S0197-0186(01)00122-X
10. Sims NR, Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta. 2010;1802(1):80–91. doi:10.1016/j.bbadis.2009.09.003
11. Le Bars PL, Katz MM, Berman N, Itil TM, Freedman AM, Schatzberg AF. A placebo-controlled, double-blind, randomized trial of an extract of Ginkgo biloba for dementia. North American EGb Study Group. Jama. 1997;278(16):1327–1332.
12. Ihl R, Tribanek M, Bachinskaya N; GOTADAY Study Group. Efficacy and tolerability of a once daily formulation of Ginkgo biloba extract EGb 761(R) in Alzheimer’s disease and vascular dementia: results from a randomised controlled trial. Pharmacopsychiatry. 2012;45(2):41–46. doi:10.1055/s-0031-1291217
13. Yang X, Zheng T, Hong H, et al. Neuroprotective effects of Ginkgo biloba extract and Ginkgolide B against oxygen-glucose deprivation/reoxygenation and glucose injury in a new in vitro multicellular network model. Front Med. 2018;12(3):307–318. doi:10.1007/s11684-017-0547-2
14. Zeng X, Liu M, Yang Y, Li Y, Asplund K. Ginkgo biloba for acute ischaemic stroke. Cochrane Database Syst Rev. 2005;4:CD003691.
15. van Beek TA, Montoro P. Chemical analysis and quality control of Ginkgo biloba leaves, extracts, and phytopharmaceuticals. J Chromatogr A. 2009;1216(11):2002–2032. doi:10.1016/j.chroma.2009.01.013
16. Nakanishi K. Terpene trilactones from Gingko biloba: from ancient times to the 21st century. Bioorg Med Chem. 2005;13(17):4987–5000. doi:10.1016/j.bmc.2005.06.014
17. Zimmermann M, Colciaghi F, Cattabeni F, et al. Ginkgo biloba extract: from molecular mechanisms to the treatment of alzheimer’s disease. Cell Mol Biol. 2002;48:613–623.
18. Biber A, Koch E. Bioavailability of ginkgolides and bilobalide from extracts of ginkgo biloba using GC/MS. Planta Med. 1999;65(2):192–193. doi:10.1055/s-2006-960467
19. Yin B, Xu Y, Wei R, et al. Ginkgo biloba on focal cerebral ischemia: a systematic review and meta-analysis. Am J Chin Med. 2014;42(4):769–783. doi:10.1142/S0192415X14500499
20. Smith JV, Luo Y. Studies on molecular mechanisms of Ginkgo biloba extract. Appl Microbiol Biotechnol. 2004;64(4):465–472. doi:10.1007/s00253-003-1527-9
21. Zhang Z, Peng D, Zhu H, Wang X. Experimental evidence of Ginkgo biloba extract EGB as a neuroprotective agent in ischemia stroke rats. Brain Res Bull. 2012;87(2–3):193–198. doi:10.1016/j.brainresbull.2011.11.002
22. Tian J, Liu Y, Chen K. Ginkgo biloba extract in vascular protection: molecular mechanisms and clinical applications. Curr Vasc Pharmacol. 2017;15(6):532–548. doi:10.2174/1570161115666170713095545
23. Zeng GR, Zhou SD, Shao YJ, et al. Effect of Ginkgo biloba extract-761 on motor functions in permanent middle cerebral artery occlusion rats. Phytomedicine. 2018;48:94–103. doi:10.1016/j.phymed.2018.05.003
24. Tulsulkar J, Glueck B, Hinds TD
25. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91.
26. Chen J, Li Y, Wang L, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32(4):1005–1011.
27. Germano AF, Dixon CE, d’Avella D, Hayes RL, Tomasello F. Behavioral deficits following experimental subarachnoid hemorrhage in the rat. J Neurotrauma. 1994;11(3):345–353.
28. Zhang R, Yang N, Ji C, et al. Neuroprotective effects of Aceglutamide on motor function in a rat model of cerebral ischemia and reperfusion. Restor Neurol Neurosci. 2015;33(5):741–759. doi:10.3233/RNN-150509
29. Dang S, Liu X, Fu P, et al. Neuroprotection by local intra-arterial infusion of erythropoietin after focal cerebral ischemia in rats. Neurol Res. 2011;33(5):520–528. doi:10.1179/016164111X13007856084287
30. Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10(2):290–293. doi:10.1038/jcbfm.1990.47
31. Gabryel B, Kost A, Kasprowska D. Neuronal autophagy in cerebral ischemia–a potential target for neuroprotective strategies? Pharmacol Rep. 2012;64(1):1–15.
32. Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(23):2861–2873. doi:10.1101/gad.1599207
33. McKnight NC, Zhenyu Y. Beclin 1, an essential component and master regulator of PI3K-III in health and disease. Curr Pathobiol Rep. 2013;1(4):231–238. doi:10.1007/s40139-013-0028-5
34. Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin1: a unique autophagy-related protein. Cell Res. 2007;17(10):839–849. doi:10.1038/cr.2007.78
35. Tian W, Li W, Chen Y, et al. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. 2015;589(15):1847–1854. doi:10.1016/j.febslet.2015.05.020
36. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi:10.1038/ncb2152
37. Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441(2):523–540. doi:10.1042/BJ20111451
38. Shacka JJ, Roth KA, Zhang J. The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front Biosci. 2008;13:718–736.
39. Springer W, Kahle PJ. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy. 2011;7(3):266–278. doi:10.4161/auto.7.3.14348
40. Hu C, Huang Y, Li L. Drp1-dependent Mitochondrial fission plays critical roles in physiological and pathological progresses in mammals. Int J Mol Sci. 2017;18(1):E144. doi:10.3390/ijms18010144
41. Farmer T, Reinecke JB, Xie S, Bahl K, Naslavsky N, Caplan S. Control of mitochondrial homeostasis by endocytic regulatory proteins. J Cell Sci. 2017;130(14):2359–2370. doi:10.1242/jcs.204537
42. Zhang P, Yang L, He H, Deng Y. Differential variations of autophagy and apoptosis in permanent focal cerebral ischaemia rat model. Brain Inj. 2017;31(8):1151–1158. doi:10.1080/02699052.2017.1298005
43. Gao Y, Chen T, Lei X, et al. Neuroprotective effects of polydatin against mitochondrial-dependent apoptosis in the rat cerebral cortex following ischemia/reperfusion injury. Mol Med Rep. 2016;14(6):5481–5488. doi:10.3892/mmr.2016.5936
44. Cao C, Han D, Su Y, Ge Y, Chen H, Xu A. Ginkgo biloba exocarp extracts induces autophagy in Lewis lung cancer cells involving AMPK/mTOR/p70S6k signaling pathway. Biomed Pharmacother. 2017;93:1128–1135. doi:10.1016/j.biopha.2017.07.036
45. Rubinsztein DC, DiFiglia M, Heintz N, et al. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1(1):11–22. doi:10.4161/auto.1.1.1513
46. Thorburn A. Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis. 2008;13(1):1–9. doi:10.1007/s10495-007-0154-9
47. Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2–11. doi:10.1128/MCB.06159-11
48. Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13. doi:10.1016/j.redox.2014.11.006
49. Tamm I, Schriever F, Dörken B. Apoptosis: implications of basic research for clinical oncology. The Lancet Oncology. 2001;2(1):33–42. doi:10.1016/S1470-2045(00)00193-5
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.