Back to Journals » OncoTargets and Therapy » Volume 16
Gilteritinib Affects the Selection of Dominant Clones in Clonal Hematopoiesis: Sequential Genetic Analysis of an FLT3-ITD Positive AML Patient with Long-Term Gilteritinib Therapy
Authors Katagiri S ![]() , Furuya N, Akahane D, Chi S, Minami Y, Harada Y, Harada H, Gotoh A
, Furuya N, Akahane D, Chi S, Minami Y, Harada Y, Harada H, Gotoh A
Received 27 April 2023
Accepted for publication 4 July 2023
Published 12 July 2023 Volume 2023:16 Pages 571—576
DOI https://doi.org/10.2147/OTT.S417137
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Nagashree Seetharamu
Seiichiro Katagiri,1 Nahoko Furuya,1 Daigo Akahane,1 SungGi Chi,2 Yosuke Minami,2 Yuka Harada,3 Hironori Harada,4 Akihiko Gotoh1
1Department of Hematology, Tokyo Medical University, Tokyo, Japan; 2Department of Hematology, National Cancer Center Hospital East, Chiba, Japan; 3Department of Clinical Laboratory, Tokyo Metropolitan Cancer and Infectious Diseases Center Komagome Hospital, Tokyo, Japan; 4Laboratory of Oncology, School of Life Sciences, Tokyo University of Pharmacy and Life Sciences, Tokyo, Japan
Correspondence: Seiichiro Katagiri, Department of Hematology, Tokyo Medical University, 6-7-1 Nishishinjuku, Shinjuku-ku, Tokyo, 160-0023, Japan, Tel +81-3-3342-6111 (ext. 5895), Fax +81-3-5381-6651, Email [email protected]
Abstract: We performed sequential molecular analyses of a 75-year-old woman with de novo FLT3-ITD positive acute myeloid leukemia (AML) who had received gilteritinib therapy for 43 months. At the time of diagnosis, her karyotype was normal; however, FLT3-ITD, NPM1, DNMT3A, and IDH2 mutations were detected. She received induction therapy with daunorubicin and cytarabine and achieved hematological complete remission (HCR). After attaining HCR, she underwent consolidation therapy with azacytidine or cytarabine, aclarubicin, and granulocyte-colony stimulating factor. However, AML relapsed eight months after the first HCR. FLT3-ITD and NPM1 mutations were persistently positive, and the patient received gilteritinib therapy. Although the FLT3-ITD clone was not detected during gilteritinib treatment, a clone harboring monosomy 7 and CBL mutations emerged. Bone marrow examinations at 15, 24, and 32 months after gilteritinib treatment revealed multi-lineage blood cell dysplasia without an increase in myeloblasts. After 33 months of treatment, gilteritinib was discontinued for two months because to ileus development, and the FLT3-ITD clone was detected again. Gilteritinib treatment was restarted, and FLT3-ITD became negative. Our analysis demonstrated that: (1) hematopoiesis derived from gilteritinib-resistant clones was generated by long-term gilteritinib treatment, and (2) FLT3-ITD clones regained clonal dominance in the absence of FLT3 inhibition. These findings suggest that gilteritinib affects the selection of dominant clones during clonal hematopoiesis.
Keywords: gilteritinib, FLT3-ITD, CBL, monosomy 7, clonal hematopoiesis
Introduction
Clonal hematopoiesis is defined that the presence of a population of cells derived from a mutated multipotent stem/progenitor cell harboring a selective growth advantage in the absence of unexplained cytopenia, hematological cancers, or other clonal disorders by the 5th edition of the World Health Organization Classification.1 It has been reported that clonal hematopoiesis increases the risk of developing myeloid neoplasms such as myelodysplastic syndromes and acute myeloid leukemia (AML).2,3
FMS-like tyrosine kinase 3 (FLT3) gene is a receptor-type tyrosine kinase, and in AML, it undergoes internal tandem duplication (ITD) mutation that duplicate and repeat part of the juxta membrane region, and tyrosine kinase domain (TKD) mutations that are point mutations in the kinase region.4 In particular, FLT3-ITD is an important prognostic factor in AML, and long-term survival is difficult to achieve in FLT3-ITD-positive AML patients who cannot undergo hematopoietic stem cell transplantation (HSCT).5
Gilteritinib is an oral, potent, selective FLT3 inhibitor with inhibitory activity against both FLT3-ITD and FLT3-TKD mutations,6 and has shown significant therapeutic efficacy in relapsed refractory FLT3-mutated AML.6,7 Gilteritinib is expected to be a therapeutic option for transplant-ineligible patients; however, the effect of long-term administration of gilteritinib on hematopoiesis is unknown. In this study, we analyzed the changes in hematopoietic clones from diagnosis to 43 months after gilteritinib treatment in an older patient with recurrent FLT3-ITD positive AML who received long-term administration of gilteritinib.
Case Presentation
A 75-year-old woman with fever was referred to our hospital and diagnosed with AML. Bone marrow (BM) examination revealed myeloperoxidase-positive leukemic cells and extensive BM necrosis (Figure 1A–C). Cytogenetic analysis of 24 metaphases showed a normal karyotype; however, molecular analysis of the patient by next-generation sequencing (NGS) conducted in Hematologic Malignancy (HM)-SCREEN-Japan-01 (UMIN000035233)8 identified FLT3-ITD, DNMT3A, IDH2, and NPM1 mutations (Table 1). She received induction therapy with daunorubicin and cytarabine and achieved hematological complete remission (HCR). She subsequently underwent two courses of azacytidine, followed by three courses of cytarabine, aclarubicin, and granulocyte-colony stimulating factor; however, AML relapsed eight months after the first HCR (Figure 1D–F). Cytogenetic analysis revealed clones containing t(3;5)(p14;p15) (Table 1). FLT3-ITD was persistently positive, and she received reinduction therapy with gilteritinib (120 mg/day). The patient achieved complete remission with incomplete count recovery on day 49 after gilteritinib treatment. However, the dose was reduced to 80 mg/day because of cytopenia. Nine months after gilteritinib therapy, her blood count recovered and she achieved a second HCR. BM examination showed no increase in myeloblasts; however, blood cells with multilineage dysplasia were observed. Cytogenetic analysis using fluorescence in situ hybridization (FISH) showed the emergence of a monosomy 7 clone. Molecular analysis of HM-SCREEN-Japan-01 did not detect FLT3-ITD or NPM1 mutations, but a new Casitas B-cell lymphoma (CBL) gene mutation was identified (Table 1). She continued gilteritinib treatment, and BM examinations were performed at 15, 24, and 32 months after starting treatment. Each BM examination revealed the continuation of multi-lineage blood cell dysplasia without an increase in myeloblasts (Figure 1G–I). Cytogenetic analysis of the three specimens showed that monosomy 7 clones gradually expanded. In the molecular analysis, samples taken 15 and 24 months after treatment were subjected to NGS analysis in the HM-SCREEN-Japan-01 study, whereas the samples taken 32 months after treatment were subjected to NGS analysis using the Komagome myeloid panel.9 All of which revealed DNMT3A, IDH2, and CBL mutations but not FLT3-ITD (Table 1). After 33 months of treatment, gilteritinib was discontinued for two months because the patient developed ileus complicated by septic shock. After the cessation of gilteritinib treatment, myeloblasts appeared in the peripheral blood. BM examination revealed 10% myeloblasts (Figure 1J–L), and FLT3-ITD was again detected by qualitative polymerase chain reaction (PCR). Gilteritinib treatment was restarted, and the peripheral blood myeloblasts disappeared. Five months after resuming treatment, BM examination showed that the myeloblast ratio had decreased to 6%. NGS performed in the HM-SCREEN-Japan-02 study (UMIN000046371)10 revealed FLT3-ITD, IDH2, and CBL mutations. However, the allele frequency of FLT3-ITD was 1.6%. Eight months after the resumption of treatment, BM examination showed multi-lineage blood cell dysplasia without myeloblast increase, and qualitative PCR demonstrated that FLT3-ITD had also become negative.
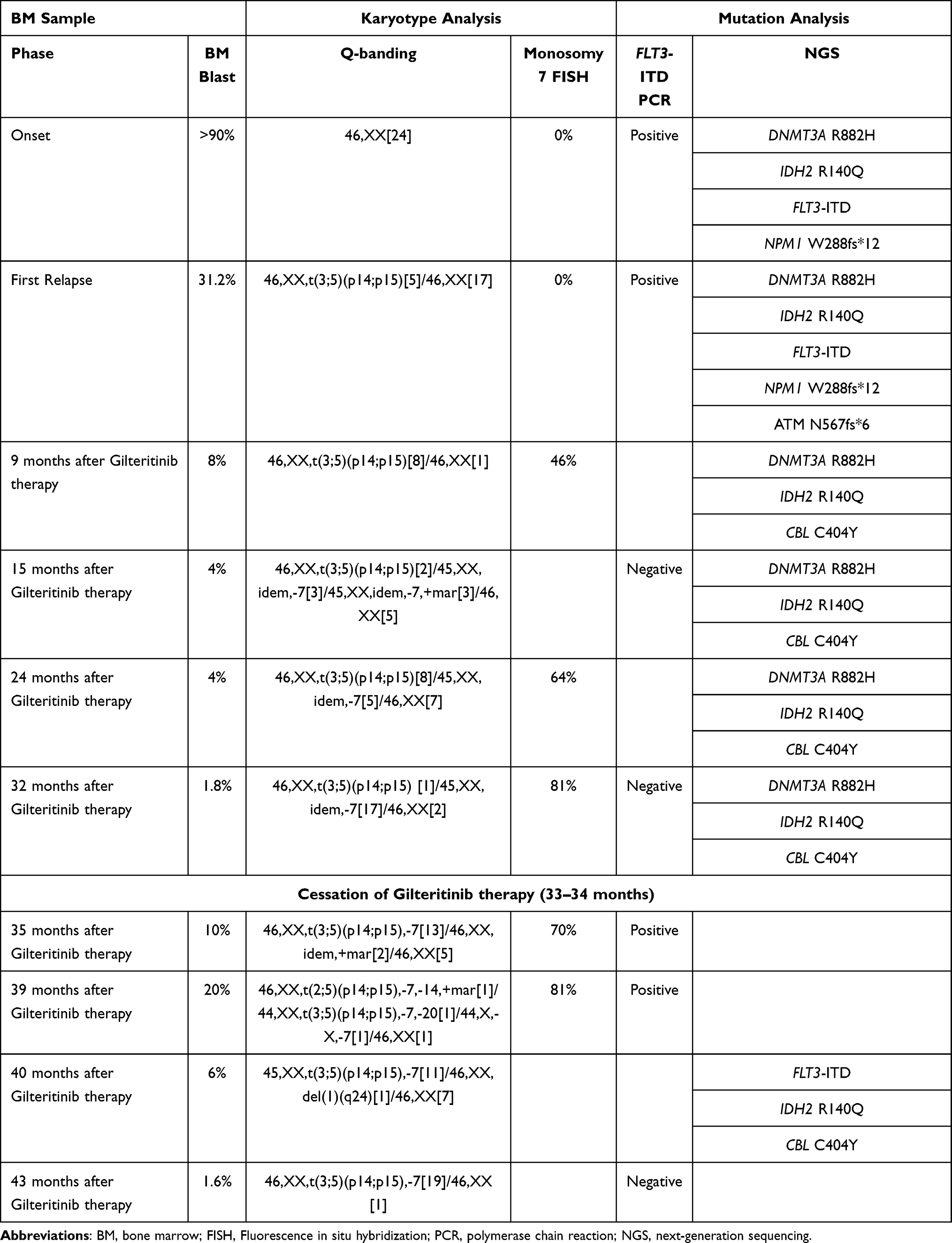 |
Table 1 Summary of Genome Profiling in a Patient with Long Term Gilteritinib Therapy |
 |
Figure 1 Bone marrow smear at onset of AML, first relapse of AML, 24 months after gilteritinib therapy, and relapse after gilteritinib cessation. (A and B) Wright–Giemsa (WG) staining at onset, showing extensive bone marrow necrosis. (C) Myeloperoxidase (MPO) staining at onset. (D–F) WG staining at first relapse, showing myeloblast increase without blood cell dysplasia. (G–I) WG staining at 24 months after gilteritinib therapy, showing multi-lineage blood cell dysplasia without myeloblast increase. (J–L) WG staining at relapse after gilteritinib cessation (35 months after gilteritinib treatment), showing a repopulation of myeloblasts against the background of multilineage dysplasia. Magnification is 100x for (A, D, G, and J), and 1000x for (B, C, E, F, H, I, K, and L). |
Discussion
Recently, the influence of chemotherapy on clonal hematopoiesis has been attracted attention. It has been shown that clonal hematopoiesis before chemotherapy is a risk factor for therapy-related myeloid neoplasia.11,12
We observed clonal hematopoiesis in a FLT3-ITD-positive AML patient at the time of diagnosis, relapse, and up to 43 months after the administration of gilteritinib. In the ADMIRAL study, eight of 247 patients in a gilteritinib cohort were confirmed to have survived in remission for more than two years without HSCT.7 However, most of them (7/8) were under the age of 65 and had a history of transplantation before participating in the study. Gilteritinib therapy can rarely be continued for more than 40 months in older AML patients.
Mutations in DNMT3A, IDH2, NPM1, and FLT3-ITD were detected at diagnosis, and DNMT3A and IDH2 were detected in all subsequent samples. Clones with these mutations were considered pre-leukemia clones.
At the time of recurrence after cytotoxic chemotherapy, NPM1 mutation and FLT3-ITD were detected, suggesting that the clone had re-expanded at the time of initial diagnosis. At the time of recurrence, chromosomal analysis revealed a clone of t(3;5)(p14;p15). Following the administration of gilteritinib, clones harboring NPM1 mutation and FLT3-ITD disappeared and a clone with CBL mutation emerged. Chromosomal analysis revealed that the clones with t(3;5)(p14;p15) had newly acquired monosomy 7.
CBL has E3 ubiquitin ligase activity and plays an inhibitory role in signal transduction by ubiquitylating activated tyrosine kinase receptors such as FLT3 and PDGFR.13 CBL mutations in myeloid malignancies are concentrated in the Linker and RING finger domains responsible for E3 ubiquitin ligase activity, impairing its activity.13,14 In recent years, activation of the RAS/MAPK pathway has attracted attention as a mechanism of resistance to gilteritinib. McMahon et al reported that 36.6% of 41 cases who resisted gilteritinib therapy showed the emergence of gene mutations related to the RAS/MAPK pathway, including CBL, after administration of gilteritinib.15 Furthermore, Schnittger et al analyzed CBL mutations in 636 cases of myelodysplastic syndromes (MDS) and MDS/myeloproliferative neoplasms and reported that CBL mutations were associated with monosomy 7 and TET2 mutation.16 This patient in the current study had multi-lineage blood cell dysplasia during gilteritinib therapy. These dysplasias are presumed to result from the expansion of gilteritinib-resistant clones with monosomy 7 and CBL mutations.
Interestingly, in the current case, the FLT3-ITD clone reappeared after the cessation of gilteritinib treatment due to ileus. Furthermore, the reemerging clones remained sensitive to gilteritinib. This observation suggests that the FLT3-ITD clone may have higher proliferative potential than the CBL mutation/monosomy 7 clone in the absence of FLT3 inhibitors. We speculated that gilteritinib suppressed long-term FLT3-ITD clones, resulting in hematopoietic formation by CBL-mutant/monosomy 7 clones.
The present study had some limitations. NGS methods for genetic analysis differ depending on the timing of the BM collection. Therefore, in this case, it may not be possible to accurately compare changes in clone size. The CBL mutation/monosomy 7 clone was not detected by NGS or FISH prior to gilteritinib administration. However, it is necessary to evaluate the possibility that this clone existed as a minor clone before the administration of gilteritinib using digital PCR.
Conclusion
The present analysis demonstrated that (1) hematopoiesis derived from gilteritinib-resistant clones was generated in a long-term gilteritinib-treated patient and (2) FLT3-ITD clones regained clonal dominance in the absence of FLT3 inhibition. These findings suggest that gilteritinib affects the selection of dominant clones during clonal hematopoiesis. Sequential clonal analyses in patients treated with long-term molecular targeted drugs, such as FLT3 inhibitors, are expected to clarify the mechanism by which hematopoietic tumors emerge from clonal hematopoiesis; however, further studies are required.
Consent to Participate
The patient’s daughter provided informed consent to publish the patient’s case details and any accompanying images.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Akihiko Gotoh reports grants, personal fees from Eisai, grants, personal fees from Ono Pharmaceutical, grants, personal fees from Taiho Pharmaceutical, grants, personal fees from Takeda Pharmaceutical, grants, personal fees from Nippon Shinyaku, grants, personal fees from Chugai Pharmaceutical, grants from MSD, grants, personal fees from Otsuka Pharmaceutical, grants, personal fees from Sumitomo Pharma, grants from Bayer, grants, personal fees from Daiichi Sankyo, grants, personal fees from Nihon Pharmaceutical, personal fees from Novartis Pharma, personal fees from Alexion Pharmaceuticals, personal fees from Kyowa Kirin, personal fees from Janssen, personal fees from Pfizer Japan, personal fees from Sanofi, personal fees from PharmaEssentia Japan, outside the submitted work. Yosuke Minami received research funding from Ono, and Honoraria from Bristol-Myers Squibb, Novartis, and Pfizer. Yuka Harada reports grants from JSPS KAKENHI Grant, grants from Clinical Research Fund of Tokyo Metropolitan Government, during the conduct of the study. Part of the NGS assay was supported by a National Cancer Research and Development Expenses Grant. The authors report no other conflicts of interest in this work.
References
1. Khoury JD, Solary E, Abla O, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36(7):1703–1719. doi:10.1038/s41375-022-01613-1
2. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16. doi:10.1182/blood-2015-03-631747
3. Abelson S, Collord G, Ng SWK, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559(7714):400–404. doi:10.1038/s41586-018-0317-6
4. Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–2439. doi:10.1182/blood.V97.8.2434
5. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221. doi:10.1056/NEJMoa1516192
6. Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381(18):1728–1740. doi:10.1056/NEJMoa1902688
7. Perl AE, Larson RA, Podoltsev NA, et al. Follow-up of patients with R/R FLT3-mutation-positive AML treated with gilteritinib in the Phase 3 ADMIRAL trial. Blood. 2022;139(23):3366–3375. doi:10.1182/blood.2021011583
8. Hosono N, Chi S, Yamauchi T, et al. Clinical utility of genomic profiling of AML using paraffin-embedded bone marrow clots: HM-SCREEN-Japan 01. Cancer Sci. 2023;114(5):2098–2108. doi:10.1111/cas.15746
9. Konishi T, Sadato D, Toya T, et al. Impact of gene alterations on clinical outcome in young adults with myelodysplastic syndromes. Sci Rep. 2023;13(1):2641. doi:10.1038/s41598-023-29794-4
10. Arai H, Chi S, Utsu Y, et al. A practice-oriented genome profiling study with the novel halo-shape annealing and defer-ligation enrichment (HANDLE) system: HM-screen-JAPAN02. Blood. 2022;140(Supplement 1):10722–10724. doi:10.1182/blood-2022-159351
11. Gillis NK, Ball M, Zhang Q, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol. 2017;18(1):112–121. doi:10.1016/S1470-2045(16)30627-1
12. Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552–555. doi:10.1038/nature13968
13. Sanada M, Suzuki T, Shih LY, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009;460(7257):904–908. doi:10.1038/nature08240
14. Dunbar AJ, Gondek LP, O’Keefe CL, et al. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008;68(24):10349–10357. doi:10.1158/0008-5472.CAN-08-2754
15. McMahon CM, Ferng T, Canaani J, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019;9(8):1050–1063. doi:10.1158/2159-8290.CD-18-1453
16. Schnittger S, Bacher U, Alpermann T, et al. Use of CBL exon 8 and 9 mutations in diagnosis of myeloproliferative neoplasms and myelodysplastic/myeloproliferative disorders: an analysis of 636 cases. Haematologica. 2012;97(12):1890–1894. doi:10.3324/haematol.2012.065375
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.