Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Giant thrombosed intracavernous carotid artery aneurysm presenting as Tolosa–Hunt syndrome in a patient harboring a new pathogenic neurofibromatosis type 1 mutation: a case report and review of the literature
Authors Conforti R, Cirillo M, Marrone V, Galasso R, Capaldo G, Giugliano T, Scuotto A, Piluso G, Melone MA ![]()
Received 12 June 2013
Accepted for publication 5 August 2013
Published 20 January 2014 Volume 2014:10 Pages 135—140
DOI https://doi.org/10.2147/NDT.S49784
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Renata Conforti,1 Mario Cirillo,2 Valeria Marrone,1 Rosario Galasso,1 Guglielmo Capaldo,3 Teresa Giugliano,4 Assunta Scuotto,1 Giulio Piluso,4 Mariarosa AB Melone3,5
1Neuroradiology Unit, Department of Clinical and Experimental Medicine and Surgery, 2Radiology Unit, Department of Medical, Surgical, Neurological, Metabolic and Aging Sciences, 3Division of Neurology, Department of Clinical and Experimental Medicine and Surgery, 4Department of Biochemistry, Biophysics and General Pathology, School of Medicine, Second University of Naples, Naples, Italy; 5Institute of Protein Biochemistry, National Research Council, Naples, Italy
Abstract: Neurofibromatosis type 1 (NF1) is a relatively common single-gene disorder, and is caused by heterozygous mutations in the NF1 gene that result in a loss of activity or in a nonfunctional neurofibromin protein. Despite the common association of NF1 with neurocutaneous features, its pathology can extend to numerous tissues not derived from the neural crest. Among the rare cerebrovascular abnormalities in NF1, more than 85% of cases are of purely occlusive or stenotic nature, with intracranial aneurysm being uncommon. Predominantly, the aneurysms are located in the internal carotid arteries (ICAs), being very rare bilateral aneurysms. This report describes a very unusual case of fusiform aneurysms of both ICAs in a Caucasian NF1 patient, with a new pathogenic intragenic heterozygous deletion of the NF1 gene, presenting at age 22 years with Tolosa–Hunt syndrome, because of partial thrombosis of the left giant intracavernous aneurysm. Medical treatment with anticoagulant therapy allowed a good outcome for the patient. In conclusion, early identification of cerebral arteriopathy in NF1 and close follow-up of its progression by neuroimaging may lead to early medical or surgical intervention and prevention of significant neurologic complications.
Keywords: neurofibromatosis type 1, NF1 gene, multiplex ligation-dependent probe amplification (MLPA), intracranial aneurysms, Tolosa–Hunt syndrome
Introduction
Von Recklinghausen neurofibromatosis or neurofibromatosis type 1 (NF1), is one of the most common autosomal dominant conditions affecting the nervous system, occurring with an estimated incidence of one in 2,500–3,000 individuals independent of ethnic group, race, and sex.1 Caused by an inactivating mutation in the NF1 gene on chromosome 17 (17q11.2), the disease is associated with increased morbidity and mortality.2,3 The NF1 gene encodes the neurofibromin, a 250 kDa cytoplasmic protein with a centrally positioned Ras-specific guanosine triphosphatase-activating domain that acts as a negative regulator of Ras/mitogen-activated protein kinase, which has a cardinal role in mitogenic intracellular signaling pathways.4 Currently, the diagnosis of NF1 is made in an individual with at least two of the following clinical features: 1) six or more café au lait spots (larger than 5 mm in greatest diameter in prepubertal individuals and larger than 15 mm in greatest diameter in postpubertal individuals); 2) skin-fold freckling in non-sun-exposed areas; 3) iris Lisch nodules (hamartomas diagnosed on slit-lamp examination); 4) two or more neurofibromas (benign peripheral nerve-sheath tumors) of any type and/or one plexiform neurofibroma; 5) optic pathway gliomas; 6) characteristic bony dysplasia of the long bones and sphenoid wing; and 7) a first-degree family relative with NF1.5 Despite the common association of NF1 with neurocutaneous features, its pathology can extend to numerous tissues not derived from the neural crest. Arteriopathy has been an underreported potential complication of NF1. The most common vascular abnormality in patients with NF1 is renal artery stenosis. When cerebrovascular abnormalities occur, they are usually occlusion and stenosis of major intracranial vessels. The association between NF1 and intracranial aneurysms has not been firmly established. Although numerous case reports and small series of patients, both in childhood and adulthood, with NF1 and intracranial aneurysms have been published, other reports have called this association into question.6–8 Here, we report a longitudinal observation through almost 2 decades in a 25 year old Caucasian patient with a new pathogenic intragenic heterozygous deletion of the NF1 gene, and with a phenotype complicated by fusiform aneurysms of both internal carotid arteries (ICAs), presenting at age 22 years with Tolosa–Hunt syndrome (THS) because of partial thrombosis of the left giant intracavernous aneurysm. A close clinical/paraclinical follow-up evaluation allowed us to act quickly in the diagnosis, and the treatment led to a rapid resolution of ptoms that are potentially very disabling.
Case report
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A 25 year old Caucasian man with skin hyperpigmentation, multiple soft-tissue masses bilaterally at the neck, and multiple café au lait spots on the trunk and limbs since 2 years of age was diagnosed with NF1 due to a mutation on the paternally derived allele. In fact, family history was remarkable for a father with clinical features consistent with NF1. The patient had been followed clinically since the age of 4 years, when a routine magnetic resonance imaging (MRI) study showed two “incidental” intracranial intracavernous ICA aneurysms: both the left and right side. There was no family history of ptomatic aneurysms. Furthermore, elective MRI screening in first-degree relatives or in other family members did not reveal aptomatic cerebral aneurysms.
The patient was therefore molecularly characterized by performing a comprehensive mutation scanning of the NF1 gene. The pathogenic mutation eluded identification at the ribonucleic acid level by direct sequencing of reverse transcription polymerase chain reaction products covering the entire NF1 transcript. Conversely, multiplex ligation-dependent probe amplification, was able to detect a new intragenic heterozygous deletion (NM_000267.3:c.1330-?_1476+?del) encompassing the adjacent exons 12 (10a) and 13 (10b) of the NF1 gene (Figure 1).9 Otherwise in good health, the patient has followed a long clinical 20-year follow-up without any special treatment. At age 22 years, the patient was admitted to our Division of Neurology for left periorbital pain, ipsilateral ocular motor nerve palsies, and diplopia. Four days prior to admission, the patient had the first episode in his life of severe left periorbital pain; 72 hours later, he also presented with limited left eye movements, ipsilateral palpebral ptosis, and horizontal diplopia. Pain did not recede after the administration of nonsteroidal anti-inflammatory drugs. Neuro-ophthalmologic examination showed left palpebral ptosis, exotropia of the primary look of the left eye, and paresis of the third, fourth, and sixth left cranial nerves. A provisional diagnosis of THS led us to perform a diagnostic workup, including routine blood tests, inflammatory markers, fasting blood glucose, lupus blood test (antinuclear antibody, anti-double-stranded deoxyribonucleic acid, cytoplasmic antineutrophil cytoplasmic antibody), and imaging studies. Laboratory tests were normal. MR brain study documented a flow-void signal of patent intracavernous side carotid siphon (arrowhead, Figure 2A) and, medially, intravascular characteristic signal modification suggestive of breakdown of hemoglobin products in the thrombus (asterisk, Figure 2A). Time-of-flight MR angiography, with 3-D maximum-intensity projection reconstruction (Figure 2B), detailed the two intracranial fusiform aneurysms, one at the left ICA as a giant fusiform intracavernous aneurysm (larger than 25 mm in diameter), and the second as a contralateral smaller aneurysm (12 mm large). Finally, an angio-computed tomography shaded surface display reconstruction image (Figure 2C), showed magnified detail of a left fusiform giant aneurysm and left selective ICA digital angiography (Figure 2D) confirmed a partially thrombosed giant intracavernous aneurysm. According to neuroradiological study, we concluded that the patient had a ptomatic form of THS.10 A conservative treatment was chosen, and the patient underwent 5 days of standard subcutaneous unfractionated heparin treatment adjusted in dose to prolong the aPTT (activated Partial Thromboplastin Time) between 2.0 and 2.5. After the fifth day of heparin therapy, oral sodium warfarin was initiated, adjusted in dose to achieve an international normalized ratio level between 2.0 and 3.0. The outcome was favorable: at week 10 the patient was aptomatic, and his neuro-ophthalmologic examination was completely normal. The patient currently has no neurological sequelae, and is still on oral anticoagulant therapy.
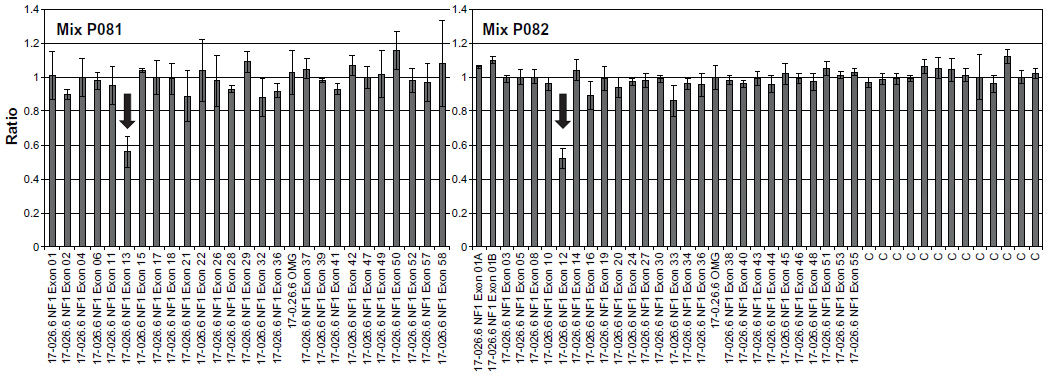 | Figure 1 Bar graph showing the result of multiplex ligation-dependent probe amplification (MLPA) analysis using the SALSA MLPA P081 and P082 NF1 kits (MRC-Holland, Amsterdam, the Netherlands). Relative amounts of probe-amplified products were compared with reference samples and data analysis was performed using the Coffalyser 9.4 package (MRC-Holland). Values under a threshold of 0.7 and over a threshold of 1.3 for multiple adjacent probes indicate the presence of a deletion or duplication, respectively. The arrows highlight the exons 12 (10a) and 13 (10b) deleted in this patient. |
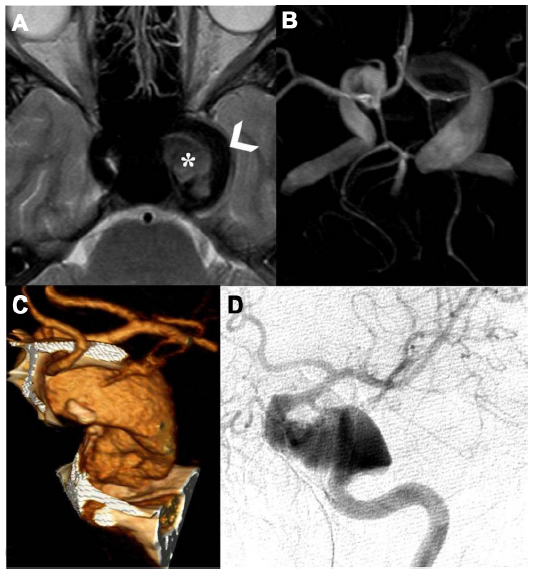 | Figure 2 Magnetic resonance (MR) axial T2-weighted cerebral study shows flow-void signal of patent intracavernous side carotid siphon (arrowhead), and, medially, intravascular characteristic signal modification to refer breakdown hemoglobin products of thrombus (asterisk) (A); time-of-flight MR angiography (TOF-MRA), using a maximum-intensity projection (MIP) algorithm with 3-D MIP reconstruction, detailed two intracranial fusiform aneurysms, one at the left internal carotid artery (ICA) as a giant fusiform intracavernous aneurysm (larger than 25 mm in diameter), and the second as a contralateral smaller aneurysm (12 mm larger) (B); angio-computed tomography (A-CT) shaded surface display reconstruction (SSD) study shows magnified detail of left partially thrombosed fusiform giant aneurysm (C); left selective ICA digital angiography anteroposterior view confirms the partially thrombosed giant intracavernous aneurysm (D). |
Discussion
Vascular disease associated with NF1 is an underrecognized complication, and includes stenoses, aneurysms, and arteriovenous malformations involving the aortic, renal, mesenteric, carotid-vertebral, subclavian-axillary, iliofemoral, intracerebral, and coronary arteries.11,12 Reubi first described the pathology of these lesions in 1945, and classified them into three types of arterial lesions according to the size of the vessel involved and the extent of the lesion – a pure intimal type, an advanced intimal type, and the intimal-aneurysmal type – and Feyrter presented an additional form: the nodular or epithelial type.13,14 A hallmark of diseased vessels in NF1 patients is lumen occlusion and intimal wall hyperplasia.13 Salyer and Salyer suggested that intimal thickening in NF1 vasculopathy is the result of a proliferation of Schwann cells within the arteries.15 This implies a pathogenic relationship between these lesions and the neurofibromas that characterize this disease. From a molecular point of view, while it is well established that aberrations in Ras signaling lead to human malignancies, emerging data generated in genetically engineered mouse models now implicate perturbations in the Ras-signaling axis in vascular smooth-muscle cells as central to the initiation and progression of neointimal hyperplasia and arterial stenosis.16
Cerebrovascular complications that are quite variable from one patient to the next can be more serious, particularly among younger patients, as reported by an analysis performed in the US from 1983 to 1995 indicating that the median age of death for NF1 patients was 15 years lower than that of the general population.3 More recent studies confirmed these observations.17,18
Among the rare cerebrovascular abnormalities in NF1, more than 85% of cases are of purely occlusive or stenotic nature, with intracranial aneurysm being uncommon. Schievink et al detected incidental intracranial aneurysms in two (5%) of 39 patients with NF1 who were hospitalized for other reasons, suggesting that patients with NF1 are at an increased risk of developing intracranial aneurysms as a vascular manifestation of NF1.7 From published case series of ptomatic intracranial aneurysms in NF1 patients, it seems there may be a wide ptomatology, either isolated or syndromic, thus underlining the importance of a clear correlation between anatomical sites and clinical presentations.6,19–21
Reported here is an unusual case of THS developing in association with a rapidly progressive thrombosis of intracavernous carotid artery aneurysm in a 25 year old NF1 patient. Because of the rarity of this association, we reviewed the literature via Ovid and PubMed in an attempt to establish possible demographic and clinical characteristics that may suggest putative mechanisms of pathogenesis. Studies from the 1940s to the present day were searched using the following terms: NF1, carotid-cavernous aneurysms, and Tolosa–Hunt syndrome. To the best of our knowledge, this is the first documented case of an association of NF1 and THS. It should be duly noted that THS accounts for <5% of cases of painful ophthalmoplegia, and affects both sexes equally and all ages, with a peak in the fifth decade. Although the etiology of the syndrome remains unclear, THS is largely considered a painful syndrome of idiopathic, self-limited inflammation of the cavernous sinus, typically responsive to corticosteroids.22,23 Diagnosis of THS involves the use of MRI scan and blood test.24,25 Diagnosis is also done based on the response of the patient suffering from this syndrome to corticosteroids or other steroidal drugs. In our case, it should be remarked that anticoagulant therapy has completely solved the ptoms without any neurological sequelae, without any need to administer corticosteroids.
Actually, there are many other causes of painful ophthalmoplegia, such as sella syndromes, cerebral aneurysms, carotid cavernous fistulae, tumors beside the cavernous or orbital sinus, diabetic neuritis, and infections from contiguous regions.10,26 Moreover, there has been report of some reversible artery stenosis in THS, and these constriction changes were considered to be caused by compression by idiopathic granulomatous inflammation or periarteritis in the cavernous sinus.22,23,27,28 Therefore, it is more likely that an etiopathogenetic linkage between THS and intracavernous vasculopathy can be found. Recently, an unusual case of THS with a reversible internal carotid dissection aneurysm and a stenosis in the intracavernous ICA segment was described in the literature.29 Usually, cavernous sinus aneurysms, which represent 5% of all intracranial aneurysms, are more frequent in the elderly and present with an indolent ophthalmoplegia. In many cases, the aneurysms were incidental findings on neuroimaging or autopsy. Intriguingly, in a recent paper, Shelton et al described the first case of cavernous sinus syndrome from an intracavernous ICA aneurysm in a baby with tuberous sclerosis, another relevant phakomatosis.30 Therefore, a hypothesis we can keep in consideration is that phakomatoses give a predisposition, by a possible developmental defect of the arterial wall, to the emergence of intracranial aneurysms, which according to their location can give rise to distinctive ptomatologies (ie, indolent ophthalmoplegia due to intracavernous ICA). In our NF1 patient, we propose that the pain associated with ophthalmoplegia may be due to an “idiopathic” inflammation of the vascular wall, whose origin cannot to our knowledge be explained by a known association with phakomatoses themselves, but at the same time cannot be excluded at all.
Conclusion
Mutations of the NF1 gene result in either a loss of function or a nonfunctional neurofibromin protein that allows for Ras activation and ultimately for uncontrolled cell proliferation. NF1 haploinsufficiency results in disease with complete penetrance and a range of clinical complications. Cerebrovascular disease appears to contribute disproportionately to mortality in children and young adults with NF1.3 On review of the literature, bilateral fusiform aneurysms in the anterior circulation, as described in our patient, are extremely uncommon in NF1 and represent a therapeutic challenge, particularly if the aneurysm is giant and intracavernously located. Furthermore, the patient’s clinical characteristics are also notable for the rapid progression of ptoms, after a clinically silent long period, and their precise anatomoclinical correlation in determining a ptomatic form of THS, emphasizing the potential dynamic nature of aneurysms. This report serves to draw attention to the management of cerebrovascular disease as a specific potential complication of NF1 that has not been specifically addressed by current published guidelines for the diagnosis and management of NF1 patients. As is well known to clinicians, unlike optic glioma, clinical examination may not identify children at risk for cerebral arteriopathy, and therefore the natural history, frequency, and pathogenesis of vascular lesion formation are not well defined.31 In September 2008, the American Heart Association published a scientific statement on the management of stroke in infants and children, and recommended that routine vascular screening may be considered in individuals with relatively common and high-risk disorders, such as NF1, Down syndrome, and sickle-cell disease.32 In particular, urgent evaluation for the presence of an intracranial aneurysm should be considered in children or young adults with NF1 or other phakomatoses who develop new cranial nerve deficits, or even other types of focal deficits. Conversely, children with aneurysms should be evaluated for the stigmata of phakomatoses. In particular, on detecting a carotid bruit, the clinician is duty-bound to investigate further, as a proportion of patients will have significant stenosis or other carotid disease, even if they are aptomatic. In contrast with current published guidelines for the diagnosis and management of NF1 patients, we recommend also the potential for universal noninvasive neuroimaging (ie, MRI and transcranial Doppler sonography as effective diagnostic tests) of young children with confirmed or probable NF1. Furthermore, targeting signaling pathways may prove to be a useful approach, because cell migration and proliferation are critical events in the development of vasculopathy. Elucidation of the role of neurofibromin in endothelial dysfunction and smooth-muscle cell migration and proliferation may lead to novel approaches to treatment of vascular disease in general, as well as in patients with NF1.
Author contributions
MABM, study concept, manuscript preparation; GC, MABM, case history, clinical data collection; RC, MC, VM, RG, AS, neuroimaging studies; TG, GP, genetic testing. All authors were involved in the critical revision of the manuscript, and read and approved the final proof before publishing.
Disclosure
The authors report no conflicts of interest in this work.
References
Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278:51–57. | |
Viskochil D, Buchberg AM, Xu G, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell. 1990;62:1887–1892. | |
Rasmussen S, Yang QH, Friedman JM. Mortality associated with neurofibromatosis 1 in the United States from 1983 to 1995: an analysis using data from death certificates. Am J Hum Genet. 1999;65:A49. | |
Gutmann DH, Collins FS. The neurofibromatosis type 1 gene and its protein product, neurofibromin. Neuron. 1993;10:335–343. | |
[No authors listed]. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575–578. | |
Rosser TL, Vezina G, Packer RJ. Cerebrovascular abnormalities in a population of children with neurofibromatosis type 1. Neurology. 2005;64:553–555. | |
Schievink WI, Riedinger M, Maya MM. Frequency of incidental intracranial aneurysms in neurofibromatosis type 1. Am J Med Genet A. 2005;134A:45–48. | |
Conway JE, Hutchins GM, Tamargo RJ. Lack of evidence for an association between neurofibromatosis type I and intracranial aneurysms: autopsy study and review of the literature. Stroke. 2001;32:2481–2485. | |
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. | |
La Mantia L, Curone M, Rapoport AM, Bussone G. Tolosa-Hunt syndrome: critical literature review based on IHS 2004 criteria. Cephalalgia. 2006;26:772–781. | |
Friedman JM, Arbiser J, Epstein JA, et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 Cardiovascular Task Force. Genet Med. 2002;4:105–111. | |
Oderich GS, Sullivan TM, Bower TC, et al. Vascular abnormalities in patients with neurofibromatosis syndrome type I: clinical spectrum, management, and results. J Vasc Surg. 2007;46:475–484. | |
Reubi F. Neurofibromatosis et lesions vasculares [Neurofibromatosis and vascular lesions]. Schweiz Med Wochenschr. 1945;75:463–465. | |
Feyrter F. über die vasculare Neurofibromatose, nach Utersuchungen am menschlichen Magen-Darmschlauch. Virchow [About the vascular neurofibromatosis, according to studies on human gastrointestinal tube]. Arch Pathol Anat. 1949;317:221–265. | |
Salyer WR, Salyer DC. The vascular lesions of neurofibromatosis. Angiology. 1974;25:510–519. | |
Li F, Munchhof AM, White HA, et al. Neurofibromin is a novel regulator of RAS-induced signals in primary vascular smooth muscle cells. Hum Mol Genet. 2006;15:1921–1930. | |
Evans DG, O’Hara C, Wilding A, et al. Mortality in neurofibromatosis 1: in North West England: an assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet. 2011;19:1187–1191. | |
Wilding A, Ingham SL, Lalloo F, et al. Life expectancy in hereditary cancer predisposing diseases: an observational study. J Med Genet. 2012;49:264–269. | |
Krishnaswami V, Gilmore J, Vahidassr D. Young stroke due to vascular anomaly from neurofibromatosis type 1. BMJ Case Rep. 2012; 2012. | |
Benatar MG. Intracranial fusiform aneurysms in von Recklinghausen’s disease: case report and literature review. J Neurol Neurosurg Psychiat. 1994;63:1279–1280. | |
Poli P, Peillon C, Ladha E, Watelet J, Testart J. [Multiple intracranial aneurysms in relation to Recklinghausen’s disease. Report of a case.] J Mal Vasc. 1994;19:253–255. French. | |
Tolosa E. Periarteritic lesions of the carotid siphon with the clinical features of a carotid infraclinoidal aneurysm. J Neurol Neurosurg Psychiatry. 1954;17:300–302. | |
Hunt WE, Meagher JN, Lefever HE, Zeman W. Painful ophthalmoplegia. Its relation to indolent inflammation of the carvernous sinus. Neurology. 1961;11:56–62. | |
Kline LB, Hoyt WF. The Tolosa-Hunt syndrome. J Neurol Neurosurg Psychiatry. 2001;71:577–582. | |
Schuknecht B, Sturm V, Huisman TA, Landau K. Tolosa-Hunt syndrome: MR imaging features in 15 patients with 20 episodes of painful ophthalmoplegia. Eur J Radiol. 2009;69:445–453. | |
Mandrioli J, Frank G, Sola P, et al. Tolosa-Hunt syndrome due to actinomycosis of the cavernous sinus: the infectious hypothesis revisited. Headache. 2004;44:806–811. | |
del Toro M, Macaya A, Vazquez E, Roig M. Painful ophthalmoplegia with reversible carotid stenosis in a child. Pediatr Neurol. 2001;24:317–319. | |
Ozawa T, Minakawa T, Saito A, Yoneoka Y, Yoshimura J, Arai H. MRA demonstration of “periarteritis” in Tolosa-Hunt syndrome. Acta Neurochir (Wien). 2001;143:309–312. | |
Zhou Z, Zhou G, Lu T, Xu G, Liu X. Tolosa-Hunt syndrome with reversible dissection aneurysm. Neurol Sci. 2010;31:777–779. | |
Shelton JB, Ramakrishnaiah R, Glasier CM, Phillips PH. Cavernous sinus syndrome from an internal carotid artery aneurysm in an infant with tuberous sclerosis. J AAPOS. 2011;15:389–391. | |
Rea D, Brandsema JF, Armstrong D, et al. Cerebral arteriopathy in children with neurofibromatosis type 1. Pediatrics. 2009;124:e476–e483. | |
Roach ES, Golomb MR, Adams R, et al. Management of stroke in infants and children: a scientific statement from a Special Writing Group of the American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young. Stroke. 2008;39:2644–2691. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.