Back to Journals » Cancer Management and Research » Volume 10
Germline BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance associated with breast/ovarian cancer: a report from North India
Authors Mehta A ![]() , Vasudevan S
, Vasudevan S ![]() , Sharma SK
, Sharma SK ![]() , Kumar D
, Kumar D ![]() , Panigrahi M
, Panigrahi M ![]() , Suryavanshi M, Gupta G
, Suryavanshi M, Gupta G ![]()
Received 13 September 2018
Accepted for publication 29 October 2018
Published 30 November 2018 Volume 2018:10 Pages 6505—6516
DOI https://doi.org/10.2147/CMAR.S186563
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Anurag Mehta,1 Smreti Vasudevan,2 Sanjeev Kumar Sharma,1 Dushyant Kumar,1 Manoj Panigrahi,1 Moushumi Suryavanshi,1 Garima Gupta2
1Department of Laboratory and Transfusion Services, Rajiv Gandhi Cancer Institute and Research Centre, Rohini, Delhi 110085, India; 2Department of Research, Rajiv Gandhi Cancer Institute and Research Centre, Rohini, Delhi 110085, India
Background: The spectrum of BRCA mutations that predispose to development of breast/ovarian cancer in Indian population remains unexplored. We report incidence and various types of pathogenic, likely pathogenic and variants of unknown significance (VUS) mutations in BRCA1 and BRCA2 genes observed at a tertiary cancer center in North India.
Materials and methods: A total of 206 unrelated breast and/or ovarian cancer patients, who met the National Comprehensive Cancer Network (NCCN) guidelines for genetic testing, were screened for germline BRCA1/BRCA 2 mutations on high-throughput sequencing platform; large genomic rearrangements were assessed by multiple ligation probe assay. Mutations were mined in mutational databases, PubMed, and discerned into classes. Furthermore, the clinicopathological correlation of BRCA mutation status with prognostic markers in breast cancer and tumor histology in ovarian cancer was performed.
Results: In total, 45/206 and 17/206 cases showed positivity for BRCA1 and BRCA2 mutations, respectively, whereas 1/206 was positive for a mutation in both the genes. Altogether, 33 distinct BRCA1 mutations were observed, among which 27 were deleterious (12 frameshifts, 8 nonsense, 1 missense, 3 splice-site variants, 2 big deletions and 1 large duplication) and 6 were VUS. Five novel BRCA1 mutations (c.541G>T, c.1681delT, c.2295delG, c.4915C>T and exon 23 deletion) were identified. Seven mutations (c.2214_2215insT, c.2295delG, c.3607C>T, c.4158_4162delCTCTC, c.4571C>A, splicesite_3 (C>T) and exon 21–23 duplication) occurred more than once, whereas 16 distinct BRCA2 mutations were noted – 9 were lethal (6 frameshifts, 2 nonsense and 1 big deletion) and 7 VUS. One unique pathogenic BRCA2 mutation (c.932_933insT) was recognized. Two mutations (c.9976A>T and c.10089A>G) recurred twice. No significant difference in hormone receptor status was observed among BRCA1 carriers, BRCA2 carriers and noncarriers.
Conclusion: We have documented various pathogenic and VUS mutations in BRCA1 and BRCA2 genes observed in the cohort. Six novel mutations were identified. The knowledge shared would assist genetic testing in enabling more focused site-specific screening for mutations in biological relatives.
Keywords: genetic screening, high-throughput sequencing, multiplex ligation-dependent probe amplification assay, novel mutations, recurrent mutations
Introduction
The breast cancer susceptibility genes BRCA1 (MIM# 113705) and BRCA2 (MIM# 600185) produce tumor suppressor proteins that participate in the repair of double-stranded DNA breaks by the “homologous recombination” (HR) DNA repair pathway that restores the DNA to its original self. The absence of BRCA proteins or dysfunctional BRCA proteins makes the HR pathway ineffective and the double-stranded breaks are thus repaired by “nonhomologous end joining”, which is mutagenic and hence carcinogenic. It is for this reason that individuals carrying deleterious germline mutations in BRCA1/BRCA2 genes have an elevated lifetime risk of developing breast cancer (by 60%–85%) and ovarian cancer (by 26%–54% for BRCA1; 10%–23% for BRCA2), when compared to 12.5% and 1% lifetime risks of breast and ovarian cancers, respectively, in women with intact BRCA genes.1–5 In mutation carrier men, the risk of developing breast cancer are 1% and 6% for mutations in BRCA1 and BRCA2 genes, respectively.6 These mutations are inherited with high penetrance in BRCA1 (>50%) and intermediate penetrance in BRCA2 (20%–50%) and account for 5%–10% of breast cancer and 15% of ovarian cancer.7,8 More than 20,000 unique variants have been identified, together for both BRCA1 and BRCA2 genes, which are spread throughout the coding, splice site and intervening sequences of these large genes.9 Mutational landscape of BRCA1/2 is a greenfield area, where novel mutations are still being identified regularly due to wider availability of sequencing facilities, the large size of these genes and ethnic and racial polymorphism. These novel mutations are analyzed in functional assays and through predictive algorithms (in silico) for their pathogenic role. Although most of these mutations are classifiable either as pathogenic (including likely pathogenic) or as benign (including likely benign), about 10% of these genetic alterations are unclassifiable now and are gathered under “variants of unknown significance” (VUS).10
The frequency and types of mutations in BRCA1 and BRCA2 genes seem to be differentially represented among the different population. The knowledge of the pathogenic and likely pathogenic mutations is useful to predict prognosis, make therapeutic decisions and apply risk reduction strategies in patients and carry out cost-effective screening in the first-degree relatives.11 Although the VUS are currently uninformative, they have a 30% likelihood of being pathogenic when functional assays, clinical data, pedigree and predictive data are fully analyzed.12 It is therefore necessary to catalog these too carefully.
The incidence and type of pathogenic mutations and VUS in BRCA1/BRCA2 genes in North Indian population that predispose to the development of breast and ovarian cancers are largely unknown, except in one publication,13 and are only now being slowly discovered. Therefore, this study is an effort to report specific damaging BRCA1/2 mutations in the North Indian population and discuss the clinicopathological features in mutation carriers.
More specifically, the aim of this study is to report the incidence and spectrum of pathogenic, likely pathogenic and VUS mutations in BRCA1 and BRCA2 genes that were observed in breast and ovarian cancer patients tested at a tertiary cancer center in North India on high-throughput sequencing platform followed by multiplex ligation probe assay for long genomic rearrangements (Big Indel) in cases who were found to be negative for pathogenic mutation on direct sequencing. Special attention has been paid to frequently recurring and some novel mutations; those were identified during the course of the study. Clinicopathological correlation with prognostic biomarker status in breast cancer and histology in ovarian cancer has also been performed.
Materials and methods
Research setting and subjects
This study was conducted in a tertiary cancer care center in North India.
A total of 206 unrelated patients with breast and/or ovarian cancer, who met the National Comprehensive Cancer Network (NCCN) recommendations,14 were comprehensively tested for germline mutations in a time frame of 3 years (2015–2018). There were 126 breast cancer subjects, 74 ovarian cancer subjects and 6 subjects diagnosed with both breast and ovarian cancers. Among the breast cancer subjects, there were three male breast cancer cases.
This study was approved by the institutional review board (Rajiv Gandhi Cancer Institute and Research Center); vide the ethical approval letter number RGCIRC/IRB/229/2018, dated September 14, 2018, for the presentation of the study. All the patients had provided written informed consent for participation in the research study and for genetic testing. This study was conducted in accordance with the Declaration of Helsinki.
Isolation of DNA from blood, next-generation sequencing (NGS) and data analysis
Genomic DNA was isolated from 0.2 mL of peripheral blood of the patients using the commercially available DNA isolation kit (Qiagen DNeasy Blood and Tissue kit; Qiagen NV, Hilden, Germany), following the manufacturer’s instructions. Isolated DNA was quantified by Qubit 3.0 Fluorometric quantitation (Thermo Fisher Scientific, Waltham, MA, USA). The library for NGS was prepared manually with 10 ng of the isolated DNA using Oncomine BRCA assay – A328400 (Thermo Fisher Scientific), containing 265 primer pairs for 100% exonic coverage with large intronic flanking regions of BRCA1 and BRCA2 genes. “Template” was prepared using the HiQ OT-200 template kit by using 100 pmol of library. The template was further enriched by employing Ion One Touch ES instrument with the help of Streptavidin MyOne beads. The barcoded and enriched template was loaded on the Ion Torrent 316/318 V2 sequencing chip for deep sequencing on personal genome machine (PGM). Data generated from the runs were assessed for quality metrics on Torrent Suite Viewer (Ion Torrent Suite 5.6; Thermo Fisher Scientific). Successively, variant calling was performed on Oncomine Ion Reporter (Ion Reporter 5.6), and the final report was generated using Oncomine Knowledge Reporter.
Multiplex ligation-dependent probe amplification (MLPA) assay
Cases tested negative for BRCA1/BRCA2 mutations were further investigated for possible large genomic rearrangements by MLPA assay. SALSA MLPA P002:BRCA1 and P090:BRCA2 kits (MRC Holland, Amsterdam, the Netherlands) were used as per the manufacturer’s instructions. Briefly, 100 ng (5 µL) of blood leukocyte DNA was denatured (98°C, 5 minutes), cooled, hybridized with BRCA1/BRCA2 probe mixture (95°C, 1 minute) and incubated overnight (60°C, 16 hours). This was followed by the addition of buffers and ligase provided with the kit (54°C, 15 minutes). Thereafter, the ligase enzyme was heat inactivated (98°C for 5 minutes). After the ligated sample attains room temperature, to 10 µL of the sample, 6 carboxyfluorescein (FAM) labeled primers, deoxyribonucleotide mixture and Taq DNA polymerase were added, and the reaction volume was made up in sterile water and amplified by PCR (35 cycles [95°C, 30 seconds; 60°C, 30 seconds and 72°C, 60 seconds]). The fragments were subjected to capillary electrophoresis on SeqStudio Genetic Analyzer (Thermo Fisher Scientific), and the data generated were analyzed using the Coffalyser.Net software (Amsterdam, the Netherlands).
Classification and identification of BRCA1 and BRCA2 variants including novel variants
The Ion Reporter Software used for variant call listed all the five classes of mutations and polymorphisms. The reported mutations were further checked in linked dbSNP15 and ClinVar databases.16 Additionally, other databases such as BRCA Exchange,9 LOVD17 and Breast Cancer Information Core (BIC)18 were mined along with PubMed publications to reaffirm the assigned class of the mutation, the level of evidence and discern novel mutations. VarSome,19 the integrated search engine, was used to access multiple databases, prediction tools and publications at a single site. In the absence of universal functional assay availability, in silico predictions by assessing phylogenetic conservation and the likelihood of severe physiochemical alterations in the protein were utilized as prediction tools, including Variant Effect Predictor (Ensembl),20 SIFT, PolyPhen, TraP, Mutation Analyzer and other prediction tools available at VarSome.19All genetic annotations and nomenclature were done on GRCh37/hg19 build. GenBank BRCA1: NM_007300 and BRCA2: NM_000059 were used as the reference sequences.
Furthermore, the cDNA position/sequence of the alteration was inputted in MutationTaster21 against Ensembl transcript ID ENST00000471181 for BRCA1 and ENST00000544455 for BRCA2 to obtain the sequence snippets of complementary DNA strand along with bioinformatics prediction for the altered sequence variant.
The variants were classified according to the American Society of Medical Genetics and Genomics (ACMG) recommendations for standards of interpretation and reporting of sequence variations. The variants were organized into five classes as follows: 1) pathogenic/Class 5, 2) likely pathogenic/Class 4, 3) variant of uncertain significance/Class 3 and 4) likely benign/Class 2 and 5) benign/Class 1.22 Without departing from the scope of this study, we have considered the pathogenic, likely pathogenic and VUS mutations.
Clinical correlation
Medical records of the subjects were curated from the “Computerized Patient Record System” maintained at the Institute. The status of breast prognostic markers, estrogen receptor (ER), progesterone receptor (PR) and HER2/neu in breast cancer subjects and tumor histopathology of the ovarian cancer subjects were retrieved from the laboratory information management system.
Statistical analysis
Descriptive statistics were used to summarize the data. Categorical variables were expressed as frequencies and corresponding percentages. Pearson’s chi-squared test of association/Fisher’s exact test was used to compare categorical data, setting the limits of statistical significance as 0.05. Statistical analysis was conducted using SPSS version 23.0 software package (IBM Corporation, Armonk, NY, USA).
Results
A total of 206 breast and/or ovarian cancer subjects were screened for mutations. The baseline characteristics of the study group are presented in Table 1. Among the cases screened, 30.1% (62/206) of the cases were positive for BRCA1/BRCA2 gene mutations, whereas one breast cancer subject was positive for a mutation in both the genes. Overall, BRCA1-positive cases exceeded BRCA2-positive cases (45/206, 21.8% vs 17/206, 8.3%). Also, among the mutation-positive cases, it was observed that BRCA mutations were about twice more common in the ovarian cancer group (42.9%, 31/74) than in the breast cancer group (22.3%, 28/126) (Table 1). Furthermore, mutation frequency was relatively higher in the dual malignancy (breast and ovarian cancers) subjects (4/6, 66.7%). These BRCA1 and BRCA2 mutations identified in the cohort were further carefully examined and profiled into appropriate classes.
 | Table 1 Baseline characteristics of the study group (n=206) |
The spectrum of BRCA1 and BRCA2 germline mutations
BRCA1 mutations
Various BRCA1 germline mutations were detected in the breast and/ovarian cancer subjects. These mutations along with their location on the chromosome, predicted variant effect, class and frequency of occurrence are enlisted in Table 2. Altogether, 33 distinct BRCA1 mutations were identified. These mutations spanned from exon 2 to exon 24 of the BRCA1 gene and were predominantly distributed around exon 10 (Table 2 and Figure 1A). One intronic indel (on IVS7) was also observed. Altogether, based on the predicted variant effect of these mutations, 12 frameshift mutations, 5 missense mutations, 8 nonsense mutations, 3 splice-site variants, 1 big duplication (doubling of exons 21–23), 2 big deletions that caused skipping of exon 23 and skipping of exon 24 and 1 synonymous substitution were observed in the exonic regions of BRCA1 gene in the subjects (Table 2). Most of the BRCA1 mutations identified were pathogenic (Class 5, 79%), one mutation was likely pathogenic (Class 4, 3%), whereas six mutations were grouped in the VUS category (Class 3, 18%) (Table 2 and Figure 1B).
 | Table 2 BRCA1 germline mutations in the breast and/or ovarian cancer group Notes: Bold indicates novel mutation. Previous reports denoted Yes are cited in the databases mentioned in the “Materials and methods” section. aReported previously by Suryavanshi et al.23 bReported by Hogervorst et al.24 cReported by Machackova et al.25 dReported by Armaou et al,26 Engert et al.27 and Sedghi et al.28 eReported by Zorrieh Zahra et al.29 |
 | Figure 1 Mutational spectrum of BRCA genes in the breast and/or ovarian cancer group. Notes: Pie chart depicting (A) exonic distribution of the distinct BRCA1 mutations (an=31). (B) Number of different types of Class 5, Class 4 and Class 3 mutations observed in BRCA1 gene (n=33). (C) Exonic distribution of distinct BRCA2 mutations (n=16). (D) Number of different types of Class 5, Class 4 and Class 3 mutations observed in BRCA2 gene (n=16). (E) Bar graph comparing the number of different Class 4/5 mutations observed in BRCA1/BRCA2 genes (n=36). aDuplication (exons 21–23) and one intronic mutation have been excluded from the pie diagram. |
BRCA2 mutations
Next, we analyzed BRCA2 sequence variants that were detected in the subjects (Table 3). Half of the mutations detected were concentrated around exon 11 (Figure 1C). In total, 16 different mutations were detected in BRCA2 gene, including 6 frameshifts, 7 missense, 2 nonsense mutations and a big deletion of exon 12. Furthermore, 7/16 (44%) of these mutations were VUS, among which 6/7 were missense mutations (Figure 1D and Table 3).
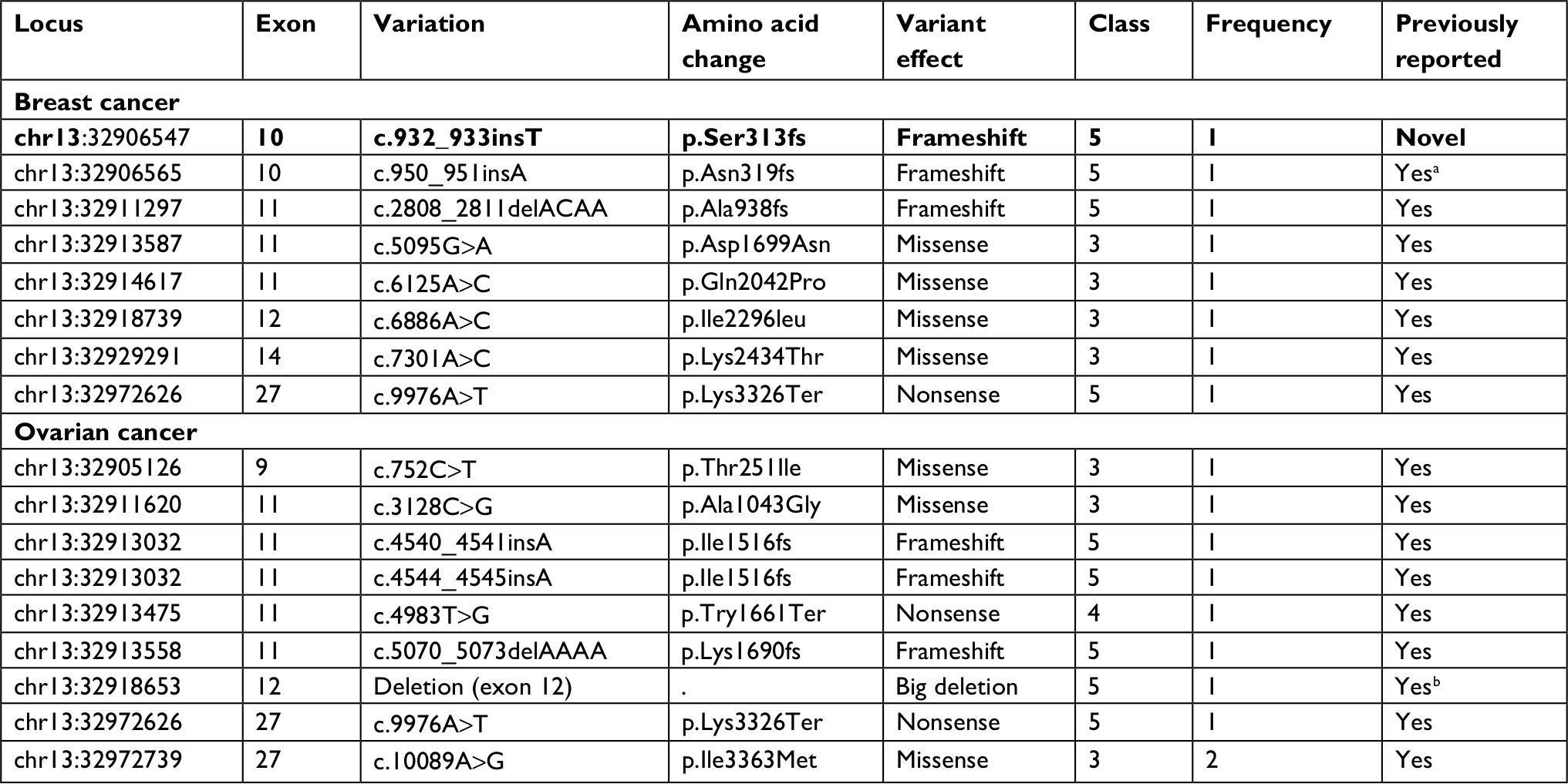 | Table 3 BRCA2 germline mutations in the breast/ovarian cancer patients Notes: Bold indicates novel mutation. Previous reports denoted Yes are cited in the databases mentioned in the “Materials and methods” section. aReported previously by Suryavanshi et al.23 bReported by Rauh-Adelmann et al.30 |
A comparison of Class 4/Class 5 BRCA1 and BRCA2 gene mutations based on the predicted variant effect, observed in the study, is shown in Figure 1E. As evident from Figure 1E, frameshift and protein decaying nonsense mutations were found to be relatively common in the cohort.
In silico analysis of novel mutations
These variants were compared in various databases and existing literature. Although most of the mutations had been recognized previously, five novel mutations (c.541G>T, c.1681delT, c.2295delG, c.4915C>T and large deletion of exon 23) in BRCA1 gene and one novel BRCA2 variant (c.932_933insT) were identified, for which previous reports were unavailable. We further sought to look into these mutations in detail. Mutation plots showing the placement of the mutation and predicted amino acid variation caused by the mutation on BRCA1/2 protein are depicted in Figure 2A and B.
 | Figure 2 Novel BRCA1/BRCA2 mutations identified in the cohort. Notes: Lollipop plots showing the distribution of germline mutations in (A) BRCA1 and (B) BRCA2 genes. Predicted amino acid change has been represented for each mutation. The plots were generated using the online tool MutationMapper – cBioPortal for Cancer Genomics31,32 (GenBank Reference BRCA1: NM_007300 and GenBank Reference BRCA2: NM_000059). |
The first novel mutation c.541G>T (E181X) is a transversion mutation, a single-nucleotide variation which results in the conversion of glutamic acid triplet codon (GAA) at position 541 to a stop codon (TAA). The second mutation c.1681delT is a frameshift mutation wherein deletion of a single base pair (T) changes the reading frame from serine (TCT) at the 561st position to leucine (CTA), thereby shuffling the frame by a single base forward, generating stop codon (TAG) after 11 codons. The third novel mutation c.2295delG is a frameshift mutation, wherein the deletion of base G shifts the frame of glutamic acid (GAG) to again glutamic acid (GAA). But this alteration further changes the next codon serine (AGT) at the 766th position to valine (GTA) and further slips the coding sequence by a single base ahead leading to the generation of a stop codon (TAG) at the 26th codon downstream of the frameshift position. The fourth novel variant identified in this study is c.4915C>T, which causes a missense mutation (H1639Y) where C>T transition replaces histidine (CAT) with tyrosine (TAT). This mutation was predicted to be VUS. Finally, the last variant is a large deletion causing skipping of exon 23, predicted to be protein-truncating at codon 1770.
In the single novel BRCA2 mutation, c.932_933insT, the insertion of a single base T modifies the frame to generate two subsequent phenylalanines (TTT and TTC) and further formats an early stop codon (TAA) at serine (TCT) at the 313th amino acid position.
Specific mutation in BRCA1/BRCA2 genes occurred frequently
Seven different genetic alterations in BRCA1 (c.2214_2215insT, c.3607C>T, c.4571C>A, c.4158_4162delCTCTC, c.2295delG, large duplication spanning exons 21–23 and splicesite_3 (C>T) variations) and two different mutations in BRCA2 (c.9976A>T and c.10089A>G) were observed in the study group, which repeated more than once in the cohort (Table 4).
 | Table 4 Recurrent BRCA1/BRCA2 gene mutations observed in the study cohort Note: Bold indicates novel mutation. Abbreviations: Br, breast; Br and Ov, both breast and ovarian cancers; Ov, ovarian. |
All the repeating BRCA1 mutations were deleterious (Class 5). Among these recurrent mutations, c.2295delG was the only novel mutation observed in two subjects (Table 4).
Clinicopathological characteristics of the study group
To determine whether there exists any relationship between BRCA1/BRCA2 mutation status and prognostic markers (ER, PR and Her2) in breast cancer or tumor histology in ovarian cancer in our study cohort, we extracted medical information from laboratory reports of the patients and analyzed the data. As shown in Table 5, the considerable fraction of BRCA1 carriers (15/24, 62.5%) were triple-negative breast cancer (TNBC) cases, whereas BRCA2 carriers were more or less equally distributed with respect to various breast cancer subtypes. However, no significant correlation was obtained between the prognostic marker status/subtype among the carriers of BRCA1 mutation, BRCA2 mutations and the noncarriers (Table 5).
 | Table 5 Correlation between BRCA mutation status and prognostic markers/tumor histopathology Notes: aOne breast cancer subject positive for a mutation in both BRCA1 and BRCA2 genes was excluded from this comparison. bHR+ include ER- and/or PR-positive subjects. Column percentage is shown in parentheses. Abbreviations: ER, estrogen receptor; HG, high grade; LG, low grade; PR, progesterone receptor. |
With respect to ovarian cancer subjects (including breast and ovarian cancer cases), almost all (34/35) BRCA1/BRCA2 mutation-positive cases had high-grade serous carcinoma histology. Endometrioid carcinoma occurred rarely, observed in three cases belonging to the noncarrier group. Furthermore, tumor histology and BRCA status failed to achieve significant correlation (Table 5).
Discussion
This study is the first to report the spectrum of germline BRCA mutations in breast and ovarian cancer patients from a large cancer care hospital in North India. All coding regions and splice sites with large flanking regions were sequenced, and large genomic rearrangements were identified using MLPA.
Forty-five and 17 patients had germline BRCA1 and BRCA2 mutations, respectively, belonging to Classes 3, 4 and 5. Among the breast cancer cases, 9 of 28 BRCA mutation carriers had a hormone receptor-positive tumor. Contrary to expectation, seven of these had BRCA1 mutation and only two of the nine cases had the BRCA2 mutation. The presence of BRCA1 mutations in ER-positive tumors is known.33–36 These investigators have reported BRCA1 mutation carrier rates of 10%–17%, whereas in this study, the incidence of BRCA1 mutations with ER-positive breast cancer is higher with a rate of 25%. This observation needs to be followed up on a larger data set as these cancers generally arise at a later age and have less aggressive biology compared to ER-negative BRCA1-mutated tumors37 and may have important implications in the use of hormone modulation for chemoprevention. In the latter study by Lips et al,37 the genomic profile of BRCA1-mutated ER-positive tumors was found to be similar to that of BRCA2-mutated ER-positive tumors. In addition, clinicopathological variables in BRCA1-mutated ER-positive cancer were similar to those of BRCA2-mutated ER-positive and sporadic ER-positive breast cancer compared to those of BRCA1-mutated ER-negative cancers. In this study, the ER-positive BRCA1-mutated cancer occurred in the age range of 32–74 years (median age being 46.5 years) and was majorly grade II (data not shown). Histologically, these revealed florid tubule formation and low mitotic activity similar to low-grade sporadic ER-positive breast cancers.
Ten cases of breast cancer tested for germline BRCA mutations were Her2 overexpressing or amplified. Of these, 5 showed deleterious BRCA mutations with 2/24 in BRCA1 and 3/7 in the BRCA2 gene. This emphasizes the need to strictly abide by the guidelines for BRCA testing and not use HR or HER2 status to guide patient selection.
Sixty-two of 206 (30.1%) breast and/or ovarian cancer patients tested showed VUS or pathogenic mutations. A similar incidence of 35% BRCA mutation among breast and/or ovarian cancer patients has been reported in another Indian study.38 To date, 510 distinct types of deleterious BRCA1/BRCA2 mutations (268 BRCA1 and 242 BRCA2) have been cataloged in Asian patients with breast cancer, most of which are frameshift or nonsense mutations.39 From the mutation listed in this compilation from Asian countries, only c.68_69delAG BRCA1 mutation was found in one of our breast cancer patients. This founder mutation in Ashkenazi Jews has also been reported in Pakistan and Malaysia. All the other Class 5 and Class 4 mutations seen by us have not been reported either in the Asian population39 or in the previous study from North Indian population.13 Likewise, the worldwide study of BRCA mutations listing five commonest mutations from several neighboring Asian countries also does not match with the five commonest BRCA1 mutations detected in this study.40
However, of the deleterious mutations identified in this study, c.2214_2215insT (p.Lys739Ter), nonsense BRCA1 mutation was the most frequent, identified in 2/126 of the breast cancer patients and 3/74 of the ovarian cancer patients. This mutation has been reported at least nine times and has the approval of the expert committee.40–42 This common mutation did not reach the proportion of a founder mutation and has not been reported from India in any other study, including the latest study by Singh et al38 and other Indian studies.5,43,44 The other four common BRCA1 mutations seen in this study were c.3607C>T (p.Arg1203Ter); c.4158_4162delCTCTC (p.Ser1387fs); splice site_3C>T and c.2295delG (p.Ser766fs). We also identified three long genomic rearrangements in our cohort of patients, and all these were located toward the 3′-end of the BRCA1 gene. These have been the duplication of exons 21–23 in two cases and deletion of exon 24 in one case. No ovarian or breast cancer cluster region was recognized. Exon 10 was the preferred location for BRCA1 mutations and was mutated in 19/45 cases.
We have identified the following five novel mutations in BRCA1, which are not found in databases and in any publication so far: nonsense mutation (c.541G>T), two frameshift mutations (c.1681delT and c.2295delG), one missense mutation (c.4915C>T) and one big deletion (del exon 23).
With respect to the BRCA2 gene, ambiguous VUS mutations were more common (7/16 of the mutation carriers). Indian population being a less tested and characterized group, a higher VUS frequency is anticipated and calls for future studies. One novel BRCA2 variant (c.932_933insT) was identified. Interestingly, this variant was observed in the subject carrying a dual mutation in both BRCA1 and BRCA2 genes. Two recurrent mutations were observed as follows: c.9976A>T (pathogenic nonsense mutation) and c.10089A>G (missense VUS), in two subjects each. Both the mutations have been reported previously. The c.9976A>T, a truncating allele mutation (creating stop codon Lys3326Ter), has been reevaluated by Thompson et al45 in familial breast cancer cases. Based on a case–control study, it was identified as a low-to-moderate risk variant that was recommended to be included in the breast cancer risk evaluating panels.45,46 This mutation has also been listed in the panel of top 20 BRCA2 mutation frequencies in the BIC database.18
Conclusion
BRCA mutations have a high lifetime risk of hereditary breast and ovarian cancers. Of all the hereditary germline mutations, BRCA mutations have a high penetrance and high incidence second only to Familial Adenomatous Polyposis and Lynch Syndrome, respectively. Identifying deleterious BRCA mutations can help both the patients and biological relatives. Creating this database is a long-drawn process and helps in identifying recurring, deleterious and VUS mutations in a population. Segregation studies and functional analysis over time can help classify VUS better. This study reports various deleterious and VUS mutations in BRCA1 and BRCA2 genes observed in North Indian population. Six novel mutations and nine recurrent mutations have been documented. The knowledge shared would help in deepening the process of screening and assist genetic services in enabling more focused site-specific screening for mutations in biological relatives.
Acknowledgment
The authors thank the patients involved in the study.
Author contributions
AM conceived the idea, supervised the experiments and bioinformatics analysis of mutations, critically evaluated the data and was involved in manuscript writing. SV performed data collection and data analysis, critically evaluated the data and organized and wrote the manuscript. SKS performed DNA extraction, DNA sequencing (NGS and MLPA) and data compilation. DK performed DNA extraction and DNA sequencing. MP performed DNA extraction and DNA sequencing. MS supervised the experiments, bioinformatics analysis of mutations and patient guiding. GG performed genetic counseling and data analysis. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Easton DF, Bishop DT, Ford D, Crockford GP. Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1993;52(4):678–701. | ||
Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002;94(18):1365–1372. | ||
Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117–1130. | ||
King MC, Marks JH, Mandell JB, New York Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302(5645):643–646. | ||
Vaidyanathan K, Lakhotia S, Ravishankar HM, Tabassum U, Mukherjee G, Somasundaram K. BRCA1 and BRCA2 germline mutation analysis among Indian women from south India: identification of four novel mutations and high-frequency occurrence of 185delAG mutation. J Biosci. 2009;34(3):415–422. | ||
Tai YC, Domchek S, Parmigiani G, Chen S. Breast cancer risk among male BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst. 2007;99(23):1811–1814. | ||
Newman B, Austin MA, Lee M, King MC. Inheritance of human breast cancer: evidence for autosomal dominant transmission in high-risk families. Proc Natl Acad Sci U S A. 1988;85(9):3044–3048. | ||
Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25(11):1329–1333. | ||
BRCA Exchange [database online]. UC Santa Cruz; 2017. Available from: http://www.brcaexchange.org/. Accessed September 9, 2018. | ||
Plon SE, Eccles DM, Easton D, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29(11):1282–1291. | ||
Mehta A. BRCA1 and BRCA2 mutations in ovarian cancer. J Curr Oncol. 2018;1(1):1–4. | ||
Lindor NM, Guidugli L, Wang X, et al. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum Mutat. 2012;33(1):8–21. | ||
Saxena S, Chakraborty A, Kaushal M, et al. Contribution of germline BRCA1 and BRCA2 sequence alterations to breast cancer in Northern India. BMC Med Genet. 2006;7:75. | ||
Daly MB, Pilarski R, Axilbund JE, et al. Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2015. J Natl Compr Canc Netw. 2016;14(2):153–162. | ||
Database of Single Nucleotide Polymorphisms (dbSNP) [homepage on the Internet]. Bethesda (MD): National Center for Biotechnology Information, National Library of Medicine; 2001. Available from: http://www.ncbi.nlm.nih.gov/SNP/. Accessed September 9, 2018. | ||
Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980–D985. | ||
Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32(5):557–563. | ||
Szabo C, Masiello A, Ryan JF, Brody LC. The breast cancer information core: database design, structure, and scope. Hum Mutat. 2000;16(2):123–131. | ||
Christos Kopanos VT, Kouris A, Chapple CE, Aguilera MA, Meyer R, Massouras A [homepage on the Internet]. VarSome: The human genomic variant search engine. BioRxiv; 2018. Available from: https://www.ensembl.org/vep. Accessed September 9, 2018. | ||
Mclaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17(1):122. | ||
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. | ||
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–423. | ||
Suryavanshi M, Kumar D, Panigrahi MK, Chowdhary M, Mehta A. Detection of false positive mutations in BRCA gene by next generation sequencing. Fam Cancer. 2017;16(3):311–317. | ||
Hogervorst FB, Nederlof PM, Gille JJ, et al. Large genomic deletions and duplications in the BRCA1 gene identified by a novel quantitative method. Cancer Res. 2003;63(7):1449–1453. | ||
Machackova E, Foretova L, Lukesova M, et al. Spectrum and characterisation of BRCA1 and BRCA2 deleterious mutations in high-risk Czech patients with breast and/or ovarian cancer. BMC Cancer. 2008; 8:140. | ||
Armaou S, Konstantopoulou I, Anagnostopoulos T, et al. Novel genomic rearrangements in the BRCA1 gene detected in Greek breast/ovarian cancer patients. Eur J Cancer. 2007;43(2):443–453. | ||
Engert S, Wappenschmidt B, Betz B, et al. MLPA screening in the BRCA1 gene from 1,506 German hereditary breast cancer cases: novel deletions, frequent involvement of exon 17, and occurrence in single early-onset cases. Hum Mutat. 2008;29(7):948–958. | ||
Sedghi M, Esfandiari E, Fazel-Najafabadi E, et al. Genomic rearrangement screening of the BRCA1 from seventy Iranian high-risk breast cancer families. J Res Med Sci. 2016;21:95. | ||
Zorrieh Zahra A, Kadkhoda S, Behjati F, et al. Mutation Screening of BRCA Genes in 10 Iranian Males with Breast Cancer. Int J Mol Cell Med. 2016;5(2):114–122. | ||
Rauh-Adelmann C, Lau KM, Sabeti N, Long JP, Mok SC, Ho SM. Altered expression of BRCA1, BRCA2, and a newly identified BRCA2 exon 12 deletion variant in malignant human ovarian, prostate, and breast cancer cell lines. Mol Carcinog. 2000;28(4):236–246. | ||
Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. | ||
Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. | ||
Robson M. Are BRCA1- and BRCA2-associated breast cancers different? Prognosis of BRCA1-associated breast cancer. J Clin Oncol. 2000;18(21 Suppl):113S–118S. | ||
Eisinger F, Charafe-Jauffret E, Jacquemier J, Birnbaum D, Julian-Reynier C, Sobol H. Tamoxifen and breast cancer risk in women harboring a BRCA1 germline mutation: computed efficacy, effectiveness and impact. Int J Oncol. 2001;18(1):5–10. | ||
King MC, Wieand S, Hale K, et al. Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: National Surgical Adjuvant Breast and Bowel Project (NSABP-P1) Breast Cancer Prevention Trial. JAMA. 2001;286(18):2251–2256. | ||
Lakhani SR, van de Vijver MJ, Jacquemier J, et al. The pathology of familial breast cancer: predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J Clin Oncol. 2002;20(9):2310–2318. | ||
Lips EH, Debipersad RD, Scheerman CE, et al. BRCA1-mutated estrogen receptor-positive breast cancer shows BRCAness, suggesting sensitivity to drugs targeting homologous recombination deficiency. Clin Cancer Res. 2017;23(5):1236–1241. | ||
Singh J, Thota N, Singh S, et al. Screening of over 1000 Indian patients with breast and/or ovarian cancer with a multi-gene panel: prevalence of BRCA1/2 and non-BRCA mutations. Breast Cancer Res Treat. 2018;170(1):189–196. | ||
Kwong A, Shin VY, Ho JC, et al. Comprehensive spectrum of BRCA1 and BRCA2 deleterious mutations in breast cancer in Asian countries. J Med Genet. 2016;53(1):15–23. | ||
Rebbeck TR, Friebel TM, Friedman E, et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum Mutat. 2018;39(5):593–620. | ||
Judkins T, Hendrickson BC, Deffenbaugh AM, et al. Application of embryonic lethal or other obvious phenotypes to characterize the clinical significance of genetic variants found in trans with known deleterious mutations. Cancer Res. 2005;65(21):10096–10103. | ||
Maistro S, Teixeira N, Encinas G, et al. Germline mutations in BRCA1 and BRCA2 in epithelial ovarian cancer patients in Brazil. BMC Cancer. 2016;16(1):934. | ||
Valarmathi MT, Sawhney M, Deo SS, Shukla NK, das SN. Novel germline mutations in the BRCA1 and BRCA2 genes in Indian breast and breast-ovarian cancer families. Hum Mutat. 2004;23(2):205. | ||
Shah ND, Shah PS, Panchal YY, et al. Mutation analysis of BRCA1/2 mutations with special reference to polymorphic SNPs in Indian breast cancer patients. Appl Clin Genet. 2018;11:59–67. | ||
Thompson ER, Gorringe KL, Rowley SM, et al. Reevaluation of the BRCA2 truncating allele c.9976A > T (p.Lys3326Ter) in a familial breast cancer context. Sci Rep. 2015;5:14800. | ||
Meeks HD, Song H, Michailidou K, et al. BRCA2 Polymorphic Stop Codon K3326X and the Risk of Breast, Prostate, and Ovarian Cancers. J Natl Cancer Inst. 2016;108(2):djv315. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.