Back to Journals » OncoTargets and Therapy » Volume 13
Genotyping on ctDNA Identifies Shifts in Mutation Spectrum Between Newly Diagnosed and Relapse/Refractory DLBCL
Authors Liu H, Yang C, Zhao X, Le J, Wu G, Wei J, Liang Y, Qian W
Received 3 August 2020
Accepted for publication 5 October 2020
Published 23 October 2020 Volume 2020:13 Pages 10797—10806
DOI https://doi.org/10.2147/OTT.S275334
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Hui Liu,1,2,* Chunmei Yang,2,3,* Xiaoyan Zhao,2,4 Jing Le,5 Gongqiang Wu,6 Juying Wei,2,3 Yun Liang,1 Wenbin Qian1,2
1Department of Hematology, The Second Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang 310009, People’s Republic of China; 2Institute of Hematology, Zhejiang University, Hangzhou, Zhejiang 31003, People’s Republic of China; 3Department of Hematology, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang 310003, People’s Republic of China; 4Department of Hematology, First Affiliated Hospital of Jiaxing University, Jianxing, Zhejiang 314000, People’s Republic of China; 5Department of Hematology, Ningbo Medical Center Lihuili Hospital, Ningbo, Zhejiang 315000, People’s Republic of China; 6Department of Hematology, Dongyang Hospital Affiliated to Wenzhou Medical University, Dongyang People’s Hospital, Dongyang, Zhejiang 322100, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wenbin Qian; Yun Liang
Department of Hematology, The Second Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou 310009, People’s Republic of China
Tel +86-571-89713674
Email [email protected]; [email protected]
Purpose: Diffuse large B cell lymphoma (DLBCL) is an aggressive B-cell malignancy with clinical and molecular heterogeneity whose genetics may have clinical implications for patient stratification and treatment. The circulating tumor DNA (ctDNA) is a novel noninvasive, real-time, and tumor-specific biomarker harboring tumor-derived genetic alterations that are identical to those of tumor cells, thus showing great promise in individualized medicine, including precise diagnosis, prediction of prognosis, response monitoring, and relapse detection for DLBCL.
Patients and Methods: In this study, we applied NGS analysis to tumor biopsies and ctDNA samples from 16 DLBCL subjects. Then, we compared the genomic alterations from 41 newly diagnosed patients and 56 relapsed/refractory (R/R) patients.
Results: Our results show that ctDNA can function as a liquid biopsy for tracking recurrently mutated genes in DLBCL (sensitivity: 87.50%). The mutational profiles of newly diagnosed and R/R DLBCL groups largely overlapped, but the frequencies of some gene mutations differ between the two cohorts. The distribution of mutations also revealed different frequencies in the two cohorts due to different signaling pathways. Genes from apoptosis pathway, immune response and BCR pathway suffered more mutations in R/R patients.
Conclusion: Overall, this study establishes ctDNA as an easily accessible source of tumor DNA for DLBCL genotyping and provides a deeper understanding of the somatic alteration spectrum for both newly diagnosed and R/R DLBCL patients.
Keywords: diffuse large B cell lymphoma, circulating tumor DNA, liquid biopsy, next-generation sequencing, mutation
Introduction
Diffuse large B cell lymphoma (DLBCL) is the most common type of non-Hodgkin’s lymphoma (NHL) and displays remarkable molecular and clinical heterogeneity.1 Most DLBCL can be subdivided into two molecular subtypes according to molecular heterogeneity through gene expression profiling (GEP): germinal center B-cell like (GCB) and activated B-cell like (ABC).2 The molecular classification of DLBCL shows a distinct correlation with the risk stratification that can have an influence on personalized treatment decisions.3
Tissue biopsy is the traditional method used in the detection of molecular features of tumor, which remains as the gold standard for GEP classification or mutation detection, but it is clinically impractical in some cases because of the invasive procedures.4 Tracking drug-resistance mutations requires serial sampling of tumors by re-biopsy, which may not be routinely feasible in clinical practice.5 Circulating tumor DNA (ctDNA) fragments are released from cancer cells into plasma with tumor-derived genetic alterations identical to those of tumor cells.6 There are definite advantages in sampling which makes it possible for the serial monitoring of disease genetics in real time. A method for DLBCL classification based on somatic mutation profiles of ctDNA has been established, and this non-invasive classification method demonstrates a high concordance rate between tumor and plasma.7 Also, several reports have shown that ctDNA can be used to track the tumor clonotypic immunoglobulin gene rearrangement in minimal residual disease monitoring.8,9
Currently, the next-generation sequencing (NGS) technology has emerged as a promising approach to mutation profiling of ctDNA due to its high-throughput, better sensitivity and specificity.5,10 In this study, we showed that ctDNA can function as a liquid biopsy for tracking recurrent mutation in DLBCL patients and reflecting the original genetics of DLBCL. Then, the mutation spectrum of the newly diagnosed and relapsed/refractory (R/R) DLBCL were compared to provide a deeper understanding on the somatic alteration spectrum of DLBCL.
Patients and Methods
Patients
Two clinical cohorts were investigated. A total of 97 patients who were diagnosed with DLBCL at the Second Affiliated Hospital, College of Medicine, Zhejiang University (Hangzhou, China) and each participating institution, respectively, from Aug 2017 to Aug 2019 were enrolled. Cohort 1 included 41 patients with newly diagnosed DLBCL, and Cohort 2 included 56 patients with R/R DLBCL who had received at least two lines of immunochemotherapy. The patients were classified into GCB or non-GCB subtype according to the Han’s algorithm. In other words, antibodies against CD10, bcl-6, and MUM1 were used to classify DLBCL into GCB and non-GCB subtypes. The study was performed with the approval of an institutional review board and written informed consent was obtained from all participants at the time of enrollment, and was conducted in accordance with the Declaration of Helsinki.
DNA Extractions
Peripheral blood samples were collected in EDTA containing tubes and then centrifuged at 820g for 10 min to separate plasma from cells within 1 h from collection. Plasma was further centrifuged at 20000g for 10 min to pellet and remove cells, and stored at −80°C until DNA extraction. QIAamp Circulating Nucleic Acid Kit was used to extract DNA according to the user’s manual. Tumor gDNA was then isolated from fresh tissue biopsies or FFPE diagnostic tissue biopsies using Qiamp DNA FFPE Tissue Kit. The Bioanalyzer (Agilent Technologies) was used to check the size of the DNA extracted from plasma.
Targeted Next-Generation Sequencing
A targeted resequencing gene panel including coding exons and splice sites of 59 genes that are recurrently mutated in DLBCL was used for this study (Shanghai Rightongene Bio-tech Co. Ltd, Shanghai, China). NGS libraries were constructed using the KAPA Library Preparation Kit (Kapa Biosystems). Multiplexed libraries were sequenced using150- or 300-bp paired-end runs on NextSeq sequencers (Illumina).
Statistical Analysis
The sensitivity and specificity of plasma ctDNA genotyping were calculated using tumor gDNA genotyping as the gold standard. The analysis was performed with SPSS Statistics version 22.0. Student’s t-test was used to determine the significance in differences of mutation distribution in newly diagnosed and R/R DLBCL according to clinical characteristics. A two-tailed significance level of 0.05 was considered statistically significant.
Result
ctDNA Genotyping Discloses Somatic Mutations in DLBCL
To provide the proof of principle that ctDNA could function as a liquid biopsy for tracking recurrently mutated genes in DLBCL, ctDNA and tissue biopsy in a discovery cohort of 16 patients were collected and subjected to NGS analysis. The results of plasma ctDNA genotyping and tumor gDNA genotyping (gold standard) were then compared. The genotyping of ctDNA collected at diagnosis identified a total of 72 somatic mutations, whereas the genotyping of the gDNA from the tissue biopsy identified 77 somatic mutations (Figure 1A and B). Nonsynonymous somatic mutations were detected in both ctDNA and tissue biopsy of all 16 patients. Biopsy-confirmed tumor mutations were detected with 87.5% sensitivity in ctDNA samples (Figure 1C).
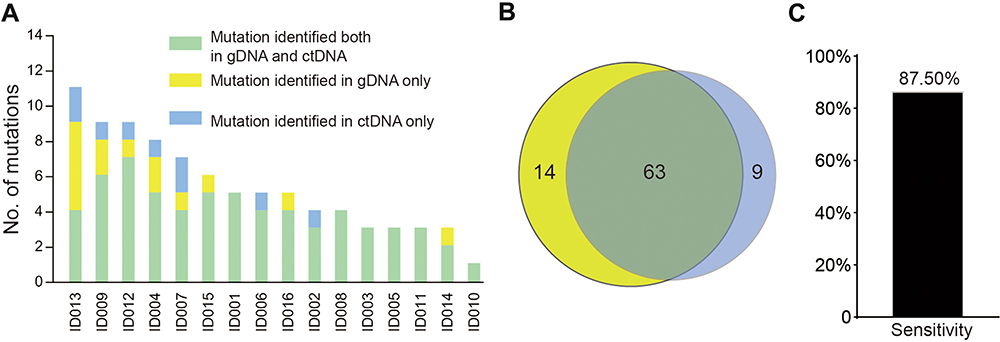 |
Figure 1 ctDNA genotyping discloses somatic mutations in DLBCL. (A). Number of mutations in a given patient detected in plasma ctDNA and/or tumor gDNA. (B). Venn diagram summarizing the overall number of mutations discovered in both plasma ctDNA and tumor gDNA. (C). The corresponding overall sensitivity of plasma ctDNA genotyping in discovering biopsy-confirmed mutations. |
Biopsy-confirmed tumor mutations not discovered in the ctDNA (n=14/77) generally had low representation in the diagnostic biopsy tissue. 87.5% of biopsy-confirmed mutations were identified in ctDNA according to the mutated allele frequency in biopsy. Consistently, using receiver operating characteristic (ROC) analysis, ctDNA genotyping showed the highest sensitivity in discovering mutations that were represented in ≥30% of the alleles of the tumor biopsy, thus demonstrating that plasma ctDNA can accurately mirror the profiles of the most abundant clones found in tumor tissues (Figure 2).
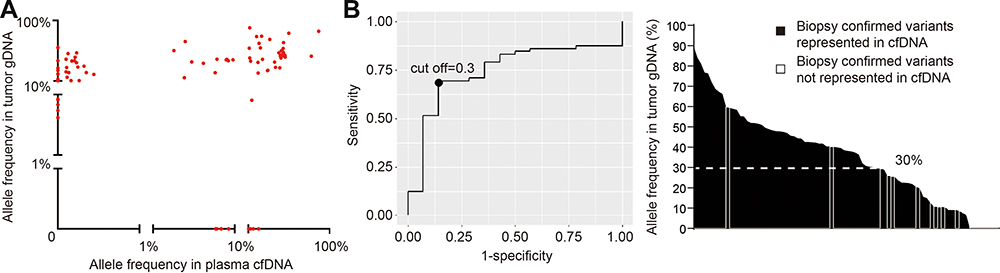 |
Figure 2 Percentage of biopsy-confirmed mutations identified in ctDNA according to mutated allele frequency in biopsy. (A). The mutation abundance in plasma cfDNA vs the mutation abundance in tumor gDNA is comparatively represented in the scatter plot for each variant identified in the discovery cohort. (B). ROC analysis illustrates the performance of plasma cfDNA genotyping in detecting biopsy-confirmed tumor variants according to the variant allele frequency of mutations in tumor gDNA in the discovery cohort. The bar graph shows the allele frequency in tumor gDNA of the variants that were discovered in plasma cfDNA (black bars) or missed in plasma cfDNA (gray bars). The dash line tracks the 30% variant allele frequency threshold. |
Genotyping of Newly Diagnosed DLBCL and Relapsed/Refractory DLBCL
We then used ctDNA to noninvasively characterize the mutational spectrum of newly diagnosed and R/R patients. Forty-one newly diagnosed patients and 56 patients with R/R DLBCL were included in this study. According to the cell-of-origin classification, 37 patients were GCB cases (38%), 58 patients were non-GCB cases (60%), and 2 patients were unclassified cases (2%). The clinicopathologic characteristics are summarized in Table 1.
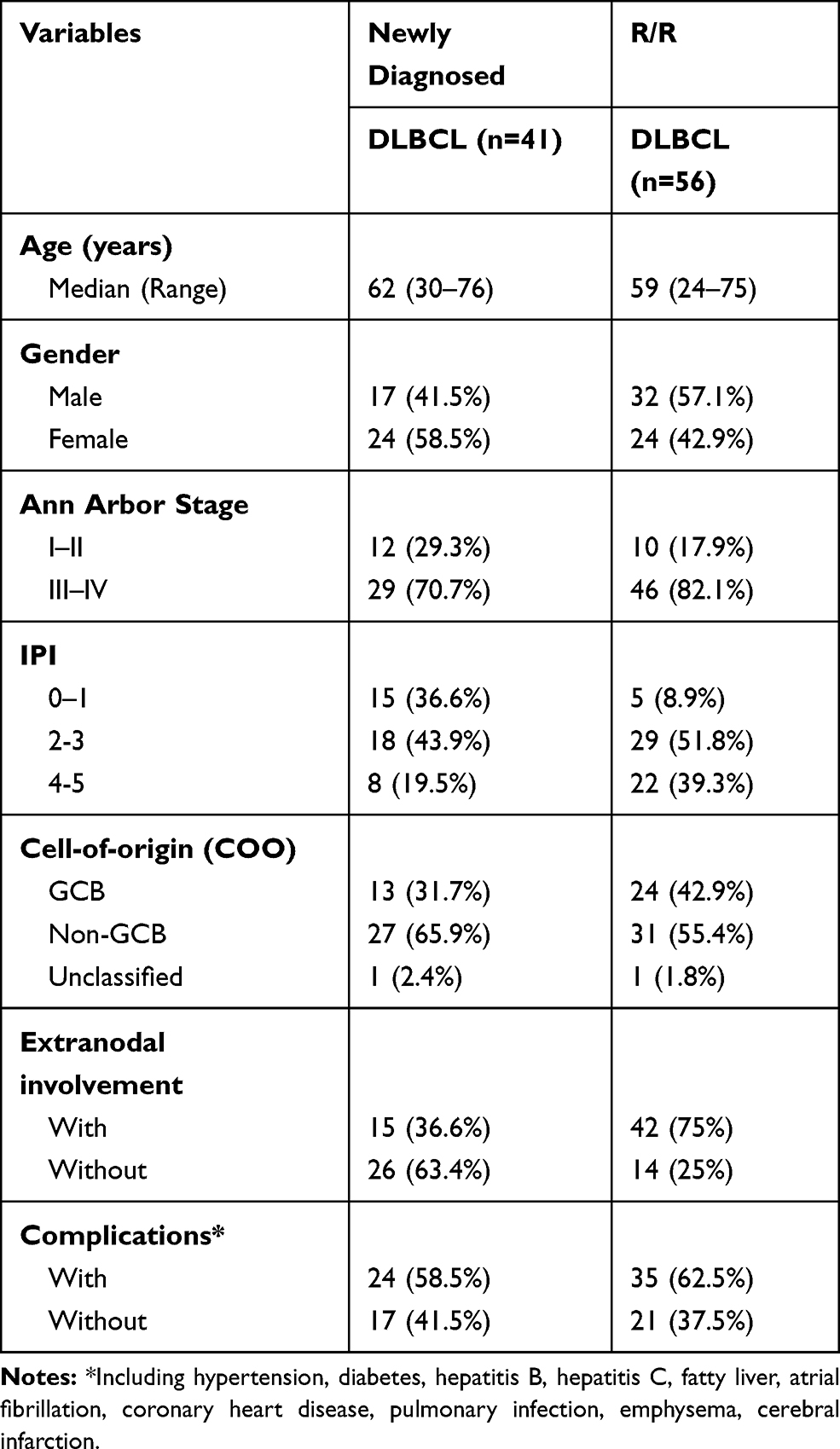 |
Table 1 Characteristics of 41 Newly Diagnosed DLBCL and 56 R/R DLBCL |
We detected nonsynonymous somatic mutations in 75.3% of patients (73/97), with an average of 3.93 mutations per case. Type and distribution of mutations are shown in Figure 3. The mutation frequencies of DLBCL-associated genes in newly diagnosed and R/R DLBCL patients are shown in Figure 4A. Genes that are recurrently affected by nonsynonymous somatic mutations with 10% or higher ratios in newly diagnosed patients include PCLO (31.3%), KMT2D (21.9%), PIM1 (21.9%), LRP1B (21.9%), SPEN (15.6%), MYD88 (12.5%), MEF2B (12.5%) and CREBBP (12.5%). The mutation of genes detected with 10% or higher ratios in patients with R/R disease include KMT2D (29.3%), PCLO (26.8%), TP53 (22.0%), PIM1 (14.6%), CCND3 (14.6%), RET (14.6%), CD58 (12.2%) and NOTCH1 (12.2%). Mutations of PIM1, PCLO and KMT2D were found in 10% or more of the patients who were both newly diagnosed and R/R DLBCL. Mutations of KMT2D (21.9% vs 29.3%), PIM1 (21.9% vs 14.6%) and PCLO (31.3% vs 26.8%) were the same in both newly diagnosed and R/R DLBCL. Mutation of genes mainly detected in newly diagnosed DLBCL were LRP1B (21.9% vs 9.8%), SPEN (15.6% vs 7.3%) and CREBBP (12.5% vs 4.9%). Mutation frequency of TP53 (3.1% vs 22.0%), CCND3 (3.1% vs 14.6%), RET (0% vs 14.6%) and NOTCH1 (3.1% vs 12.2%) increased in patients with R/R DLBCL. A comparison of the mutations between the two cohorts showed that the newly diagnosed and R/R DLBCL groups were on different parts of the somatic alteration spectrum.
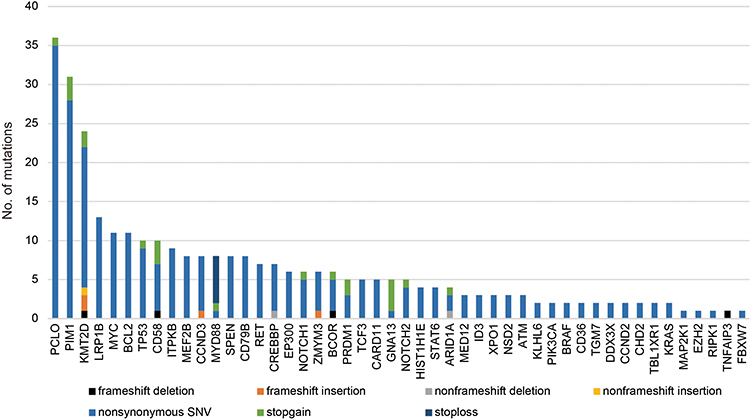 |
Figure 3 Number and type of nonsynonymous somatic mutations identified in each gene. |
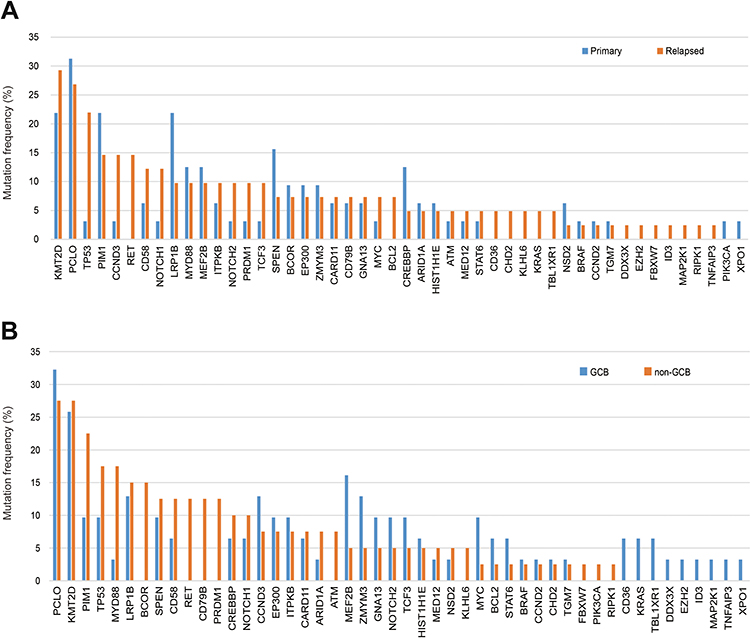 |
Figure 4 Mutation distribution among DLBCL patients. (A). Distribution of mutations identified in primary and relapse/refractory DLBCL samples. (B). Distribution of mutations identified in DLBCL samples according to COO classification. |
We then explored the gene mutation distribution according to the cell-of-origin (COO) molecular classification. The result showed that in genes with mutation frequencies ≥10% in either GCB or non-GCB, the mutation proportion of PCLO, KMT2D, LRP1B, SPEN, CREBBP, NOTCH1, CCND3, EP300 and ITPKB were similar in both. Mutations of MEF2B, ZMYM3, GNA13, NOTCH2, TCF3 and MYC were mainly detected in the GCB type. Mutations of PIM1, TP53, MYD88, BCOR, CD58, RET, CD79B and PRDM1 significantly increased in no-GCB DLBCL patients (Figure 4B).
Distribution of Mutations According to Signaling Pathways
We also identified the pathways that are recurrently mutated in newly diagnosed and R/R DLBCL. We subdivided the genes into eleven specific pathways (Supplementary Table 1). The results showed that epigenetic regulation pathway was the most affected by mutation, followed by other, apoptosis pathway, NF-κB pathway transcription factors, and NOTCH pathway, etc. (Figure 5). According to signaling pathways, the distribution of gene mutations showed different frequencies in newly diagnosed and R/R disease. The frequencies of mutated genes in epigenetic regulation pathway, NF-κB pathway, NOTCH pathway, JAK-STAT pathway, transcription factors, MAP-kinase pathway, PI3K-AKT-mTOR pathway and RNA splicing were approximately equal in both cohorts. Nevertheless, genes in apoptosis pathway, immune response and BCR pathway suffered more mutations in R/R patients than newly diagnosed patients, indicating that these pathways play an important role in treatment resistance.
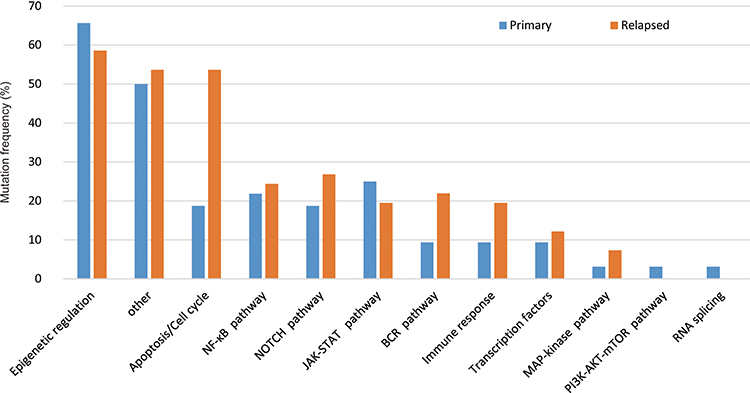 |
Figure 5 Distribution of mutations according to signaling pathways in primary and relapse/refractory DLBCL samples. |
Distribution of Mutations According to Clinical Characteristics
We then explored the mutation distribution in newly diagnosed and R/R DLBCL, according to International Prognostic Index (IPI ≤2 and IPI ≥3), complications, Ann Arbor stage (stage Ⅰ-Ⅱ and stage III-Ⅳ), extranodal involvement and subtype (Figure 6).
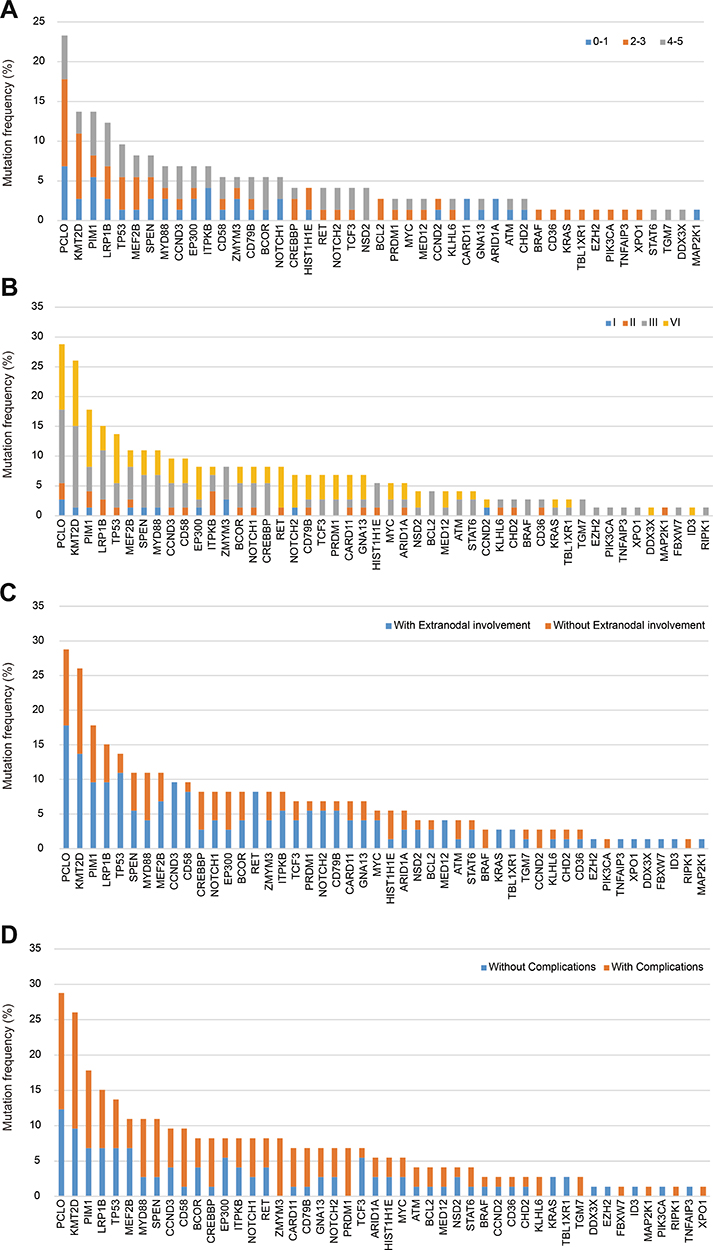 |
Figure 6 Distribution of mutations according to clinical characteristics. (A). Distribution of mutations according to IPI. (B). Distribution of mutations according to complications. (C). Distribution of mutations according to Ann Arbor stage. (D). Distribution of mutations according to extranodal involvement. |
For patients with IPI ≥3, newly diagnosed patients harbored more mutations of BCOR (p=0.023) and GNA13 (p=0.044) than R/R patients. In patients with complications, R/R DLBCL patients suffered more TP53 mutations than newly diagnosed patients (p=0.022). For Ann Arbor stage III and IV, mutations of TP53 were mainly detected in R/R DLBCL (p=0.0017), and mutations of PCLO were mainly detected in newly diagnosed patients (p= 0.0279). For patients without extranodal involvement, mutations of PCLO were mainly detected in newly diagnosed patients (p=0.008). Meanwhile, for patients with extranodal involvement, mutations of TP53, RET and NOTCH2 were mainly detected in R/R DLBCL (p=0.003, p= 0.032, and p= 0.044, respectively). In non-GCB DLBCL, mutations of TP53 were mainly detected in R/R patients (p=0.037).
We also explored the mutation distribution in patients with different clinical characteristics in newly diagnosed and R/R DLBCL groups. In newly diagnosed DLBCL, PCLO mutations were mainly detected in IPI ≤2 patients (p=0.011). MEF2B mutations were mainly detected in patients without complications (p=0.041). Patients with extranodal involvement harbored more mutations of MYD88 than patients without extranodal involvement (p=0.022). In R/R DLBCL, mutations of KMT2D (p=0.00009), TP53 (p=0.0017) were mainly detected in stage III-IV patients than in stage I-II patients. Mutations of PCLO (p=0.015), CCND3 (p=0.017), RET (p=0.031) were mainly detected in patients with extranodal involvement.
Discussion
ctDNA can be used as a reliable source of tumor DNA for the identification of cancer mutations, clonal evolution and genetic mechanisms of resistance. In DLBCL, malignant cell VDJ gene sequences have been demonstrated to be detected in the serum of the patients, which suggests that ctDNA, a specific molecular biomarker, can function as a useful test for disease surveillance.8,9,11 Rossi D et al5, investigated ctDNA genotyping of untreated-DLBCL and results have proven that ctDNA genotyping is as accurate as genotyping of the diagnostic biopsy through detection of no-immunoglobulin somatic mutations and can be used as a non-invasive tool to monitor the emergence of treatment-resistant clones. Another study documented that the amount of ctDNA at diagnosis has an independent predictive value for the outcome of DLBCL patients, and ctDNA genotyping can not only classify transcriptionally defined DLBCL subtype but also facilitate non-invasive detection of minimal residual disease as well as emergent resistance.7 These studies suggest that ctDNA analysis reveals molecular-biological factors that affect clinical outcomes and provides the foundation for individualized therapy. However, there are relatively few studies investigating the ctDNA genotyping of DLBCL patients with relapsed/refractory disease.
In the current study, we analyzed ctDNA genotyping from a newly diagnosed DLBCL cohort and an R/R DLBCL cohort. We observed that recurrently mutated genes in the newly diagnosed cohort include PCLO, KMT2D, PIM1, LRP1B, SPEN, MYD88, MEF2B and CREBBP, etc. Many of these affected genes had been previously identified as highly mutated in DLBCL.3,5,12,13 ctDNA analysis also showed that mutations of PIK3CA and XPO1 were found in the newly diagnosed, but not in R/R, patients. On the other hand, mutations of RET, BCL2, CD36, CHD2, KLHL6, KRAS and TBL1XR1 were only detected in the R/R cohort. In addition, mutation frequencies of TP53 (3.1% vs 22.0%), CCND3 (3.1% vs 14.6%), RET (0% vs 14.6%) and NOTCH1 (3.1% vs 12.2%) were significantly increased in the patients with R/R DLBCL. These data suggest a divergence of ctDNA genotyping between the newly diagnosed and R/R patients.
TP53 is implicated functionally as a tumor-suppressor gene in DLBCL.13 In a large cohort of de novo DLBCL patients treated with R-CHOP, TP53 mutations have been demonstrated to be correlated with disease progression and poor survival.14 More importantly, TP53 is a strong predictor of clinical outcome and is independent of IPI, COO or double-hit status in DLBCL patients. Therefore, mutant TP53 can be used as an important biomarker to better stratify DLBCL patients. It should be noted that in the current study, mutations of TP53 were mainly detected in R/R DLBCL and were in correlation with adverse prognostic factors such as patients with complications, extranodal involvement, Ann Arbor stage III-Ⅳ and non-GCB type, which also indicate that TP53 is significant in prognostic evaluation.15
The mutations in CCND3 in hot spots have been shown to increase protein stability, and CCND3 was also found to have clonal enrichment between diagnosis and relapse. It was suggested that CCND3 plays an important role in therapeutic resistance owing to its significantly greater prevalence in R/R DLBCL.16 Schmitz et al14 have identified four genetic subtypes in DLBCL, namely MCD, BN2, EZB and N1. N1 subtype was identified based on NOTCH1 mutations that were primarily in ABC cases (95%). Given its poor response to R-CHOP and its prominent T-cell gene-expression signature, immune-checkpoint inhibitors could be studied in the subtype, which may improve the clinical outcome.
RET mutations are seen in medullary thyroid cancer. Aberrant activation of RET mainly resulted from fusion with partner genes, mutation and overexpression. However, mutations of RET have not been reported in DLBCL. In this study, RET mutations increased in R/R DLBCL. This indicates that RET mutations may be related to disease progression and resistance to therapy, which will require further investigations.
PCLO mutations are observed very commonly in DLBCL, encoding a protein that functions as part of the presynaptic cytoskeletal matrix, which was thought to be involved in regulating neurotransmitter release. A role for PCLO in calcium sensing has also been suggested, but a role in lymphoma has not been reported before. The local rate of mutation at the PCLO locus is unusually high, giving rise to passenger mutations of no functional consequence in DLBCL. Additional work is clearly needed to resolve the role, if any, of PCLO mutations in DLBCL and other cancers.17 In this study, we showed that mutations of PCLO were dominant in newly diagnosed patients, correlating with earlier stage (stage I and II), and were mainly detected in patients without extranodal involvement.
It has been reported that MYD88 mutations occur frequently in variable B cell lymphoma, with L265P being the most prevalent variant. MYD88 mutations frequently coincided with mutations of CD79B. Schmitz et al14 classified cases with MYD88 L265P and CD79B mutations as MCD subtypes that were dominantly non-GCB. In patients with extranodal involvement, mutations of MYD88 were commonly detected in central nervous system lymphoma and testicular lymphoma. It has been shown that patients with MYD88/CD79B dual mutations were significantly more responsive to ibrutinib, a BTK inhibitor. In this study, MYD88 mutations were mainly detected in newly diagnosed patients with complications, who may benefit from the BTK inhibitor.
The lysine-specific histone methyltransferase KMT2D has emerged as one of the most frequently mutated genes in DLBCL. Integrative genomic analyses indicate that KMT2D affects H3K4 methylation and expression of a specific set of genes, including those in the CD40, JAK-STAT, Toll-like receptor, and B cell receptor pathways. This suggests that KMT2D acts as a tumor-suppressor gene whose early loss facilitates lymphomagenesis by remodeling the epigenetic landscape of the cancer precursor cells.18 Our data show that in R/R DLBCL, mutations of KMT2D were mainly detected in stage ш-Ⅳ patients, implying that it may be associated with disease progression.
MEF2B mutations were predominantly detected in DLBCL cases without MYC translocation (18%) and were rare or absent in other subgroups.19 The expression of MEF2B protein is important for the growth of GCB-DLBCL cells and is an essential component of the BCL6 gene transcriptional complex for the regulation of DLBCL growth.20
In this study, we performed NGS in patients with both ctDNA and tissue biopsy. Mutations were detected in ctDNA with 87.5% sensitivity in comparison with gDNA derived from tumor biopsy, which was consistent with previous reports.5 Mutations presented in ≥30% of the alleles of the tumor biopsy can be detected in ctDNA with a much higher sensitivity. However, some mutations of genes which are present in tumor biopsy could not be detected in ctDNA, which may be due to the low allele frequency of mutation.5 Because of the genomic heterogeneity within tumors, tissue biopsy might not be representative of the entire tumor genetics. Indeed, different areas of the same tumor may show different genetic profiles while biopsies of different parts of the tumor may miss mutations occurring in subclones residing in anatomically distant sites.21,22 Previous studies suggest that small subclones have a clinical impact on treatment resistance and ctDNA can provide a powerful way to mirror both the clonal and subclonal composition of the tumor.23 Here, our results show that extra mutations can be detected in ctDNA but are absent in tissue biopsy. This means that ctDNA is also able to detect potentially relevant mutations that are not identified within the original tissue biopsy, which suggests that ctDNA could act as a liquid biopsy to provide complementary information in DLBCL.
Overall, our findings highlight ctDNA as an alternative to mirror tissue biopsy for the detection of mutated genes in DLBCL and the shifts in mutational composition between newly diagnosed and R/R patients. However, there were some limitations in this study. The number of patients was relatively small and the primary efficacy endpoint was not achieved. Thus, a study with a larger sample size and further follow-up is necessary.
Data Sharing Statement
The datasets and material used and/or analyzed during the current study are available from the corresponding author upon request.
Ethics Approval
This experiment was approved by an independent ethics committee of the Second Affiliated Hospital, College of Medicine, Zhejiang University.
Acknowledgments
We appreciate the patients for their peripheral blood samples.
Author Contributions
All authors made substantial contributions to conception and design, acquisition, analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed on the journal to which the article will be submitted; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Co-first author: Hui Liu and Chunmei Yang.
Co-correspondence: Yun Liang and Wenbin Qian.
Funding
The research was supported by National Natural Science Foundation of China (No. 81830006, and 81670178), Zhejiang Provincial Natural Science Foundation (No.LY18H160008), and the Science Technology Department of Zhejiang Province (No. LGF19H080001).
Disclosure
The authors declare that they have no competing interests.
References
1. Menon MP, Pittaluga S, Jaffe ES. The histological and biological spectrum of diffuse large B-cell lymphoma in the World Health Organization classification. Cancer J. 2012;18(5):411–420. doi:10.1097/PPO.0b013e31826aee97
2. Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. doi:10.1038/35000501
3. Sun P, Chen C, Xia Y, et al. Mutation Profiling of Malignant Lymphoma by Next-Generation Sequencing of Circulating Cell-Free DNA. J Cancer. 2019;10(2):323–331. doi:10.7150/jca.27615
4. Lenz G, Wright GW, Emre NC, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105(36):13520–13525. doi:10.1073/pnas.0804295105
5. Rossi D, Diop F, Spaccarotella E, et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood. 2017;129(14):1947–1957. doi:10.1182/blood-2016-05-719641
6. Bardelli A, Pantel K. Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell. 2017;31(2):172–179. doi:10.1016/j.ccell.2017.01.002
7. Scherer F, Kurtz DM, Newman AM, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8(364):364ra. doi:10.1126/scitranslmed.aai8545
8. Roschewski M, Dunleavy K, Pittaluga S, et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: a correlative biomarker study. Lancet Oncol. 2015;16(5):541–549. doi:10.1016/S1470-2045(15)70106-3
9. Roschewski M, Staudt LM, Wilson WH. Dynamic monitoring of circulating tumor DNA in non-Hodgkin lymphoma. Blood. 2016;127(25):3127–3132. doi:10.1182/blood-2016-03-635219
10. Dubois S, Jardin F. The role of next-generation sequencing in understanding the genomic basis of diffuse large B cell lymphoma and advancing targeted therapies. Expert Review of Hematology. 2016;9(3):255–269. doi:10.1586/17474086.2016.1130616
11. Faham M, Zheng J, Moorhead M, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120(26):5173–5180. doi:10.1182/blood-2012-07-444042
12. Kurtz DM, Green MR, Bratman SV, et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood. 2015;125(24):3679–3687. doi:10.1182/blood-2015-03-635169
13. Kurtz DM, Scherer F, Jin MC, et al. Circulating Tumor DNA Measurements As Early Outcome Predictors in Diffuse Large B-Cell Lymphoma. J Clin Oncol. 2018;36(28):2845–2853. doi:10.1200/JCO.2018.78.5246
14. Schmitz R, Wright GW, Huang DW, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med. 2018;378(15):1396–1407. doi:10.1056/NEJMoa1801445
15. Farinha P, Ennishi D, Mottok A, et al. TP53 Expression Correlates with TP53 Mutations and Is an Independent Predictor of Clinical Outcome in Patients with DLBCL Treated with R-CHOP. Blood. 2019;134(Supplement_1):3964. doi:10.1182/blood-2019-121943
16. Wu F-T, Lu L, Xu W, Li J-Y. Circulating tumor DNA: clinical roles in diffuse large B cell lymphoma. Ann Hematol. 2019;98(2):255–269. doi:10.1007/s00277-018-3529-9
17. Lohr JG, Stojanov P, Lawrence MS, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109(10):3879–3884. doi:10.1073/pnas.1121343109
18. Ortega-Molina A, Boss IW, Canela A, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21(10):1199–1208. doi:10.1038/nm.3943
19. Momose S, Weißbach S, Pischimarov J, et al. The diagnostic gray zone between Burkitt lymphoma and diffuse large B-cell lymphoma is also a gray zone of the mutational spectrum. Leukemia. 2015;29(8):1789–1791. doi:10.1038/leu.2015.34
20. El Jamal SM, Grada Z, El Dinali MH, et al. MEF2B is a member of the BCL6 gene transcriptional complex and induces its expression in diffuse large B-cell lymphoma of the germinal center B-cell-like type. Lab Invest. 2019;99(4):539–550. doi:10.1038/s41374-018-0152-2
21. Rasi S, Monti S, Zanni M, et al. Liquid Biopsy As a Tool for Monitoring the Genotype of Diffuse Large B-Cell Lymphoma. Blood. 2015;126(23):127. doi:10.1182/blood.V126.23.127.127
22. Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi:10.1056/NEJMoa1113205
23. Spina V, Rossi D. Liquid biopsy in tissue-born lymphomas. Swiss Med Wkly. 2019;149:w14709.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.